
Abstract
The organization of mammalian genomes into sub-megabase sized Topologically Associated Domains (TADs) has recently been revealed by techniques derived from Chromosome Conformation Capture (3 C), such as High Chromosome Contact map (Hi-C). Disruption of this organization by structural variations can lead to ectopic interactions between enhancers and promoters, and to alteration of genes expression patterns. This mechanism has already been described as the main pathophysiological mechanism in several syndromes with congenital malformations. We describe here the case of a fetus with a severe multiple congenital anomalies syndrome, including extensive polydactyly of the four limbs. This fetus carries a de novo deletion next to the IHH gene, encompassing a TAD boundary. Such an IHH TAD boundary deletion has already been described in the Dbf mouse model, which shows a quite similar, but less severe phenotype. We hypothesize that the deletion harbored by this fetus results in the same pathophysiological mechanisms as those of the Dbf model. The description of this case expands the spectrum of the disruption of chromatin architecture of WNT6/IHH/EPHA4/PAX3 locus, and could help to understand the mechanisms of chromatin interactions at this locus.
Similar content being viewed by others


Introduction
The study of 3D genome organization by High Chromosome Contact map, derived from Chromosome Conformation Capture, revealed megabase to sub-megabase sized chromatin domains named Topologically Associated Domains (TADs) [1,2,3]. These domains are fundamental structural units of the genome, and are essential to genes’ expression regulation. TADs allow interactions of enhancers with their target promoters located in the same TAD, and prevent interactions with promoters located in neighboring TADs. Disruptions of TAD boundaries by chromosomal rearrangements can lead to abnormal interactions between gene promoter and enhancer. The deregulation of the transcriptional pattern of these genes can result to Mendelian disorders [4,5,6]. Several loci affected by such disruption of chromatin architecture have already been described, such as the WNT6/IHH/EPHA4/PAX3 locus [7].
The EPHA4 gene encodes a protein-tyrosine kinase receptor of the ephrin subfamily, implicated in central nervous system development. During development, Epha4 is expressed in various neural tissues, such cortex, corpus callosum, cortico-spinal tract and diencephalon [8], but also in limbs, where it is required for normal innervation [9].
WNT6, IHH and PAX3 genes belong to 3 well-known gene families playing critical roles during embryonic development. Especially, IHH belongs to the hedgehog family, and is required for endochondral bone formation by regulating the proliferation and differentiation of chondrocytes [10]. During development IHH is predominantly expressed in prehypertrophic chondrocytes.
Structural variants encompassing the TAD boundaries at the WNT6/IHH/EPHA4/PAX3 locus can lead to three different phenotypes, each characterized by specific abnormalities of the extremities.
The pathophysiological mechanism is a misexpression of WNT6, IHH or PAX3 genes, due to an interaction with EPHA4 distal limb enhancers [7].
We report here a fetus carrying a heterozygous de novo deletion encompassing a boundary of the IHH gene TAD. The fetus presented with a severe phenotype with extensive polydactyly of the four limbs. This case expands the spectrum of developmental defects associated with WNT6/IHH/EPHA4/PAX3 locus rearrangements.
Materials and methods
Array-CGH and Quantitative PCR
See Supplementary data for extended Materials and Methods. Array-CGH were performed using microarrays 4 × 180K, G2565C scanner, and Cartagenia Bench Lab CNV platform for interpretation (Agilent Technologies, Santa Clara, CA, USA), according to the manufacturer’s instructions.
Thirteen quantitative PCR were performed in breakpoint uncertainty intervals, using SYBR Green Mix on the Light Cycler 480 System, under standard conditions, in triplicate.
Breakpoint spanning PCR
A primer pair was designed in close non-deleted regions previously determined by qPCR: Forward primer 1 5′-TTGGGGCTGAACTGCTTGTA-3′ and Reverse primer 1 5′-GCATTTGAGGCTATGGCTATTTTCT-3′. A main PCR product at around 1300 bp was amplified. A hemi-nested PCR was then performed using the same PCR mix replacing the Forward primer 1 by Forward primer 2 5′-ATAGGCCGGCCCGTTTGTT-3′ in a closer non-deleted region. An 800 bp PCR product was amplified and Sanger sequenced. Data has been analyzed using CutePeaks software [11].
Results
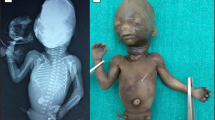
We report here the case of a female fetus. The mother was 23 years old, and she reported in her medical history a previous early spontaneous miscarriage, without further examination. This second pregnancy was interrupted at 15 gestational weeks because of a severe fetal phenotype with a large cystic hygroma, polydactyly, phocomelia, hyperechoic kidneys and megacisterna magna. The fetus was macerated, with epidermolysis on 80% of the skin surface, and was hypotrophic. External examination showed a bilateral cleft lip and palate, and ambiguous external genitalia, with an incomplete fusion of urethral folds. Rhizomelic micromelia of all four limbs, and pterygium colli were observed. Examination of the four limbs showed polydactyly, with 8 fingers on each hand with a right mirror hand, 7 toes on the left foot and 6 toes on the right foot with an enlarged hallux (Fig. 1). On visceral examination, the gall bladder was missing, and there was polysplenia. The internal genitalia were female, but abnormal, with a hypoplastic uterus. Dissection of the heart showed a single great vessel. The two atria were visible, with normal situs. There was an overriding aorta, with a large interventricular communication. There was no identifiable pulmonary artery trunk and no pulmonary artery emerging from the single artery trunk. Examination of the aortic arch showed a possible aortopulmonary collateral artery from the aortic isthmus reaching the right lung, and another one from the aorta reaching the left lung. The venous pulmonary return seemed normal. Microscopic examination showed significant autolytic alterations. However we were able to see that the pulmonary parenchyma was at the canalicular stage. On kidney slides, there were corticomedullary cystic tubular dilations, but the gestational term and the maceration rendered the examination difficult. The brain was completely autolyzed.

Right and left hands of the fetus, and fetal X-rays: We observe an extensive polydactyly, with 8 fingers on each hand, and a right mirror hand, associated with phocomelia
X-rays showed a bilateral humeral and femoral agenesis, associated with a bilateral tibiofibular hypoplasia, and abnormal scapulae.
180 K array-CGH performed on chorionic villus sample showed a de novo 2q35 deletion with a maximal size range of 1019 kb:
GRCh37 NC_000002.11 g.(219912758_219928949)_(220900731_220931554)del (ISCN 2016 nomenclature: arr[GRCh37] 2q35(219912758 × 2,219928949_220900731 × 1,220931554 × 2)dn)
Thirteen qPCRs were performed to reduce the region of uncertainty, then the breakpoints of the deletion were defined at a molecular level by spanning PCR sequencing as:
GRCh37 NC_000002.11 g.219925666_220914504delinsTAGTGCCTCGA.
This deletion encompasses 988,839 base pairs, and sequence analysis showed an additional insertion of 11 base pairs between the two breakpoints. The chromosomal rearrangement has been submitted to the Clinvar database (Accession: SCV000786655).
The centromeric breakpoint is located 429 basepairs upstream the IHH gene transcription start site.
This chromosomal rearrangement was not present in the CNV (Copy Number Variation) databases (Decipher [12], ISCA [13] and DGV [14]). This interval includes 25 RefSeq genes.
Previous studies [7, 15] determined that this chromosomal region is organized in three different TADs: a centromeric TAD encompassing WNT6 and IHH, a median TAD encompassing EPHA4 and a cluster of enhancers regulating its expression, and a telomeric TAD encompassing PAX3 (Fig. 2). The identified deletion encompasses the boundary between the WNT6/IHH TAD and the EPHA4 TAD.

Disruption of chromatin architecture of IHH/EPHA4 locus. a 2q35 region visualized with the 3D Genome Browser [23]. On top, Hi-C data from Rao et al. [15] (GM12878 cell line, 25 kb resolution). This region encompasses several TADs, shown as alternating blue and yellow lines. The region between block dots is represented in Fig. 2b. b On top, representation of the IHH/EPHA4 locus. The deletion of the Dbf mouse model removes the TAD boundary between the IHH and the EPHA4 TADs, allowing an interaction between an enhancer cluster, and the IHH gene. The deletion of the fetal case also encompasses this TADs boundary, but the cluster enhancer too. However, the EPHA4 TAD involved other putative enhancers and could lead to the same deregulation mechanism. In particular one of the predicted enhancer of the FANTOM5 database [22] (coordinates: NC_000002.1: g.(221360545_221360555)) is significantly overrepresented in chondrocytes, and could be involved in this mechanism. On bottom, Sanger sequence of the breakpoints spanning PCR. The deletion is 988,839 base pairs, with an additional insertion of 11 base pairs between the two breakpoints. The coordinate of this deletion/insertion is: GRCh37 NC_000002.11 g.219925666_220914504delinsTAGTGCCTCGA
Discussion
The fetal case reported here carried a de novo heterozygous deletion encompassing the telomeric WNT6/IHH TAD boundary, without involving the IHH gene.
The pathophysiological mechanisms of chromosomal rearrangements at the WNT6, IHH, EPHA4, and PAX3 genes locus in the mouse model have been delineated by Lupiáñez et al. [7]. Deletions, duplications or inversions involving one of the median TAD boundaries lead to an ectopic interaction between WNT6, IHH or PAX3 and the cluster of enhancers located in the EPHA4 TAD, and thus to a specific phenotype. For example, heterozygous duplications encompassing IHH and the boundary between the telomeric and the median TADs lead to a phenotype mimicking the acrocallosal syndrome in human, with polysyndactyly of the hands and feet, agenesis of the corpus callosum, dysmorphism and severe intellectual disability [16]. A deletion involving the same TAD boundary has been identified in the Doublefoot mice model (Dbf) [17, 18]. This mutant mouse strain shows a similar phenotype, with preaxial polydactyly with 6–9 triphalangeal digits in all four limbs, tibial hypoplasia, broadened skull, hydrocephalus, and thickened, kinked tail.
Interestingly, the clinical features of the fetus are quite similar to the phenotype of the Dbf mice model and to the acrocallosal-like syndrome due to IHH duplication. In particular the polydactyly of this fetus are the same, with mirror configuration of the fingers. Therefore, we can hypothesize that the main pathophysiological effect of the deletion of this fetus is the same: an ectopic interaction between the IHH gene and enhancers in the EPHA4 TAD.
Even if this hypothesis is supported by the current knowledge of the chromatin architecture at this locus, we cannot rule an effect of the 25 deleted genes. However, the implication of 24 of them is unlikely according to literature and loss-of-function mutations frequency in databases (Supplemental data). The remaining gene, ATG9A, encodes a subunit of the adaptor protein 4 complex. This complex contains other subunits encoded by genes involved in autosomal recessive spastic paraplegia [19]. The KO mice model for ATG9A shows hindlimb clasping, decreased motor coordination, weak grip strength and thin corpus callosum, without any other congenital malformations. The heterozygous mice do not present any clinical features. Furthermore there is no other data in the literature arguing for an implication of the heterozygous deletion of this gene in the phenotype of the fetus.
The centromeric breakpoint of the deletion is close to IHH transcription start site, and thus encompasses part of the IHH promoter. However, IHH is thought to be still expressed. Indeed, annotation by GPMiner software [20] shows that the TATA box is still present in the promoter, located at −206 bp. Furthermore, Ushijima et al [21]. showed by promoter deletion analysis that the C/EBPβ and RUNX2 responsive element of the IHH promoter, responsible of the IHH expression in chondrocytes, is located between −214 and −210 bp, and thus is not encompassed by the deletion.
The deletion of the fetus is larger than the Dbf deletion and encompassed the cluster of limb enhancers identified in the EPHA4 TAD by Lupiáñez [7] (Fig. 2).
However, the EPHA4 TAD involved several other putative enhancers, as identified by CAGE experiments by the FANTOM5 consortium project [22], with one of them significantly overrepresented in chondrocytes (Fig. 2).
The different number of enhancers involved in this pathophysiological mechanism between the Dbf model mouse and this fetus could explain his more severe phenotype, with phocomelia, and several visceral malformations.
In conclusion, we report here the first human case of deletion of IHH TAD boundary, which showed a similar but more severe phenotype than the Dbf mouse model. The delineation of the diverse phenotypes associated with CNVs at this specific WNT6/IHH/EPHA4/PAX3 locus could help to understand the mechanisms of chromatin interactions at this locus.
References
Lieberman-Aiden E, van Berkum NL, Williams L, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–93.
Dixon JR, Selvaraj S, Yue F, et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–80.
Dixon JR, Gorkin DU, Ren B. Chromatin domains: the unit of chromosome organization. Mol Cell. 2016;62:668–80.
Matharu N, Ahituv N. Minor loops in major folds: enhancer–promoter looping, chromatin restructuring, and their association with transcriptional regulation and disease. PLoS Genet. 2015;11:e1005640.
Krijger PHL, de Laat W. Regulation of disease-associated gene expression in the 3D genome. Nat Rev Mol Cell Biol. 2016;17:771–82.
Acemel RD, Maeso I, Gómez-Skarmeta JL. Topologically associated domains: a successful scaffold for the evolution of gene regulation in animals: topologically associated domains. Wiley Interdiscip Rev: Dev Biol. 2017;6:e265.
Lupiáñez DG, Kraft K, Heinrich V, et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell. 2015;161:1012–25.
Greferath U, Canty AJ, Messenger J, Murphy M. Developmental expression of EphA4-tyrosine kinase receptor in the mouse brain and spinal cord. Gene Expr Patterns. 2002;2:267–74.
Helmbacher F, Schneider-Maunoury S, Topilko P, Tiret L, Charnay P. Targeting of the EphA4 tyrosine kinase receptor affects dorsal/ventral pathfinding of limb motor axons. Development. 2000;127:3313–24.
Vortkamp A, Lee K, Lanske B, Segre GV, Kronenberg HM, Tabin CJ. Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science. 1996;273:613–22.
Labsquare Team, Schutz S, Gueudelot O. Cutepeaks 0.1.0: A simple Sanger trace file. 2017. https://doi.org/10.5281/zenodo.824555.
Firth HV, Richards SM, Bevan AP, et al. DECIPHER: database of chromosomal imbalance and phenotype in humans using Ensembl resources. Am J Human Genet. 2009;84:524–33.
Kaminsky EB, Kaul V, Paschall J et al. An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities: Genet Med. 2011; 13: 777–84.
MacDonald JR, Ziman R, Yuen RKC, Feuk L, Scherer SW. The database of genomic variants: a curated collection of structural variation in the human genome. Nucleic Acids Res. 2014;42:D986–992.
Rao SSP, Huntley MH, Durand NC, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–80.
Yuksel-Apak M, Bögershausen N, Pawlik B, et al. A large duplication involving the IHH locus mimics acrocallosal syndrome. Eur J Hum Genet. 2012;20:639–44.
Lyon MF, Quinney R, Glenister PH, Kerscher S, Guillot P, Boyd Y. Doublefoot: a new mouse mutant affecting development of limbs and head. Genet Res. 1996;68:221–31.
Babbs C, Furniss D, Morriss-Kay GM, Wilkie AOM. Polydactyly in the mouse mutant Doublefoot involves altered Gli3 processing and is caused by a large deletion in cis to Indian hedgehog. Mech Dev. 2008;125:517–26.
De Pace R, Skirzewski M, Damme M, et al. Altered distribution of ATG9A and accumulation of axonal aggregates in neurons from a mouse model of AP-4 deficiency syndrome. PLoS Genet. 2018;14:e1007363.
Lee T-Y, Chang W-C, Hsu JB-K, Chang T-H, Shien D-M. GPMiner: an integrated system for mining combinatorial cis-regulatory elements in mammalian gene group. BMC Genom. 2012;13(Suppl 1):S3.
Ushijima T, Okazaki K, Tsushima H, Ishihara K, Doi T, Iwamoto Y. CCAAT/enhancer binding protein β regulates expression of indian Hedgehog during chondrocytes differentiation. PLoS ONE. 2014; 9: e104547.
Andersson R, Gebhard C, Miguel-Escalada I, et al. An atlas of active enhancers across human cell types and tissues. Nature. 2014;507:455–61.
Wang Y, Zhang B, Zhang L et al. The 3D genome browser: a web-based browser for visualizing 3D genome organization and long-range chromatin interactions. 2017. https://doi.org/10.1101/112268.
Acknowledgements
The authors thank the patients for participating in this study, and the Centre of Biology and Pathology of Bordeaux University Hospital for its full support. This study makes use of data generated by the DECIPHER community. A full list of centers who contributed to the generation of the data is available from http://decipher.sanger.ac.uk and via email from decipher@sanger.ac.uk. Funding for the project was provided by the Wellcome Trust.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical statement
Written informed consent was received for a pathological examination of fetal and placental tissues. The authors adhere to the Declaration of Helsinki Principles.
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Trimouille, A., Tingaud-Sequeira, A., Pennamen, P. et al. Deletion in 2q35 excluding the IHH gene leads to fetal severe limb anomalies and suggests a disruption of chromatin architecture. Eur J Hum Genet 27, 384–388 (2019). https://doi.org/10.1038/s41431-018-0290-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-018-0290-4
