
Abstract
The expansion of modern humans to the American continent after the Last Glacial Maximum led the way to the present-day distribution of American aborigines. Recent advances in autosomal DNA research and expanded testing of mtDNA lineages has provided a clearer picture of the number and timing of founding lineages. However, both autosomal DNA and mtDNA research have provided unresolved competing theories between the short-term and the long-term models of the Beringian standstill hypothesis. Further, the source of founding paternal lineages of American aborigines and their relationship with ancient Siberia populations remains ambiguous. In this study, we reanalyzed a 7.0 Mbp region of 132 paternal Y-chromosome sequences, including 39 newly reported ones, of male samples from American aborigines and Eurasian populations. Among Eurasian samples, we identified Y-chromosome branches that are most closely related to known American aborigine founding lineages, that is, Q1-L804 links to Q1-M3, Q1-L330 links to Q1-Z780, Q1-M120 links to Q1-B143, and C2-F1756 links to C2-P39. The revised phylogenetic tree and age estimates indicate a narrow timeframe (~15.3–14.3 kya) for the upper time limit of human entry to the American continent. Our analysis suggests that the in situ differentiation of Q-M242 in Central Eurasia and South Siberia region gave rise to numerous sub-lineages older than 15.3 kya, and the founding of Paleo-Indian paternal lineages is part of the great Q1-L53 diffusion throughout the Eurasia after the Last Glacial Maximum. The results of our study will assist in future studies of the history of modern populations in Eurasia and the Americas.
Similar content being viewed by others
Introduction
The origin and diffusion of American aborigines has long fascinated linguists, archeologists, and geneticists. Generally, American aborigines are categorized into three groups: Amerindian, Na-Dene populations, and Eskimo-Aleut [1]. Both archeological and genetic evidence suggests that Eskimo-Aleut populations entered the North American subcontinent in a late wave of migration several thousand years ago [2, 3]. Therefore, only Amerindian and Na-Dene populations are considered descendants of the first group of modern humans on the American continent [1]. In this study, the term “Native American” refers to these two groups.
Subjects of interest to population genetics include the location of Eurasian origin, the date of first colonization, the number and path of migrations, and the age of surviving archeological sites in the Americas. Genetics has for some time been used to explore these questions. Methods used have included mtDNA, autosomal DNA, and the non-recombining part of the Y-chromosome, NRY. From the mtDNA perspective, there are five established founding lineages (mtDNA haplogroups A2, B2, C1, D1, and X2a) [4,5,6]. The first four of these mtDNA lineages have deep connections to East Asia while the fifth has a less certain origin. Recent improvements in autosomal DNA testing technology for ancient DNA samples have revealed additional connections to Eurasia [7,8,9,10,11]. Meanwhile, there are at least four founding Y-chromosome lineages Q1-M3 [12, 13], Q1-Z780 [14, 15], C2-P39 [13, 16], Q-NWT01 (AKA Q-F746) [16, 17], and several unclear minor lineages. [12, 13, 16,17,18,19,20] Of these, the Q-NWT01 lineage is Eskimo-Aleut and not considered part of the Native American group as defined in this paper.
It is generally accepted that the Native American founding population is genetically closely related to ancient populations in Siberia [7, 10]. Recent ancient DNA studies suggest that ancient northern Eurasians, represented by 24,000-year-old MA1, contributed substantially to the gene pool of modern Native Americans [7, 10]. Additionally, the expansion age of Native American peoples has long been used as a calibration point for the whole tree of human mtDNA and Y-chromosomes [21, 22]. However, it is possible that the paternal and maternal lineages of Native Americans diverged before their entry to the Americas. The assumption that paternal lineages of Native Americans diverged at about the same time as the initial peopling of the continent has yet to be verified by both modern samples and ancient DNA.
At one time, Clovis Culture (~13.0–12.6 thousand years ago, hereafter kya) archeological sites were considered the oldest remains on the American continent [23]. However, increasing archeological evidence supports pre-Clovis human occupation of the Americas as early as 15,000 years ago [24, 25]. Further, although, the Anzick-1 boy was initially used to link the Clovis Culture to the expansion of Y-chromosome lineage Q-M242 in the Americas, its refined placement on the tree in this study indicates that it is only the progenitor of the Q1-FGC47532 sub-lineage, which expanded from Q1-Z780 after colonization [26, 27].
In the past decade, the “Beringian standstill hypothesis” has been proposed to explain the early differentiation of ancestors of American aborigines from their close relatives in Siberia [5]. There are two parts to this hypothesis. The first part is the “Long-term Beringian standstill model” which argues that a Native American founding population reached Beringia during or before the Last Glacial Maximum (LGM) (dated to ca. 18,000–24,000 cal BP), where they were genetically isolated for up to ~3–9 kya before moving south across the ice-sheets in North America at about 15 kya [5]. The second part is the “Short-term Beringian standstill model” which suggests that an ancestor group of Native American arrived at Beringia after the LGM (aka. after 18 kya) and stayed there for a short period before moving south at about 15,000 years ago [6].
Recent studies of the maternal gene pool through mtDNA can support both parts of the “Beringian standstill hypothesis” [5, 10, 28]. The split times of four of the founding mtDNA lineages (A2, B2, C1, and D1) of Native Americans and their close relatives in Siberia are between 24.9 and 18.4 kya [28]. Additionally, mtDNA suggests that the Native American founding population experienced a reduction in size that lasted for 3000–4000 years [5]. This reduction may be linked to the deteriorating conditions of Beringia at the LGM. An alternative route has been proposed along the western coast of North America ~15–17 kya [4, 29]. Archeological support for the Beringian standstill hypothesis is scarce [30]. At present, there is no unequivocal archeological evidence to support such an entry to the American continent earlier than 15 kya [24]. Archeological sites in the Beringia region are not dated to earlier than 14 kya. A possible exception is the Bluefish Caves site in the Yukon Territory of Canada (dating to 24 kya), which is still in doubt [31, 32]. Therefore, additional demographic modeling and ancient DNA data are needed to explain the early differentiation between the Native American founding population and close relatives in Siberia.
Previous genetic studies of Y-chromosome have identified a number of founding paternal lineages for American aborigines, including Y-chromosome haplogroup Q1-M3 [12, 13], C2-P39 [13, 16], and Q-NWT01 (AKA Q-F746) [16, 17]. There are also several unclear minor lineages. [12, 13, 16,17,18,19,20] Haplogroup Q1-M3 is found at high frequencies in nearly all American aborigine populations. Additionally, ~9-ky-old Kennewick Man and the remains of a male from Alaska (ONYKCM) belong to lineage Q1-M3 [9, 33]. Scholars have proposed that the expansion of this lineage corresponds to the initial colonization of humans throughout the America continent [12,13,14]. Haplogroup Q-L330, a closely related Y-chromosome lineage of Q1-M3, was discovered among South Siberia populations [34,35,36], indicating the ancient link between populations in two distant regions. Haplogroup C2-P39 scatters randomly in the North America [13, 16] and a number of C2*-M217 samples were found in Colombian and Ecuador populations in South America [13, 19]. Scholars have also suggested that haplogroup C2-P39 and C2*-M217 may represent another two minor founding paternal lineages of American aborigines [13, 16]. The samples of haplogroup Q-NWT01 were mainly found in Inuvialuit and other populations near the North Circumpolar Region [16, 17]. The research of Karmin et al.[35] indicated that lineage Q-B143 is a sub-branch of Q-NWT01. Ancient DNA analysis suggests that the ~4-ky-old Saqqaq male belongs to the Q1-B143 lineage [3]. Therefore, this lineage may represent a later wave of human migration near the North Circumpolar Region, rather than being a founding lineage of Native Americans [16, 17]. Furthermore, ancient DNA studies have shown that the ~12.6-ky-old Anzick-1 boy belongs to the Q1-FGC47532 sublineage of haplogroup Q1-Z780 [8]. However, few works have mentioned haplogroup Q-L804 [37] and Q-Z780 [14]. Previous studies focused on the internal diversity of paternal gene pool of American aborigines and little is known about the differentiation process of founding paternal lineages of American aborigines from their close relatives in Siberia and more broadly in Eurasia.
In this study, we collected Y-chromosome sequences from Eurasian populations that are related to paternal lineages of American aborigines, especially those in North Asia. Our first objective was to determine the most closely related lineage of each Native American paternal lineage in Eurasian populations by phylogenetic analysis. The second goal was to estimate the split time of all studied samples from their common ancestor to study the population differentiation process in Siberia, which was directly related to the emergence of the founding group of Native Americans (hereafter called “Paleo-Indians”). In particular, we were interested in re-examining the Beringian standstill hypothesis based on new evidence from the perspective of the paternal Y-chromosome. Overall, we used a refined Y-chromosome phylogenetic tree with accurate dating to explore the origin of paternal lineages of Native American populations.
Materials and methods
Detailed information is described in Supplementary text, Materials and Methods. We reviewed published literature and from it collected previously sequenced Y-chromosome DNA results from American aborigines and their relatives from Eurasia populations [14, 35, 38,39,40]. We then selected suitable samples from our lab for additional Y-chromosome sequencing. Some sequences from our lab were phylogenetically close to samples from American aborigines while others were only used to provide a better resolution for the upstream structure of the whole-phylogenetic tree. In collaboration with a group of citizen scientists, 15 men that tested at Family Tree DNA provided their Y-chromosome sequences. Six of those men trace their ancestry to Eurasia and the other nine men are descendants of American aborigines. Methods for collaboration with citizen scientists and informed consent for these samples follows Balanovsky et al.[41]. The demographic and phylogenetic information for the 132 samples involved in this study, including 56 samples from American aborigines, is listed in Supplementary Table S1. All individuals signed informed consent forms before their participation. The ethics committee for biological research at the School of Life Sciences in Fudan University approved the study. Mapping of reads and SNP calling were conducted with the human reference hg37 using GATK packages and standard procedures for next-generation sequencing. Bayesian evolutionary analyses were implemented in BEAST (v2.4.3) [42].
Two approaches were used to calculate the splitting time of the phylogenetic tree with BEAST software. First, we applied the point mutation rate of 0.74 × 10−9 per site per year which was inferred from the genome of 12.6-ky-old Anzick-1 Boy [8, 35]. Second, two calibration points were used for age calculation [8, 43]. More detailed descriptions of the sequence data processing, genotyping and filtering, studied samples, phylogenetic inference, and choosing of dating approaches are included in the Supplementary text, Materials and Methods.
The raw sequence data reported in this paper has been deposited in the Genome Sequence Archive [44] in BIG Data Center, Beijing Institute of Genomics (BIG) [45], Chinese Academy of Sciences, under accession numbers PRJCA000419, PRJCA000420, and PRJCA000430 that are publicly accessible at http://bigd.big.ac.cn/gsa. The sequences of the 15 citizen science testers from Family Tree DNA were deposited in the NCBI GenBank database under accession number SRP135954.
Results
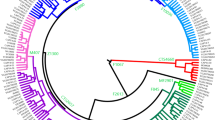
A schematic phylogenetic tree and the location of studied samples are shown in Fig. 1. Detailed sample information and a detailed phylogenetic tree with markers of important clades are provided in Supplementary Table S1 and Figure S1, respectively. Age and posterior support estimates for each clade on Figure S1 are reported in Table S5. The age estimations of two approaches are close to each other. As shown in Figs. 2 and 3, we identified the most closely related Eurasian lineage for each paternal lineage of American aborigines (also see Figure S1).

Phylogenetic tree and the location of studied samples

Founding paternal lineages of American aborigines and their most closely related lineages among Eurasia populations

Proposed paternal origin of Paleo-Indians in Siberia
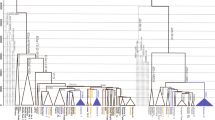
As shown in Fig. 2, Y-chromosome C2-M217 has a number of sub-lineages and the split time between C2-P39, C2-B77, and C2-F1756 was ~14.27 kya (95% CI = 13.35–15.11 kya). The sub-lineage C2-B77 was assigned to a Koryaks sample [35]. Samples of sub-lineage C2-F1756 (DYS448del) have mainly been found in Mongolic-speaking and Turkic-speaking populations [46]. We observed a continuous differentiation of C2-M217 between 15.3 kya and 14.3 kya (Fig. 2, Figure S1, and Table S5). Differentiation during this period gave rise to all sub-lineages of C2-M217 in Siberia and America, including C2-M48, C2-F1918 (also known as the “Star cluster” [47]), C2-F1756 (DYS448del), C2-P39, and three other minor lineages.
The divergence time between Y-chromosome Q1-B143 and Q1-M120 was about 15.25 kya (95% CI = 13.86–16.86 kya). The Y-chromosome of the Saqqaq male belonged to lineage Q1-B143, a predominant lineage in the Koryak population [3, 35]. In a previous study, Y-chromosome Q1-NWT01 (the ancestral lineage of B143 and M120) was also found at high frequencies in Eskimo-Aleut populations, but was absent from Native American populations [16, 17]. It is highly possible that Q1-NWT01 samples in Eskimo-Aleut populations also belong to lineage Q1-B143. However, owing to the low sequence coverage of the Saqqaq male, we were unable to determine the split time between the Saqqaq male and other Koryak samples. An ancient DNA analysis indicated that the lineage Q1-B143 may be related to the human expansion later than 6 kya near the Arctic Circle [3]. The most closely related lineage of Q1-B143 in Eurasian populations is Q1-M120, a frequently observed lineage in East Asia [48].
The Y-chromosome Q1-Z780 lineage is exclusively found in Native American populations [14, 15]. It is represented in ancient DNA samples by Anzick-1. Previous studies have linked the Y-chromosome of Anzick-1 to the greater Q-M242 and Q1-Z780 founding lineages in the Americas [8, 49]. We find in tree analysis that the Anzick-1 boy belonged to the Q-FGC47532, sub-lineage of Q1-Z780 (see Figure S1 and Table S4). The most recent common ancestor of all available Q1-Z780 samples was dated to ~15.00 kya (95% CI = 14.12–15.88 kya). The Anzick-1 boy shared SNPs Y32176 (hg19: 21559812, T>A) and FGC47532 (hg19: 16071882, T>C) with two living Native Americans (FDY-005 and FDY-014) from Mexico. These shared SNPs confirmed the close relationship between the ancient population of the Anzick-1 boy and modern Native Americans.
The discovery of Q1-L804 provides insight into the origin of Q1-M3, the predominant paternal lineage in Native American populations [14]. To date, most Q1-L804 samples have been found by personal DNA testing, done by citizen science testers, at commercial institutes like Family Tree DNA; an exception is Eng-hgQ-1 reported by Hallast et al.[50]. According to data obtained from www.yfull.com, a citizen science website, samples of lineage Q1-L804 are restricted to Northwest Europe. The split time between Q1-L804 and Q1-M3 was ~16.29 kya (95% CI = 15.33–17.25 kya). Additionally, the split time between Q1-M1107 and Q1-L330, a predominant paternal lineage in Altai populations, was about 17.24 kya (95% CI = 16.25–18.22 kya). As proposed in previous studies [20], the lineage Q1-M3 underwent a successful expansion in the American continent (Figure S1). The most recent common ancestor of all available Q1-M3 samples was dated to ~14.58 kya (95% CI = 13.76–15.37, Figure S1 and Table S5).
Discussion
We identified a number of Y-chromosome lineages in Eurasian populations that are closely related to the founding paternal lineages of American aborigines, including C2-P39, Q1-Z780, and Q1-M3. The lineage C2-P39 formed at ~14.3 kya, while the ages of the most recent common ancestor of Q1-Z780 and Q1-M3 were ~15.0 kya and 14.6 kya, respectively. We also described a large number of newly defined subclades and variants (Table S4). Genotyping these newly discovered variants for large samples of Siberia populations and American aborigines will help clarify the demographic history of these populations.
Differentiation and diffusion in Paleolithic Siberia
Based on the phylogenetic analyses and the current distributions of relative sub-lineages, we propose that the prehistoric population differentiation in Siberia after the LGM (post-LGM) provided the genetic basis for the emergence of the Paleo-Indian, American aborigine, population. According to the phylogenetic tree of Y-chromosome haplogroup C2-M217 (Fig. 2 and Figure S1), eight sub-lineages emerged in a short period between 15.3 kya and 14.3 kya (Table S5). Within these sub-lineages, haplogroups C2-M48, C2-F1918, and C2-F1756 are predominant paternal lineages in modern Altaic-speaking populations [46, 51, 52]. Samples of haplogroups C2-F8535 and C2-P53.1 were found in two Turkic- and Mongolic-speaking minorities in China (Table S1). Both archeological and genetic data suggest that Altaic-speaking populations are results of population expansion in the past several thousand years in the Altai Mountain, Mongolia Plateau, and Amur River region [51,52,53,54]. By contrast, three other sub-lineages, C2-B79, C2-B77, and C2-P39, appear only in Koryaks and Native Americans [16, 35]. The latitude of the Altai Mountain, the Mongolia Plateau, and Amur River region are much lower than that of Beringia, where the ancestors of Native Americans finally separated from their close relatives in Siberia. Therefore, the phylogeographic patterns of sub-lineages of C2-M217 in this study reveal a major splitting event between populations in a lower latitude region of Siberia and ancestors of Koryaks and Native Americans during the post-LGM period.
The sub-lineages of the Y-chromosome Q-M242 haplogroup were found in populations throughout the Eurasia continent. According to available data, the Q1-L804 lineage is exclusively found in Northwest Europe, while Q1-M120 is primarily restricted to East Asia [48]. Additionally, the lineage Q1-L330 is the predominant paternal lineage in Altai, Tuva, and Kets in South Siberia [34,35,36, 55]. A number of Q1-M242 samples have also been found in ancient remains from South Siberia and adjacent regions [56, 57]. Other sub-lineages of Q-M242 are scattered widely in different geographic regions of Eurasia, including Q1-L275, Q1-M25, and Q1-Y2659 [14, 35, 37, 58]. Additionally, the Y-chromosome of a 6000–5100 BCE sample (I4550) from Zvejnieki, Latvia has been identified as Q1-L56 [59]. These findings suggest that the sub-lineages of Q-M242 started to diffuse throughout Eurasia in a very ancient period.
Emergence of Paleo-Indian populations
The revised phylogenetic tree of Y-chromosome haplogroup Q-M242 in this study provides clues regarding the origin of Native American lineages Q1-M3 and Q1-Z780 (Fig. 3). According to our estimates, haplogroup Q1-L54 expanded rapidly between 17.2 kya and 15.0 kya and finally gave rise to two major founding paternal lineages of Native American populations, known as Q1-Z780 and Q1-M3. Ancient DNA studies indicate that the early population in South Siberia, represented by MA1 genomes, had a genetic influence on both modern western European and Native American populations [7]. Therefore, we conclude that the accumulated diversity of sub-lineages of Q-M242 before 15.3 kya resulted from the in situ differentiation of Q-M242 in Central Eurasia and South Siberia since the Paleolithic Age, and the appearance of the Paleo-Indian population is part of the great human diffusion throughout the Eurasia after the Last Glacial Maximum.
It is also interesting to note the strong bottleneck effect during the migration of Paleo-Indian populations toward Beringia and America (Fig. 3). Among the eight observed sub-lineages of C2-M217 before 15 kya in Siberia, only three sub-lineages (C2-B79, C2-B77, and C2-P39) were involved in the migration toward Beringia and only one (C2b-P39) ultimately reached the America continent. Among the ten observed sub-lineages of Q-M242 before 15 kya in Siberia, only three sub-lineages (Q1-B143, Q1-Z780, and Q1-M3) were involved in the migration toward Beringia and only two (Q1-Z780 and Q1-M3) reached the American continent before 14 kya.
Age of entry into America
The age of the split between founding Y-chromosome lineages of Native Americans (C2-P39, Q1-Z780, and Q1-M3) and their most closely related lineages in Eurasia can be considered the upper time limit of the time of entry into America. The observed expansion of unique paternal lineages of Native American lineages (Q1-Z780 and Q1-M3 in this study) can be considered the lower time limit for the founding Paleo-Indian population, either before or after the entry into America. In general, our Y-chromosome analysis provides a narrow timeframe (~15.3–14.3 kya) for the upper limit of human entry into the American continent. The divergence time between Q1-B143 and Q1-M120 (~15.25 kya) was also consistent with this differentiation pattern. Our calculations are consistent with the expansion time of Native American mtDNA lineages [28, 60] and available archeological evidence [23,24,25].
A Y-chromosome perspective on the “Beringian standstill hypothesis”
The “Beringian standstill hypothesis” argues that the ancestral population of Native Americans reached the Beringia region before 18 kya (or up to 30 kya) and experienced long-term isolation before dispersal into America [5, 6, 30,31,32, 61]. This hypothesis is largely based on the diversity of founding maternal mtDNA lineages of Native Americans [5, 6]. Additionally, remains from the Yana Rhinoceros Horn site in Northeastern Siberia (~32 kya) [5] and Bluefish Caves in Yukon Territory of Canada (~28 kya) [32] were thought to support this hypothesis. Nevertheless, it is worth noting that there is a gap of more than 13,000 years between the dates of these two sites and the earliest known remains related to Native Americans after 15 kya [23,24,25]. As discussed in the Introduction section, the “Beringian standstill hypothesis” is comprised of two parts, aka the “Long-term Beringian standstill model” and the “Short-term Beringian standstill model” [5, 6, 30,31,32, 61].
Recently, a new ancient DNA research published by Moreno-Mayar et al.[11] continues to support the “Long-term Beringian standstill model”. The authors concluded that the DNA results support a single ancestor group of Native Americans and the Beringian ‘standstill model’. However, the authors proposed another scenario which suggests that all major genetic events of their autosomal DNA “Native American clade” before 18 kya happened in northeast Asia, rather than the Beringia region [11]. The authors also mentioned that “scenario 1 best fits the archeological and Paleoecological evidence, as the earliest securely dated sites in Beringia are no older than around 15–14 kya, and the LGM cold period is unlikely to be associated with northward-expanding populations” [11]. Nevertheless, the authors abandoned this scenario in their discussion and prefer the “Long-term Beringian standstill model” although they admit to reservations.
Y-chromosome sequences data in this study tends to refute the hypothesis that the ancestral group of Native American populations arrived in Beringia before 18 kya and remained isolated there for 3–9 thousand years [62]. The split time between the founding paternal lineages of Native Americans (C2-P39, Q1-Z780, and Q1-M3) and their most closely related lineages in Siberia is between 17.2 kya and 14.3 kya (Fig. 2). More importantly, all Y-chromosome differentiation events likely took place in low latitude regions of Siberia. Thus, our conclusions prefer the “Short-term Beringian standstill model” and concur that the ancestor group of Native American arrived at Beringia after the LGM (aka. after 18 kya) and stayed there for a short period before expanding in the Americas at about 15,000 years ago [23,24,25].
Alternatively, we propose several hypotheses which may fulfill the gap between ancient genomes studies and our Y-chromosome study.
(1) As estimated by Moreno-Mayar’s paper [11], the gene flow from ancient north Eurasians into all Native Americans happened 25–20 kya. The ancient north Eurasian population was represented by Mal’ta Boy (~24 kya) near Baikal Lake and Afontova Gora-2 near Yenisei River [7]. Therefore, the living area of the Native American founding population during this period should be close to that of ancient north Eurasians, aka the Southern region of Siberia. It is unreasonable to deduce that this kind of gene flow happened in the Beringia region.
(2) The period between 20 kya (median of 22–18.1 kya) and 16 kya (median of 17.5–14.5 kya) estimated by Moreno-Mayar’s paper [11] may corresponds to the long-distance migration of the Native American founding population from the Southern region of Siberia to the Alaskan part of Beringia. Special branches of mtDNA lineages, Y-chromosome lineages, and special components in the autosomal gene pool may have emerged during this period. The age of Dyuktai culture (17–13 kya) in Yakutia Republic fits with the period of northward migration we proposed [63]. On the other hand, the earliest securely dated sites in Beringia are no older than around 15–14 kya [23,24,25].
(3) The internal expansion times of Y-chromosome Q1-Z780 and of Q1-M3 in this study are <15 ka. These estimates can be considered as the first phase of expansion of the Native American founding population on the American continent. Thus, our Y-chromosome results are consistent with the description of this period in Moreno-Mayar’s paper [11] and with existing archeological evidence [23,24,25].
(4) It is highly possible that the Native American founding populations before 15 ka were heterogeneous [11] and contained a series of small families-clans-groups which had different origins and were somehow isolated from each other. Even though there may have been continuous gene flow between these small groups, they had not become a completely homogeneous population group. As we can see from evidence of Y-chromosome, there are at least four founding paternal lineages for Native American (Q1-M3, Q1-Z780, Q1-B143, and C2-P39). These founding lineages are of different origins and separated from each other for 17–50 ky. The current calculation mode of ancient genomes studies may have combined several admixture events to a single one and thus caused some bias [10, 11]. Due to higher chance of admixture, the full genomes and mtDNA data cannot provide such a clear differentiation scenario for the Native American founding populations. Also, given the available evidence of the Y-chromosome, there may be no common and homogeneous ancestor group for all modern Native American populations.
(5) The observed diversity in the maternal and paternal gene pool of Paleo-Indians may have been accumulated during their migration from South Siberia toward Beringia, rather than during long-term isolation in Beringia. Previous studies also suggest that the age of the most recent common ancestor of mtDNA lineages in human population tends to be older than that of the Y-chromosome lineages in the same population [22, 35, 64]. Therefore, we conclude that the age of founding mtDNA lineages of Native Americans [5, 6] cannot be treated as solid evidence in support of the long-term model of the Beringian standstill hypothesis.
However, we cannot exclude the possibility that there is an ancestor group who has left autosome gene traces, but not a Y-chromosome trace, in modern Native American populations. This ancestor group, as proposed by Moreno-Mayar’s paper [11] and other scholars [7,8,9,10], could have reached Beringia during or before the Last Glacial Maximum (LGM) (dated to ca. 18,000–24,000 cal BP). However, proposals like this lack support in currently available archeological evidence and the known trend of human migration during the LGM [23,24,25, 29, 31, 32]. The earliest securely dated sites in Beringia are no older than around 15–14 kya [23,24,25]. More solid archeological evidence and ancient DNA analyses are needed to qualify the “Long-term Beringian standstill model” and verify our deduction in this study.
The Solutrean hypothesis
The Solutrean hypothesis suggests that Upper Paleolithic Solutrean culture in Europe is ancestral to Clovis culture, the first culture of modern human in the American continent [65]. The restricted distribution of Y-chromosome lineage Q1-L804 in Northwest Europe might support the Solutrean hypothesis from the Y-chromosome perspective. However, recent studies of ancient DNA suggested that ancient northern Eurasians, represented by 24,000-year-old MA1, contributed substantially to the gene pool of both Paleolithic human remains in America and modern American aborigines [7,8,9]. We demonstrated that Y-chromosome haplogroups Q-M242 and C2-M217 experienced a significant expansion in Siberia during the post-LGM period and some of the newly emerging sub-lineages migrated toward Beringia and America. Y-chromosome lineage Q1-L804 in Northwest Europe may be a relic of a prehistoric migration from South Siberia or descendants of an unknown male from Asia in recent history. More ancient DNA analyses from remains of Solutrean culture are needed to falsify or verify the direct connection between ancient Solutrean and Paleo-Indian populations.
In conclusion, our genetic study provides insights into the paternal founding lineages for Native Americans and puts forward a new theory for the origin of Native Americans. We determined the split time between founding lineages of Native Americans and their most closely related lineages in Eurasia, especially those from Siberia. We have proposed that continuous population differentiation in Siberia during the post-LGM period eventually gave rise to Paleo-Indian population, which migrated toward Beringia and eventually arrived at the American continent. Additional sequences of samples from Siberia and ancient DNA studies will help clarify the detailed demographic history of Siberian populations and the Native American founding populations in the future.
References
Greenberg JH, CGT II, Zegura SL, et al. The Settlement of the Americas: a comparison of the linguistic, dental, and genetic evidence [and comments and reply]. Curr Anthropol. 1986;27:477–97.
Harritt RK. Paleo-eskimo beginnings in North America: a new discovery at Kuzitrin lake, Alaska. Études/Inuit/Studies 1998;22:61–81.
Rasmussen M, Li Y, Lindgreen S, et al. Ancient human genome sequence of an extinct Palaeo-Eskimo. Nature. 2010;463:757–62.
Perego UA, Achilli A, Angerhofer N, et al. Distinctive Paleo-Indian migration routes from Beringia marked by two rare mtDNA haplogroups. Curr Biol. 2009;19:1–8.
Tamm E, Kivisild T, Reidla M, et al. Beringian standstill and spread of Native American founders. PLoS ONE. 2007;2:e829.
Mulligan CJ, Kitchen A, Miyamoto MM. Updated three-stage model for the peopling of the Americas. PLoS ONE. 2008;3:e3199.
Raghavan M, Skoglund P, Graf KE, et al. Upper Palaeolithic Siberian genome reveals dual ancestry of Native Americans. Nature. 2014;505:87–91.
Rasmussen M, Anzick SL, Waters MR, et al. The genome of a Late Pleistocene human from a Clovis burial site in western Montana. Nature. 2014;506:225–9.
Rasmussen M, Sikora M, Albrechtsen A, et al. The ancestry and affiliations of Kennewick Man. Nature. 2015;523:455–8.
Raghavan M, Steinrucken M, Harris K, et al. Genomic evidence for the Pleistocene and recent population history of Native Americans. Science. 2015;349:aab3884.
Moreno-Mayar JV, Potter BA, Vinner L, et al. Terminal Pleistocene Alaskan genome reveals first founding population of Native Americans. Nature. 2018;553:203–7.
Lell JT, Sukernik RI, Starikovskaya YB, et al. The dual origin and Siberian affinities of Native American Y chromosomes. Am J Hum Genet. 2002;70:192–206.
Zegura SL, Karafet TM, Zhivotovsky LA, Hammer MF. High-resolution SNPs and microsatellite haplotypes point to a single, recent entry of Native American Y chromosomes into the Americas. Mol Biol Evol. 2004;21:164–75.
Poznik GD, Xue Y, Mendez FL, et al. Punctuated bursts in human male demography inferred from 1,244 worldwide Y-chromosome sequences. Nat Genet. 2016;48:593–9.
Lippold S, Xu H, Ko A, et al. Human paternal and maternal demographic histories: insights from high-resolution Y chromosome and mtDNA sequences. Investig Genet. 2014;5:13.
Dulik MC, Owings AC, Gaieski JB, et al. Y-chromosome analysis reveals genetic divergence and new founding native lineages in Athapaskan- and Eskimoan-speaking populations. Proc Natl Acad Sci USA. 2012;109:8471–6.
Olofsson JK, Pereira V, Borsting C, Morling N. Peopling of the North Circumpolar Region--insights from Y chromosome STR and SNP typing of Greenlanders. PLoS ONE. 2015;10:e0116573.
Mezzavilla M, Geppert M, Tyler-Smith C, Roewer L, Xue Y. Insights into the origin of rare haplogroup C3* Y chromosomes in South America from high-density autosomal SNP genotyping. Forensic Sci Int Genet. 2015;15:115–20.
Roewer L, Nothnagel M, Gusmao L, et al. Continent-wide decoupling of Y-chromosomal genetic variation from language and geography in native South Americans. PLoS Genet. 2013;9:e1003460.
Battaglia V, Grugni V, Perego UA, et al. The first peopling of South America: new evidence from Y-chromosome haplogroup Q. PLoS ONE. 2013;8:e71390.
Cann RL, Stoneking M, Wilson AC. Mitochondrial DNA and human evolution. Nature. 1987;325:31–6.
Poznik GD, Henn BM, Yee MC, et al. Sequencing Y chromosomes resolves discrepancy in time to common ancestor of males versus females. Science. 2013;341:562–5.
Waters MR, Stafford TW Jr. Redefining the age of Clovis: implications for the peopling of the Americas. Science. 2007;315:1122–6.
Goebel T, Waters MR, O’Rourke DH. The late Pleistocene dispersal of modern humans in the Americas. Science. 2008;319:1497–502.
Waters MR, Stafford TW Jr., McDonald HG, et al. Pre-Clovis mastodon hunting 13,800 years ago at the Manis site, Washington. Science. 2011;334:351–3.
Kivisild T. The study of human Y chromosome variation through ancient DNA. Hum Genet. 2017;136:529–46.
Fiedel SJ. The Anzick genome proves Clovis is first, after all. Quat Int. 2017;444:4–9.
Llamas B, Fehren-Schmitz L, Valverde G, et al. Ancient mitochondrial DNA provides high-resolution time scale of the peopling of the Americas. Sci Adv. 2016;2:e1501385.
Dillehay TD, Ocampo C, Saavedra J, et al. New archaeological evidence for an early human presence at Monte Verde, Chile. PLoS ONE. 2015;10:e0141923.
Hoffecker JF, Elias SA, O’Rourke DH, Scott GR, Bigelow NH. Beringia and the global dispersal of modern humans. Evol Anthropol. 2016;25:64–78.
Borrero LA. Ambiguity and debates on the early peopling of South America. PaleoAmerica. 2016;2:11–21.
Bourgeon L, Burke A, Higham T. Earliest human presence in North America dated to the last glacial maximum: new radiocarbon dates from Bluefish Caves, Canada. PLoS ONE. 2017;12:e0169486.
Kemp BM, Malhi RS, McDonough J, et al. Genetic analysis of early holocene skeletal remains from Alaska and its implications for the settlement of the Americas. Am J Phys Anthropol. 2007;132:605–21.
Dulik MC, Zhadanov SI, Osipova LP, et al. Mitochondrial DNA and Y chromosome variation provides evidence for a recent common ancestry between Native Americans and Indigenous Altaians. Am J Hum Genet. 2012;90:229–46.
Karmin M, Saag L, Vicente M, et al. A recent bottleneck of Y chromosome diversity coincides with a global change in culture. Genome Res. 2015;25:459–66.
Flegontov P, Changmai P, Zidkova A, et al. Genomic study of the Ket: a Paleo-Eskimo-related ethnic group with significant ancient North Eurasian ancestry. Sci Rep. 2016;6:20768.
Huang YZ, Pamjav H, Flegontov P, et al. Dispersals of the Siberian Y-chromosome haplogroup Q in Eurasia. Mol Genet Genom. 2018;293:107–17.
Mallick S, Li H, Lipson M, et al. The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature. 2016;538:201–6.
Lu D, Lou H, Yuan K, et al. Ancestral origins and genetic history of Tibetan highlanders. Am J Hum Genet. 2016;99:580–94.
Wong LP, Ong RT, Poh WT, et al. Deep whole-genome sequencing of 100 southeast Asian Malays. Am J Hum Genet. 2013;92:52–66.
Balanovsky O, Gurianov V, Zaporozhchenko V, et al. Phylogeography of human Y-chromosome haplogroup Q3-L275 from an academic/citizen science collaboration. BMC Evol Biol. 2017;17:18.
Bouckaert R, Heled J, Kuhnert D, et al. BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput Biol. 2014;10:e1003537.
Wei LH, Yan S, Yu G, et al. Genetic trail for the early migrations of Aisin Gioro, the imperial house of the Qing dynasty. J Hum Genet. 2016;62:407–11.
Wang Y, Song F, Zhu J, et al. GSA: Genome Sequence Archive. Genom, Proteom Bioinform. 2017;15:14–8.
BIGDC. The BIG Data Center: from deposition to integration to translation. Nucleic Acids Res. 2017;45:D18–24. Members
Wei LH, Huang YZ, Yan S, et al. Phylogeny of Y-chromosome haplogroup C3b-F1756, an important paternal lineage in Altaic-speaking populations. J Hum Genet. 2017;62:915-18.
Zerjal T, Xue Y, Bertorelle G, et al. The genetic legacy of the Mongols. Am J Hum Genet. 2003;72:717–21.
Zhao YB, Zhang Y, Li HJ, Cui YQ, Zhu H, Zhou H. Ancient DNA evidence reveals that the Y chromosome haplogroup Q1a1 admixed into the Han Chinese 3,000 years ago. Am J Hum Biol. 2014;26:813–21.
Adamov D, Guryanov V, Karzhavin S, Tagankin V, Urasin V. Defining a new rate constant for Y-chromosome SNPs based on full sequencing data. Russ J Genet Geneal. 2015;7:68–89.
Hallast P, Batini C, Zadik D, et al. The Y-chromosome tree bursts into leaf: 13,000 high-confidence SNPs covering the majority of known clades. Mol Biol Evol. 2015;32:661–73.
Malyarchuk B, Derenko M, Denisova G, et al. Phylogeography of the Y-chromosome haplogroup C in northern Eurasia. Ann Hum Genet. 2010;74:539–46.
Wei LH, Yan S, Lu Y, et al. Whole-sequence analysis indicates that the Y chromosome C2*-Star Cluster traces back to ordinary Mongols, rather than Genghis Khan. Eur J Human Genet. 2018;26:230–7.
Sinor D. The Cambridge History of Early Inner Asia. ume I. Cambridge: Cambridge University Press; 1990.
Balaresque P, Poulet N, Cussat-Blanc S, et al. Y-chromosome descent clusters and male differential reproductive success: young lineage expansions dominate Asian pastoral nomadic populations. Eur J Human Genet. 2015;23:1413–22.
Karafet TM, Osipova LP, Gubina MA, Posukh OL, Zegura SL, Hammer MF. High levels of Y-chromosome differentiation among native Siberian populations and the genetic signature of a boreal hunter-gatherer way of life. Hum Biol. 2002;74:761–89.
Hollard C, Keyser C, Giscard PH, et al. Strong genetic admixture in the Altai at the Middle Bronze Age revealed by uniparental and ancestry informative markers. Forensic Sci Int Genet. 2014;12:199–207.
Allentoft ME, Sikora M, Sjogren KG, et al. Population genomics of Bronze Age Eurasia. Nature. 2015;522:167–72.
Malyarchuk B, Derenko M, Denisova G, et al. Ancient links between Siberians and Native Americans revealed by subtyping the Y chromosome haplogroup Q1a. J Hum Genet. 2011;56:583–8.
Mathieson I, Alpaslan Roodenberg S, Posth C, et al. The Genomic History of SoutheasternEurope. bioRxiv. Nature. 2018;555:197–203.
Soares P, Ermini L, Thomson N, et al. Correcting for purifying selection: an improved human mitochondrial molecular clock. Am J Hum Genet. 2009;84:740–59.
Mulligan CJ, Szathmáry EJE. The peopling of the Americas and the origin of the Beringian occupation model. Am J Phys Anthropol. 2017;162:403–8.
Graf KE, Buvit I. Human dispersal from Siberia to Beringia: assessing a Beringian standstill in light of the archaeological evidence. Curr Anthropol. 2017;58:S583–603.
Yi S, Clark G, Aigner JS, et al. The “Dyuktai Culture” and New World Origins [and Comments and Reply]. Curr Anthropol. 1985;26:1–20.
Cruciani F, Trombetta B, Massaia A, Destro-Bisol G, Sellitto D, Scozzari R. A revised root for the human Y chromosomal phylogenetic tree: the origin of patrilineal diversity in Africa. Am J Hum Genet. 2011;88:814–8.
Bradley B, Stanford D. The North Atlantic ice-edge corridor: a possible Palaeolithic route to the New World. World Archaeol. 2004;36:459–78.
Acknowledgements
We are grateful to all sample donors. We thank the Family Tree DNA lab team including Brent Manning and Michael Sager and the Yfull team for their help and comments. The administrators of the haplogroup Q-M242 and the Nordic Q citizen science projects helped in collecting and analyzing these data. This study was supported by the National Natural Science Foundation of China (91731303 to S.X. and H.L., 31222030 and 31671297 to H.L., 31401060 to Y.S., 31271338 to L.J., 31501011 to Y.L., 31525014, 31771388 and 31711530221 to S.X.), MOE Scientific Research Project (113022A to H.L.), Shanghai Shuguang Project (14SG05 to H.L.), Ministry of Science and Technology of China (MOST) (2016YFC0900300 to H.L.). S.X. acknowledges financial support from the Strategic Priority Research Program (XDB13040100) and Key Research Program of Frontier Sciences (QYZDJ-SSW-SYS009) of the Chinese Academy of Sciences (CAS), the Program of Shanghai Academic Research Leader (16XD1404700), and the National Key Research and Development Program (2016YFC0906403). L.-H.W. was supported by Future Scientists Project of China Scholarship Council. C.-C.W was supported by the Nanqiang Outstanding Young Talents Program of Xiamen University. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author contributions
L.-H.W., L.-X.W., Q.-S.W., and R.C. carried out the molecular genetic studies and drafted the manuscript. S.Y., Q.-S.W., R.C., V.G., S.W., A.B., A.O., Y.L., and C.Z. collected the samples. Q.-S.W., S.Y., Y.L., and C.Z. participated in the experiments. Y.-Z.H., V.G., A.B., A.O., C.-C.W., and L.J. participated in the statistical analysis. S.-H.X. and H.L. designed the study and revised the manuscript. All authors read and approved the final manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Wei, LH., Wang, LX., Wen, SQ. et al. Paternal origin of Paleo-Indians in Siberia: insights from Y-chromosome sequences. Eur J Hum Genet 26, 1687–1696 (2018). https://doi.org/10.1038/s41431-018-0211-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-018-0211-6
This article is cited by
-
Phylogenetic analysis of the Y-chromosome haplogroup C2b-F1067, a dominant paternal lineage in Eastern Eurasia
Journal of Human Genetics (2020)
