
Abstract
Split-hand–split-foot malformation (SHFM) is a rare condition that occurs in 1 in 8500–25,000 newborns and accounts for 15% of all limb reduction defects. SHFM is heterogeneous and can be isolated, associated with other malformations, or syndromic. The mode of inheritance is mostly autosomal dominant with incomplete penetrance, but can be X-linked or autosomal recessive. Seven loci are currently known: SHFM1 at 7q21.2q22.1 (DLX5 gene), SHFM2 at Xq26, SHFM3 at 10q24q25, SHFM4 at 3q27 (TP63 gene), SHFM5 at 2q31 and SHFM6 as a result of variants in WNT10B (chromosome 12q13). Duplications at 17p13.3 are seen in SHFM when isolated or associated with long bone deficiency. Tandem genomic duplications at chromosome 10q24 involving at least the DACTYLIN gene are associated with SHFM3. No point variant in any of the genes residing within the region has been identified so far, but duplication of exon 1 of the BTRC gene may explain the phenotype, with likely complex alterations of gene regulation mechanisms that would impair limb morphogenesis. We report on 32 new index cases identified by array-CGH and/or by qPCR, including some prenatal ones, leading to termination for the most severe. Twenty-two cases were presenting with SHFM and 7 with monodactyly only. Three had an overlapping phenotype. Additional findings were identified in 5 (renal dysplasia, cutis aplasia, hypogonadism and agenesis of corpus callosum with hydrocephalus). We present their clinical and radiological findings and review the literature on this rearrangement that seems to be one of the most frequent cause of SHFM.
Similar content being viewed by others

Introduction
Ectrodactyly or split-hand/split-foot malformation (SHFM) is a rare condition that occurs in 1 in 8500–25,000 newborns and accounts for around 15% of all limb reduction defects. It is a limb malformation affecting the central rays of the autopod involving syndactyly, median clefts of the hands and feet, aplasia and/or hypoplasia of phalanges, metacarpals and metatarsals [1]. SHFM is extremely variable in its phenotypic expression between families, within families and even between limbs of a single patient, ranging from syndactyly and oligodactyly to the most severe expression-monodactyly with only a single phalanx [2].
Monodactyly is a rare malformation of the extremities, with agenesis of the four preaxial rays of the hand and foot. It has been considered as part of the SHFM spectrum since the first publication in 1916 [3]. There are two main anatomic varieties of SHFM. Type 1 split-hand/split-foot presents as a ‘lobster claw’, with the absence of the central rays. This is generally associated with syndactyly between the digits on each side of the cleft. Type 2 split-hand/split-foot is associated with a preaxial ray deficiency and therefore there is no cleft. Usually, only the fifth digit is present, and thus the term monodactyly is used [4].
SHFM and monodactyly are clinically heterogeneous and can be either isolated, associated with other malformations or part of syndromic entities, such as Ectrodactyly-Ectodermal dysplasia-Cleft (EEC) [5], Cornelia-De Lange [6,7,8] and Smith–Lemli–Opitz syndromes [9].
Genetics of SHFM is complex, with 7 known loci and 3 causative genes (SHFM1 at 7q21.3 (DYNC11 and DLX5), SHFM2 at Xq26, SHFM3 at 10q24.3, SHFM4 (TP63) at 3q27 [10, 11], SHFM5 at 2q31, SHFM6 at 12q13.11-q13 (WNT10B), SHFM/SHFLD at 17p13.3). Tandem 10q24 (SHFM3) duplications created by an unequal recombination between sequences of the centromeric region and sequences of the DACTYLIN gene region were associated with ectrodactyly in 2003 by de Mollerat et al., who pointed to the extreme clinical variability of this phenotype [12]. Of note, in a four-generation Chinese family carrying the duplication of the SHFM3 region, one of the affected individuals presented with fibular monodactyly, while others showed classical SHFM [13]. Deletions and translocations at the 7q21 region have been associated with syndromic SHFM [14,15,16]. Large chromosomal deletions at 2q31 region (SHFM5) including the HOXD cluster have also been described in association with split-hand split-foot or monodactyly [17,18,19].
SHFM3 in humans has been located at 10q24 and the naturally occurring Dactylaplasia mouse is the animal model for this condition [20, 21]. The two existing Dac alleles result from MusD-insertions upstream of or within Dactylin (Fbxw4). 325–570 kb tandem genomic duplications at chromosome 10q24 involving at least the DACTYLIN (FBXW4) gene have been found in SHFM3 patients [22,23,24]. No causal sequence alterations have been found, although two interesting genes (FGF8 and FBXW4) reside within the critical locus [24]. Duplications always seem to include BTRC and POLL, whereas FBXW4 can be only partially included. A poster on 5 cases presenting with 10q24 duplication had shown that part of BTRC and the whole of the POLL gene up-regulation alone were insufficient to cause SHFM3 as patients carrying duplications did not present with any limb defect [25]. In 2015, Li et al. identified the minimal critical region responsible for the SHFM3 phenotype in a series of 42 patients gathered from DECIPHER, previously published cases (PubMed) and a family with 6 affected individuals. Interestingly, they concluded that duplication of sequence in exon 1 of BTRC could be sufficient to the development of the SHFM3 phenotype and suggested that this may be via cis-acting or trans-acting effects on genes or regulatory sequences involved in the limb development pathway [26]. It is also interesting that patients harbouring wider duplications might also present with other limb malformations such as bilateral femoral hypoplasia but no SHFM [27]. No point variant in any of the genes residing within the duplicated region has been reported so far, and it is still not clear how the duplication leads to the SHFM3 phenotype. Indeed, complex alterations of gene regulation mechanisms that would impair limb morphogenesis are likely. We report on 32 new index cases of 10q24 duplication (22 SHFM including 3 with preaxial polydactyly, 7 monodactylies and 3 patients presenting overlapping phenotypes) and describe the first prenatal case of SHFM associated with this chromosomal rearrangement.
Case reports
Patient’s phenotypes
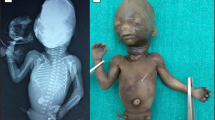
All patients were examined by experienced clinical geneticists. Their phenotypes are summarised in Table 1 and photographs of their hands and feet, when available are presented in Fig. 1. X-rays of their hands and feet, when available, are presented in Fig. 2. For some patients, variants in the coding part of TP63 had been excluded by direct sequencing. Written consent was obtained from all patients and/or their legal guardians for publication of the images, as well as clinical and radiological data.

Photographs of patients’ hands and feet. 1 Case 1. Prenatal ultrasound revealing bilateral split hands. Left hand. Note 1–2 and 3–4 syndactyly as well as excessive tissue between 2 and 3. Right hand. Note 1–2 and 3–4 syndactyly. Bilateral split feet. 2 Case 2. Note bilateral monodactyly of hands and feet. 3 Case 3. Note bilateral monodactyly of hands and feet. 4 Case 4. Note bilateral monodactyly of hands and feet. 5 Case 5. Note ectrodactyly of 4 limbs. 6 Case 6. Note ectrodactyly of 4 limbs. 7 Case 7. Note ectrodactyly of 4 limbs. 8 Case 8. Note monodactyly of hands. 9 Case 9. Note split hands, right split-foot and left 2–3–4 toe syndactyly. 10 Case 10. Note normal hands and split feet. 11 Case 11. Note ectrodactyly/syndactyly of 4 limbs. 12 Case 12. Note ectrodactyly of 4 limbs. 18 Case 18. Note ectrodactyly of 4 limbs. 19 Case 19. Note ectrodactyly of 4 limbs. 20 Case 20. Note ectrodactyly of 3 limbs. The right is very mildly affected with a triphalangeal thumb and a radial deviation of the index. 21 Case 21. Note ectrodactyly of 4 limbs. 22 Case 22. Note ectrodactyly of feet and one hand and preaxial polydactyly on other hand. 23 Case 23. Note ectrodactyly of feet. 24 Case 24. Note ectrodactyly and monodactyly of hands and feet. 25 Case 25. Note ectrodactyly of 4 limbs. 26 Case 26. Note ectrodactyly of 4 limbs following surgery for a right hand preaxial polydactyly

X-rays of patients’ hands and feet. 1 Case 1. Note skin syndactyly of hands and split feet. X-rays performed at 12 months. 2 Case 2. Note monodactyly of hands and feet. X-rays performed at 1 month. 5 Case 5. Note split hands. X-rays performed at 32 years. 6 Case 6. Note ectrodactyly of 4 limbs. X-rays performed at 5 months. 8 Case 8. Note monodactyly of 4 limbs. X-rays performed at 25 years. 11 Case 11. Note skin syndactyly and absent distal phalanges. X-rays performed at 18 months. 12 Case 12. Note ectrodactyly of 4 limbs. X-rays performed at 5 years. 20 Case 20. Note ectrodactyly of the left hand and both feet. Note camptodactyly of the right index. X-rays performed at 30 months. 21 Case 21. Note ectrodactyly of 4 limbs. X-rays performed at 24 weeks’ gestation. 22 Case 22. Note ectrodactyly of feet and preaxial polydactyly of hand. X-rays performed at age 3 months. 23 Case 23. Note ectrodactyly of feet. X-rays performed at age 12 months. 25 Case 25. Note ectrodactyly of feet and monodactyly of hands. X-rays performed at age 39 years. 26 Case 26. Note ectrodactyly of feet and polydactyly/ectrodactyly and distal amputations of hands. X-rays performed at age 3 years. 28 Case 28. Note monodactyly of hands and ectrodactyly of feet. X-rays performed at age 18 years
Patient’s material
Peripheral blood cell DNA from patients and their parents when available was obtained after informed consent following standard protocols.
Array CGH analysis
Different types of array-CGH were performed as patients were tested in different genetics department in France, Lebanon, Switzerland, UK and Poland.
Detection of gene copy number was performed by array-CGH following the manufacturer’s recommendations (AgilentTM, Agilent Technologies, Santa Clara, CA) using either 244K, 180K, 60K or 44K oligo probes approximately spaced at 35–40 kb intervals across the genome (Human Genome 18 or 19). Commercial (PromegaTM) or non-commercial female or male genomic DNA were used as reference in hybridizations. Array-CGH results were extracted with Feature extraction software and analysed with the DNA-analytics software by applying an ADM2 segmentation algorithm to identify chromosome aberrations. Copy-number gains and losses were determined using a threshold of 0.3 and −0.3, respectively. Aberrant signals obtained with three or more neighbouring oligonucleotide probes were considered indicative of genomic aberrations. Data (array-CGH and phenotype) was submitted in Decipher or in the ClinVar database for all cases.
High-resolution array-CGH was performed on genomic DNA and analysed versus reference DNA (Kreatech, Amsterdam, The Netherlands). NimbleGen 135k or 1.4 M CGH microarray was used with a calculated functional resolution of 0.2 Mb (95% confidence limits, Human Genome 19). The DNA samples were labelled (test with Cy3 and reference with Cy5) and co-hybridised to the microarray in accordance with the manufacturer’s instructions (NimbleGen Arrays User’s Guide: CGH and CGH/LOH Arrays v9.1, Roche NimbleGen, Madison, WI, USA). The microarray was washed and then scanned on an Axon GenePix 4400A Scanner using GenePix Pro 7 software (Molecular Devices, Sunnyvale, CA, USA). Raw data was normalized, LOESS correction applied and the data ratios calculated using DEVA v1.01 Software (Roche NimbleGen). The normalized data was processed using Infoquant Fusion v6.0 software (Infoquant, London, UK) with analysis call settings of 3 consecutive probes ± 0.4 Cy3/Cy5 ratio.
Real-time quantitative PCR (qPCR) analysis
To evaluate 10q24 duplication by qPCR, a set of different primer pairs located within the SHFM3 locus was designed (including exon 1 and exon 15 of BTRC gene) using the Primer 3 v0.4.0 software (http://primer3.ut.ee) (Primer sequences are available upon request). qPCR was performed in a total volume of 25 μl containing 12.5 μl of SYBR Green PCR Master Mix (Applied Biosystems), 10 ng of genomic DNA and 0.25 μl of primers (100 mM each). Samples were run on the Applied Biosystems 7900HT or ViiA7 Real-Time PCR System in triplicate in separate reactions to permit the quantification of the target sequences normalized to the RPPH1 and SALL4 genes or ALB and F8 genes. PCR conditions were as follows: 2 min activation step at 50 °C, 10 min initial denaturation step at 95 °C followed by 40 cycles at 95 °C for 15 s and 60 °C for 1 min. By use of calibrator samples of normal control DNA, the gene copy number was estimated on the basis of the comparative ΔΔCt method. The experiments were repeated twice. A cut-off of relative gene copy numbers of 1.3 was used for duplication.
Data for patients who only had qPCR analysis was entered in the LOVD database.
MLPA
A custom probe: 10_103216238 was used for the MLPA [28].
Results
Results are summarised in Table 1 and Fig. 3 for all patients [29].

Array CGH 10q24 duplications are displayed for all cases tested. All positions have been converted to the build hg38 for this figure. The TAD region shown has been detected in human embryonic stem cells and overlaps with the minimal duplicated region across all cases. Inversion of POLL, DPCD, FBW4 and FGF8 which occurred during vertebrate evolution is shown. All coding and non-coding Ensembl v87 genes overlapping the region chr10:101121147–102503988 are shown, including SUFU and FGF8. The phastCons conservation score for 29 mammals aligned to the human genome was downloaded from UCSC. For every 1000 base window, a mean conservation score between 0 and 1 was calculated indicating the probability of negative selection
Twenty-two index patients presented with SHFM (including 3 with preaxial polydactyly), 7 with monodactylies affecting 3 or all 4 limbs and 3 patients with a mixed phenotype comprising ectro and monodactyly. Seventeen cases were familial and 15 occurred de novo. Parental samples were tested for all de novo cases except cases 28 and 32, by qPCR and none of them carried the duplication. Segregation studies for familial cases were only performed for cases 22, 25, 29 and 31 and confirmed that all affected individuals were carrying the duplication, and that unaffected relatives did not. There was a very wide range of clinical variability, even between individuals from the same family. When possible (n = 35), sex was recorded for singletons, index and familial cases. The male to female ratio was 15/20 in this cohort. Five cases presented with additional findings (2 cutis aplasia, 1 renal hypoplasia, 1 hypogonadism and 1 agenesis of corpus callosum with hydrocephalus). One case was identified during the pregnancy following detection of ectrodactyly involving all 4 limbs on scans, and the array-CGH had been performed on foetal DNA extracted from the amniotic fluid.
Ten patients were screened with Agilent 180K array-CGH, nine patients with 44K array-CGH, two with Agilent 244K array-CGH, one with Agilent 60K array-CGH [hg 18 or hg 19] as well as 2 with Nimblegen 135K and 1 with Nimblegen 1.4 M array-CGH [hg 19]. Breakpoints for patients 1–7, 11, 13, 15, 16, 19–32 are summarised in Table 1 and Fig. 3 shows the sizes of the duplication for cases 1–7, 11, 13, 15, 16 and 19–32. The minimum size duplication was 291 kb and the maximum size duplication was 597 kb. All cases tested by array-CGH had duplication of at least BTRC and POLL genes. Seven cases had duplication of LBX1 gene as well and 12 patients had duplications comprising the FBXW4 gene (although partially for cases 24 and 27). Twenty-six patients had a qPCR that either confirmed the array-CGH result (19 cases) or that was done as a first line diagnostic test (7 cases). One patient had an MLPA that confirmed the array-CGH result.
Discussion
SHFM3 is caused by duplication at the 10q24 locus. DACTYLIN (FBXW4), LBX1, SUFU, BTRC and FGF8 genes are located in this region [12, 24, 30]. According to previously reported series, SHFM3 seems to be one of the most common causes of SHFM when an underlying cytogenetic or molecular mechanism has been identified (12% against 13% with 17p13.3 duplication [30]; 20% [2]). SHFM3 is inherited in an autosomal dominant manner and to our knowledge, the phenotype is fully penetrant but there is a very wide range of clinical variability, even between individuals from the same family [31]. In case 22, the father only presented with cutaneous syndactyly of the feet (bilateral 1st and 2nd toes and 3rd and 4th toes). Hands were normal. Radiographs of the hands and feet had shown no skeletal involvement. This patient was diagnosed because his daughter presented with typical SHFM. In case 10, there was ectrodactyly of the lower limbs only with normal hands. Preaxial polydactyly is a relatively common finding in SHFM3 [2, 30, 32], but infrequent in other SHFM types. The preaxial involvement can range from preaxial and central ray aplasia to milder preaxial involvement (i.e. preaxial polydactyly), reminiscent of Holt-Oram or Okihiro syndromes which can also present with either absent thumbs or triphalangeal thumbs. In 2012, Klopocki et al. reported a series of 10 patients with 10q24 duplication [2]. Only 1 patient presented with monodactyly and most cases manifested ectrodactyly, ranging from 1 to all 4 limbs. One case presented additionally with learning difficulties and oligodontia, one with intellectual disability and 2 with preaxial polydactyly. Dimitrov et al. in 2010 suggested that the 10q24.31q24.32 duplication cause a syndromic form of SHFM [24]. They reported associated common facial dysmorphic features in their 6 patients, however we did not observe similar findings in our patients and there has been no subsequent report showing identical appearance. It seems therefore that this is mostly an isolated form of SHFM as very few cases present with additional findings and it is not certain that these additional features are linked with the limb defects (5/32 in our series, 2/10 in Klopocki et al. [2]).
The naturally occurring Dactylaplasia (Dac) mouse is an animal model for human SHFM3. It has absent central digits, hypoplasia or aplasia of metacarpal/metatarsal bones and syndactyly. The phenotype results from disruption of the Dactylin gene and the defect is inherited in an autosomal semi-dominant manner, where heterozygotes present with classical SHFM whereas homozygotes show monodactyly. In addition, the variable phenotype depends also on homozygosity for a recessive mdac modifier allele that appears only in certain inbred strains [33]. The reduced expression of the Dactylin gene is thought to play a central role in the pathogenesis of SHFM3 and this was supported by a possible underlying gene dosage mechanism [34]. However, the relationship between gene dosage and phenotype is complex and one of the hypotheses is based on diminished phenotype for both increased and decreased gene dosage, indicating either multi-subunit complexes with a single component that has a tight stoichiometry (gene balance hypothesis) or specific regulatory imbalances as a consequence of under- (insufficient amount hypothesis) and/or over-expression [1, 35, 36]. The loss of digits in Dac mutants involves increased cell death in a specific portion of the apical ectodermal ridge (AER) and SHFM3 is due to a disruption of the AER which is characterized by regulatory complexity [37]. It has also been suggested that epigenetics could be involved in the pathogenesis of SHFM3 [38]. Recently, Li et al. have shown that duplication of the first exon of the BTRC gene could be responsible for the SHFM3 phenotype and that the highest density of conserved non-coding elements is found in the BTRC gene [26]. However, further functional analyses will be needed to confirm this finding. BTRC had always been thought to be of particular interest as it functions as a ubiquitination factor of proteins involved in several signalling transduction pathways involved in limb development [39], but it is possible that conserved non-coding elements within the BTRC gene point to another example of disturbed cis-regulation such as topological associated domains (TADs) [29]. There does not seem to be a correlation between severe clinical involvement and larger duplications. Indeed, wider duplications involving the FGF8 gene are not associated with the SHFM phenotype, despite comprising the first exon of the BTRC gene [27]. FGF8 induces and regulates the limb bud patterning via AER signalling and Fgf8 inactivation in mouse models in the early limb ectoderm causes hypoplasia/aplasia of specific distal skeletal elements [40].
In conclusion, SHFM3 is one of the most common types of SHFM known to date. We report on a cohort of 32 index patients gathered through various collaborations across Europe. When such patients are seen in Genetics Clinics, and if an autosomal dominant inheritance is the most likely, array CGH should be the first-line test when available, but qPCR or MLPA for SHFM3 (10q24) or SHFM/SHFLD (17p13) loci could be offered as an alternative [41, 42]. Prenatal diagnosis based on scans and invasive testing in pregnancy can allow better genetic counselling and management in the context of non-syndromic SHFM. Future studies with animal models containing sequence from exon 1 of BRTC may help exploring its effect on the AER, as well as refining the breakpoints at a nucleotide level [26].
References
Velinov M, Ahmad A, Brown-Kipphut B, Shafiq M, Blau J, Cooma R et al. A 0.7 Mb de novo duplication at 7q21.3 including the genes DLX5 and DLX6 in a patient with split-hand/split-foot malformation. Am J Med Genet. 2012;158:3201–6.
Klopocki E, Lohan S, Doelken SC, Stricker S, Ockeloen CW, Soares Thiele de Aguilar R et al. Duplications of BHLHA9 are associated with ectrodactyly and tibia hemimelia inherited in non-Mendelian fashion. J Med Genet. 2012;49:119–25.
Brachmann W. Ein Fall von symmetrischer Monodaktylie durch Ulnadefekt, mit symmetrischer Flughautbildung in den Ellenbeugen, sowie anderen Abnormalitäten (Zwerghaftigkeit, Halsrippen, Behaarung). Jb Kinder. 1916;84:225–35.
Temtamy SA, McKusick VA. The genetics of hand malformations. Birth Defects Orig Artic Ser. 1978;14:1–619.
Duijf PHG, van Bokhoven H, Brunner H. Pathogenesis of split-hand/split-foot malformation. Hum Mol Genet. 2003;12:R51–60.
Maruiwa M, Nakamura Y, Motomura K, Murakami T, Kojiro M, Kato M et al. Cornelia de Lange syndrome associated with Wilms’ tumour and infantile haemangioendothelioma of the liver: report of two autopsy cases. Virchows Arch A Pathol Anat Histopathol. 1988;413:463–8.
Pfeiffer RA, Correll J. Hemimelia in Brachmann-de Lange syndrome (BDLS): a patient with severe deficiency of the upper and lower limbs. Am J Med Genet. 1993;47:1014–7.
Urban M, Hartung J. Ultrasonographic and clinical appearance of a 22-week-old fetus with Brachmann-de Lange syndrome. Am J Med Genet. 2001;102:73–5.
Singer LP, Marion RW, Li JK. Limb deficiency in an infant with Smith–Lemli–Opitz syndrome. Am J Med Genet. 1989;32:380–3.
Bujdoso G, Lenz W. Monodactylous split hand-split foot. A malformation occurring in three distinct genetic types. Eur J Pediatr. 1980;133:207–15.
van Bokhoven H, Hamel BC, Bamshad M, Sangiorgi E, Gurrieri F, Dujif PH et al. p63 Gene mutations in EEC syndrome, limb-mammary syndrome, and isolated split hand-split foot malformation suggest a genotype–phenotype correlation. Am J Hum Genet. 2001;69:481–92.
de Mollerat XJ, Gurrieri F, Morgan CT, Sangiorgi E, Everman DB, Gaspari P et al. A genomic rearrangement resulting in a tandem duplication is associated with split hand–split foot malformation 3 (SHFM3) at 10q24. Hum Mol Genet. 2003;12:1959–71.
Yang W, Hu ZJ, Yu XF, Li QH, Zhang AJ, Deng X et al. A DNA duplication at chromosome 10q24.3 is associated with split-hand split-foot malformation in a Chinese family. Zhonghua Yi Xue Za Zhi. 2006;86:652–8.
Ignatius J, Knuutila S, Scherer SW, Trask B, Kere J. Split hand/split foot malformation, deafness, and mental retardation with a complex cytogenetic rearrangement involving 7q21.3. J Med Genet. 1996;33:507–10.
Marinoni JC, Stevenson RE, Evans JP, Geshuri D, Phelan MC, Schwartz CE. Split foot and developmental retardation associated with a deletion of three microsatellite markers in 7q21.2–q22.1. Clin Genet. 1995;47:90–5.
McElveen C, Carvajal MV, Moscatello D, Towner J, Lacassie Y. Ectrodactyly and proximal/intermediate interstitial deletion 7q. Am J Med Genet. 1995;56:1–5.
Del Campo M, Jones MC, Veraksa AN, Curry CJ, Jones KL, Mascarello JT et al. Monodactylous limbs and abnormal genitalia are associated with hemizygosity for the human 2q31 region that includes the HOXD cluster. Am J Hum Genet. 1999;65:104–10.
Goodman FR, Majewski F, Collins AL, Scambler PJ. A 117-kb microdeletion removing HOXD9–HOXD13 and EVX2 causes synpolydactyly. Am J Hum Genet. 2002;70:547–55.
Bijlsma EK, Knegt AC, Bilardo CM, Goodman FR. Increased nuchal translucency and split-hand/foot malformation in a fetus with an interstitial deletion of chromosome 2q that removes the SHFM5 locus. Prenat Diagn. 2005;25:39–44.
Sidow A, Bulotsky MS, Kerrebrock AW, Birren BW, Altshuler D, Jaenisch R et al. A novel member of the F-box/WD40 gene family, encoding dactylin, is disrupted in the mouse dactylaplasia mutant. Nat Genet. 1999;23:104–7.
Friedli M, Nikolaev S, Lyle R, Arcangeli M, Duboule D, Spitz F et al. Characterization of mouse Dactylaplasia mutations: a model for human ectrodactyly SHFM3. Mamm Genome. 2008;19:272–8.
Raas-Rothschild A, Manouvrier S, Gonzales M, Farriaux JP, Lyonnet S, Munnich A. Refined mapping of a gene for split hand-split foot malformation (SHFM3) on chromosome 10q25. J Med Genet. 1996;33:996–1001.
Lyle R, Radhakrishna U, Blouin J-L, Gagos S, Everman DB, Gehrig C et al. Split-hand/split-foot malformation 3 (SHFM3) at 10q24, development of rapid diagnostic methods and gene expression from the region. Am J Med Genet. 2006;140:1384–95.
Dimitrov B, de Ravel T, Van Driessche J, de Die-Smulders C, Toutain A, Vermeersch JR et al. Distal limb deficiencies, micrognathia syndrome (OMIM 246560) and syndromic forms of split hand foot malformation (SHFM) are caused by chromosome 10q genomic rearrangements. J Med Genet. 2010;47:103–11.
Bergbaum A, Irving M, Mansour S, Homfray T, Dimitrov B, Flinter F et al. BTRC upregulation alone is insufficient to cause SHFM3—evidence from 5 new cases. In: European human genetics conference. 2011; P03.079.
Li CF, Angione K, Milunsky JM. Identification of critical region responsible for split hand/foot malformation type 3 (SHFM3) phenotype through systematic review of literature and mapping of breakpoints using microarray data. Microarrays. 2015;5:2.
Socha M, Sowinska-Seidler A, Mundlos S, Spielmann M, Jamsheer A. A 10q24.32 duplication causes bilateral femoral hypoplasia through formation of a novel sub-TAD. In: European human genetics conference. 2017; C05.4.
Hills A, Ahn JW, Donaghue C, Thomas H, Mann K, Ogilvie CM. MLPA for confirmation of array CGH results and determination of inheritance. Mol Cytogenet. 2010;3:19.
Gehrke AR, Shubin NH. Cis-regulatory programs in the development and evolution of vertebrate paired appendages. Semin Cell Dev Biol. 2016;57:31–9.
Everman DB, Morgan CT, Lyle R, Laughridge ME, Bamshad MJ, Clarkson KB et al. Frequency of genomic rearrangements involving the SHFM3 locus at chromosome 10q24 in syndromic and non-syndromic split-hand/foot malformation. Am J Med Genet. 2006;140:1375–83.
Tayebi N, Jamsheer A, Flöttmann R, Sowinska-Seidler A, Doelken SC, Oehl-Jaschkowitz B et al. Deletions of exons with regulatory activity at the DYNC1I1 locus are associated with split-hand/split-foot malformation: array CGH screening of 134 unrelated families. Orphanet J Rare Dis. 2014;9:108.
Elliott AM, Evans JA, Chudley AE. Split hand split foot malformation (SHFM). Clin Genet. 2005;68:501–5.
Johnson KR, Lane PW, Ward-Bailey P, Davisson MT. Mapping the mouse dactylaplasia mutation, Dac, and a gene that controls its expression, mdac. Genomics. 1995;29:457–64.
Basel D, De Paepe A, Kilpatrick MW, Tsipouras P. Split hand foot malformation is associated with a reduced level of Dactylin gene expression. Clin Genet. 2003;64:350–4.
Kondrashov FA, Koonin EV. Increased nuchal translucency and split-hand/foot malformation in a fetus with an interstitial deletion of chromosome 2q that removes the SHFM5 locus. Trends Genet. 2004;20:287–90.
Deutschbauer AM, Jaramillo DF, Proctor M, Kumm J, Hillenmeyer ME, Davis RW et al. Mechanisms of haploinsufficiency revealed by genome-wide profiling in yeast. Genetics. 2005;169:1915–25.
Seto ML, Nunes ME, MacArthur CA, Cunningham ML. Pathogenesis of ectrodactyly in the Dactylaplasia mouse: aberrant cell death of the apical ectodermal ridge. Teratology. 1997;56:262–70.
Kano H, Kurosawa K, Horii E, Ikegawa S, Yoshikawa H, Kurahashi H et al. Genomic rearrangement at 10q24 in non-syndromic split-hand/split-foot malformation. Hum Genet. 2005;118:477–83.
Maniatis T. A ubiquitin ligase complex is essential for the NF-kappaB, Wnt/Wingless and Hedgehog signaling pathways. Genes Dev. 1999;13:505–10.
Lewandoski M, Sun X, Martin GR. Fgf8 signalling from the AER is essential for normal limb development. Nat Genet. 2000;26:460–3.
Sowińska-Seidler A, Socha M, Jamsheer A. Split-hand/foot malformation—molecular cause and implications in genetic counseling. J Appl Genet. 2014;55:105–15.
Carter TC, Sicko RJ, Kay DM, Browne ML, Romitti PA, Edmunds ZL et al. Copy-number variants and candidate gene mutations in isolated split hand/foot malformation. J Hum Genet. 2017;62:877–84.
Acknowledgements
Aleksander Jamsheer was supported by the Polish National Science Centre Grant UMO-2016/22/E/NZ5/00270 as well as by the Polish National Centre for Research and Development (Grant no. LIDER/008/431/L-4/12/NCBR/2013). Anna Sowinska-Seidler was supported by the Polish National Science Centre Grant UMO-2016/21/D/NZ5/00064. Magdalena Socha was supported by the Polish National Science Centre Grant UMO-2016/23/N/NZ2/02362.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Holder-Espinasse, M., Jamsheer, A., Escande, F. et al. Duplication of 10q24 locus: broadening the clinical and radiological spectrum. Eur J Hum Genet 27, 525–534 (2019). https://doi.org/10.1038/s41431-018-0326-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-018-0326-9
This article is cited by
-
Combinatorial effects on gene expression at the Lbx1/Fgf8 locus resolve split-hand/foot malformation type 3
Nature Communications (2023)
-
SHFLD3 phenotypes caused by 17p13.3 triplication/ duplication encompassing Fingerin (BHLHA9) invariably
Orphanet Journal of Rare Diseases (2022)
-
Genome sequencing in families with congenital limb malformations
Human Genetics (2021)
