
Abstract
Trio based whole exome sequencing via the Deciphering Developmental Disorders (DDD) study has identified three individuals with de novo frameshift variants in the Suppressor of Variegation, Enhancer of Zeste, and Trithorax (SET) gene. Variants in the SET gene have not previously been recognised to be associated with human developmental disorders. Here we report detailed phenotypic information and propose that SET is a new Intellectual Disability/Developmental Delay (ID/DD) gene.
Similar content being viewed by others


Introduction
De novo pathogenic (or likely pathogenic) variants are an important cause of moderate and severe intellectual disability (ID). The Deciphering Developmental Disorders (DDD) study [1] recruited nearly 14,000 patients with developmental delay and other features. To date 14 novel ID genes have been identified through the DDD study [2]. In other published larger scale exome sequencing projects, there is only one reported case of a predicted function-affecting variant in SET in association with ID/DD. [3]
SET (Suppressor of Variegation, Enhancer of Zeste, and Trithorax) codes for a phosphoprotein which is recognised to be important in a various nuclear functions including apoptosis, transcription, nucleosome assembly and histone chaperoning [4]. It is widely expressed in human and mouse tissues and is located in the cell nucleus and also found in the endoplasmic reticulum. SET is thought to play a role in mitosis by blocking cyclin B-CDK1 [5]. SET protein forms a complex with Prothymosin (alpha), a histone H1-binding protein, and thus has a role in the decondensation of compacted chromatin fibres [6, 7] and therefore in regulation of gene expression. Isoform 2 anti-apoptotic activity is mediated by inhibition of the GZMA-activated DNase, NME1. In the course of cytotoxic T-lymphocyte (CTL)-induced apoptosis, GZMA cleaves SET, disrupting its binding to NME1 and releasing NME1 inhibition. Isoform 1 and isoform 2 are potent inhibitors of protein phosphatase 2 A. Isoforms 1 and 2 also inhibit EP300/CREBBP and PCAF-mediated acetylation of histones (HAT) and nucleosomes, most probably by masking the accessibility of lysines to the acetylases. The predominant target for inhibition is histone H4. HAT inhibition leads to silencing of HAT-dependent transcription and prevents active demethylation of DNA [4].
Here we describe the cases and summarise key features and phenotypic similarities, as supporting evidence for SET as a gene important in ID.
Methods
The three individuals were recruited via UK NHS Regional Genetics Services to the DDD study [1]. DDD recruited via Genetic centres throughout the UK and Republic of Ireland. Using microarray and whole exome sequencing, DDD aims to provide diagnoses for children and adults with previous undiagnosed developmental disorders.
13,632 families were recruited. Exome sequencing was performed on the affected individual and their parents, as previously described [8]. The affected individuals also had high‐resolution analysis for copy number abnormalities using array‐based comparative genomic hybridisation (aCGH). Potentially causative de novo variants were identified using the DeNovoGear software [9]. Targeted Sanger sequencing was then used to validate these putative pathogenic variants. Data for these are available via the publically accessible DECIPHER database (decipher.sanger.ac.uk, patient IDs 259410, 263897, and 265149), which provides positional genomic information together with phenotype descriptive terms. This study makes use of data generated by the DECIPHER community. A full list of centres who contributed to the generation of the data is available from http://decipher.sanger.ac.uk and via email from decipher@sanger.ac.uk. Funding for the project was provided by the Wellcome Trust.
Consent was obtained for publishing publication of photographs from legal guardians.
An additional case was identified from a Canadian exome sequencing study. Hamdan et al. performed exome sequencing in 41 trios consisting of probands with moderate to severe ID and their unaffected parents [3]. They identified 12 de novo variants, proposed to affect function, in genes not previously associated with ID. One of these was a de novo deletion in SET resulting in the creation of a premature stop codon. This case has been included as patient 4.
Phenotypic features were collected from responsible clinicians or from ‘De Novo Mutations in Moderate or Severe Intellectual Disability’ by Hamdan et al. for patient 4. Growth parameter percentiles and z scores were calculated from the UK WHO growth charts [10].
Results
Clinical features
The patients’ ages were between 10 and 17 years at diagnosis, with three males and one female. Table 1 shows a summary of the clinical features for each case.
Craniofacial
A range of mild dysmorphic features were reported in the the cases (Fig. 1).

Facial features of patients. Patient 1 at 2 years and 10 years (a, b); Patient 2 at 2 years, 4 years, 7 years 8 months and 17 years (c–f);Patient 3 at 9 years (g). Patient 1 has a depressed nasal bridge, hypertrichosis and synophrys. Patient 2 has a broad nasal base, anteverted nares, widely spaced teeth, smooth upper lip and mild occipital plagiocephaly. Patient 3 has a number of described features including narrowing of the bi-frontal regions, prominent and broad forehead, hypertelorism, striking blue eyes, flat nasal tip and thick lower lip vermillion
When the images of the three patients identified through DDD were reviewed by expert Dysmorphologists at the DDD Collaborators meeting, it was agreed that although there are similarities in facial appearance, these are not specific enough to make this an easily recognisable dysmorphic syndrome. All the patients have in common a wide mouth with thick lower lip vermillion, nose with a broad base and widely spaced teeth. No photographs or facial features are reported for the Hamdan et al. patient.
Growth
Birth weights of the cases varied between −1.603 and 0.790 SD. Patients 1 and 2, who have identical variants, had similar patterns of growth with a relatively low birth weight and height. Postnatal head size was varied between the cases from −3.896 SD in patient 4 at 9 years to 0.834 SD in patient 3 at the same age.
Development
All cases had delayed motor development with Patients 1 and 2 walking around their 3rd birthday and Patient 3 and 4 at around 2 years. They all had speech and language delay. First words noted between 24 months and 48 months. Patients 1–3 attended Special Schools, with patient 3 also spending some time in Mainstream education.
Behaviour
Behavioural and psychiatric problems were only reported in patient 2, who had significant problems with temper tantrums. As he got older, he displayed hyperphagia. As a 15 year old, he was diagnosed with schizophrenia. Patient 4 had attention deficit without hyperactivity.
Other features
Seizures were not reported in any case. 2 out of 3 were reported to have low tone in infancy. Patient 3 had changes on MRI at 1 year 10 months described as ‘consistent with periventricular high signal on the left side’ but this may be variant of normal. Patients 1, 2 and 3 are reported as having generalised joint laxity. Patient 1 has crowded and curly toes and a slightly hairy back. Patient 2 had a lower right tracheal bronchus, which was identified on bronchoscopy after investigations for chronic cough. Patient 3 has an area of increased pigmentation over his lower thoracic and lumber spine which has a leathery texture. He also has short 5th fingers with 5th finger clinodactaly and square finger tips. In both patients 1 and 2, a diagnosis of Williams’ syndrome was considered. Patient 3 had been investigated for Pitt Hopkins syndrome. No additional features were reported for patient 4.
Variants
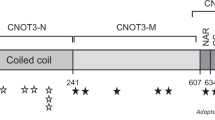
Through the DDD study, de novo frameshift variants were identified in three patients. In two unrelated patients identical variants were found (c.167_170delACAG p.(Arg57Leufs*10)). In the third patient the variant was c.459_460delCA p.(Lys154ArgfsTer6). These were reported in transcript NM_001122821.1, which corresponds to ENST 00000372692. These three cases were found from the 4323 families where analysis has been completed, giving a frequency of 0.069%. The Hamdan et al. case had a de novo deletion resulting in a premature stop codon in the same transcript (c.699_701del p(Tyr233*)). Patient 2 also has a chromosome 2q 35 deletion (0.20 to 0.33 Mb) which was found to be paternally inherited, present in other unaffected relatives and thought not to be significant. Fig. 2 provides a schematic representation of the protein with the position of the variants demonstrated.

Representation of the SET protein showing functional domains and approximate location of reported variants to date (adapted from DECIPHER, decipher.sanger.ac.uk [14])
Discussion
This collection of patients with de novo frameshift variants in SET all have similar patterns of delayed development and some similarities in facial appearance. The DDD study reported 14 genes achieving genome-wide significant statistical evidence without previous compelling evidence for association with DDs, of which SET was one such gene (p value 1.2 × 10−7) [2]. Bioinformatic data support the hypothesis that de novo variants in SET are disease causing. SET has a low haploinsufficiency score (2.03), [11] and a pLI score of 0.96 [12] suggesting that SET is extremely loss of function (LOF) intolerant.
GnomAD reports only 2 LOF variants in SET; a splice donor variant (c.112 + 1 G > A) and a frameshift variant (c.112delG) with an allele count of 1 in both cases and allele frequencies of 0.00003230 and 0.000006650 respectively [13].
DECIPHER reports 6 other variants (uncertain significance or not yet determined) in SET in addition to those described in this paper, 2 of which are predicted to be LOF variants (1 frameshift and 1 start_lost variant) [14]. Unfortunately the inheritance pattern is not known for either of these two additional LOF variants. In the case of the start_lost variant, the responding clinicians feel an alternative variant found is the more likely explanation for the phenotype. Of the remaining four variants (which are all missense variants) two of these are paternally inherited and two are missense variants of unknown inheritance and are reported in patients with one and two other variants respectively. Clinvar reports three somatic missense variants in SET only [15].
The DECIPHER database allows review of copy number variation at this locus [14]. Ten cases are reported with a loss including SET, varying in size from 703 kb to 4.13 Mb. five of these are de novo, three unknown inheritance and two paternally inherited. In six of the eight cases where phenotype features are reported, ID is included. Reviewing the genes deleted by these copy number losses, SET appears a good candidate gene to explain the phenotype based on haploinsufficiency and pLi scores. There are no other clear candidates within the Developmental Disorders Genotype-Phenotype Database genes (DDG2P genes) [2] to account for the ID phenotype associated with these copy number losses. The only other monoallelic DDG2P gene common to all these losses is associated with Early Infantile Encephalopathy (SPTAN1) and none of the copy number loss cases include seizures in the phenotype.
Variants in SET have not been widely recognised as a cause for developmental delay. SET was first isolated and characterised as an oncogene in 1992 [16]. Wang et al. used a proteomic screen to identify the oncoprotein SET as a major cellular factor that profoundly inhibits p53 transcriptional activity in unstressed cells and whose binding with p53 is dependent on C-terminal domain acetylation status [17]. The protein SET belongs to a family of acidic domain-containing proteins that interact with the lysine-rich domains of transcriptional regulators in an acetylation-dependent manner and inhibit their function [17].
There is evidence to suggest SET is required for both neuronal development and survival. Kim et al. demonstrated that the knockdown of SET/TAF-Iβ by si-RNA induces neuronal cell differentiation, thus implicating SET/TAF-Iβ as a negative regulator of neuronal differentiation [18]. SET protein also appears to be involved in neuronal survival through the neuronal apoptotic pathway which is up-regulated in Alzheimer’s disease [19].
The protein SET has been identified as an important binding partner of Microcephalin (MCPH1) [20]. MCPH1 and SET interact and participate in the regulation of chromosome condensation. Classically, in MCPH1 related microcephaly, premature chromosome condensation (PCC) is seen. Leung demonstrated that in knockdown of SET in mouse and human cell lines, the same PCC phenotype resulted, confirming that SET acts with MCPH1 in the regulation of chromosome condensation [20].
Hamden et al. suggested phenotypic similarities between cases with MCPH1 variants and the individual they describe with a de novo SET indel [3]. Interestingly, while the patient reported by Hamdan et al. did have congenital microcephaly, microcephaly was not present in our patients, who all had head size within the normal range, suggesting phenotypic heterogeneity. We have not looked for evidence of PCC in our patients.
Looking at the interactions of the SET protein, we can further appreciate how SET-related neurodevelopmental disorder may have similarities with previously described syndromes. Isoform 2 of SET protein is a component of the SET complex, composed of at least ANP32A, APEX1, HMGB2, NME1, SET and TREX1, but not NME2 or TREX2. Within this complex, SET protein directly interacts with ANP32A, NME1, HMGB2 and TREX1. SET protein also interacts with APBB1, CHTOP, SETBP1, SGO1 [4]. Of these genes, so far SETBP1 and TREX1 are the only 2 which have been linked to ID. De novo variants in SETBP1 were first identified in 12 patients with Schinzel-Giedion syndrome [21]. The variants were clustered to a highly conserved 11-bp exonic region, suggesting a gain-of-function or dominant negative effect. Haploinsufficiency or LOF variants in SETBP1 result in a different phenotype characterised by a less severe degree of learning disability without the typical dysmorphic features of Schinzel-Giedion [22]. There are overlapping phenotypic features between LOF SETBP1 variants and the patients with SET frameshift variants, but the absence of distinctive features in either group makes it difficult to draw any conclusions other than that LOF variants in both genes lead to ID. TREX1 variants have been seen in association with Aicardi-Goutieres syndrome, which presents with a profound ID, microcephaly and a period of encephalopathy and then regression of development. ANP32A is known to play an important role in brain development [23, 24]. Although there are as yet no reports of variants in ANP32A associated with ID there is a single research variant in the DECIPHER database in a patient with ID [14].
In summary, our case series describes phenotypic similarities between cases of de novo heterozygous frameshift SET variants. There is evidence to support the assumption that LOF variants in SET can cause ID. SET related neurodevelopmental disorder adds to the already extensive list of disorders associated with defects in chromatin remodelling. The genes involved in the SET complex and those interacting with SET should be a focus of further study as a potential cause of ID. Further cases are required for delineation, but this is unlikely to be a well-defined easily recognisable phenotype and strengthens the case for routine whole exome or genome sequencing in this patient group.
References
Firth HV, Wright CF. The Deciphering Developmental Disorders (DDD) study. Dev Med & Child Neurol. 2011;53:702–3.
Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature. 2017;542:433–8.
Hamdan FF, Srour M, Capo-Chichi J-M, Daoud H, Nassif C, Patry L, et al. De Novo Mutations in Moderate or Severe Intellectual Disability. PLoS Genet 2014;10:e1004772.
UNIPROT. http://www.uniprot.org/uniprot/Q01105. Accessed 1 Dec. 2016.
Canela N, Rodriguez-Vilarrupla A, Estanyol JM, et al. The SET protein regulates G2/M transition by modulating cyclin B-Cyclin-dependent kinase 1 Activity. J Biol Chem. 2003;278:1158–64.
Miyamoto S, Suzuki T, Muto S, et al. Positive and negative regulation of the cardiovascular transcription factor KLF5 by p300 and the oncogenic regulator SET through interaction and acetylation on the DNA-binding domain. Mol Cell Biol. 2003;23:8528–41.
Karetsoua Z, Martica G, Tavoularia S, et al. Prothymosin α associates with the oncoprotein SET and is involved in chromatin decondensation. FEBS Lett. 2004;577:496–500.
Wright CF, Firth HV, Study DDD, et al. Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. Lancet. 2015;385:1305–14.
Ramu A, Noordam MJ, Schwartz RS, et al. DeNovoGear: De novo indel and point mutation discovery and phasing. Nat Methods. 2013;10:985–7.
UK-WHO growth charts. www.rcpch.ac.uk/growthcharts.
Huang N, Lee I, Marcotte EM, Hurles ME. Characterising and predicting haploinsufficiency in the human genome. PLoS Genet. 2010;6:e1001154.
Firth H, Richards SM, Bevan P, et al. DECIPHER: database of chromosomal imbalance and phenotype in humans using ensembl resources. Am J Human Genet. 2009;84:524–33.
Lek M, Karczewski KJ, Exome Aggregation Consortium, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
DECIPHER. decipher.sanger.ac.uk. Accessed 20 Mar. 2018.
National Center for Biotechnology Information, Clinvar. www.ncbi.nlm.nih.gov/clinvar/. Accessed 12 April 2018.
von Lindern, M, van Baal, S, Wiegant, J, Raap, A, Hagemeijer, A, Grosveld, G, ‘Can,’ a putative oncogene associated with myeloid leukemogenesis, may be activated by fusion of its 3-prime half to different genes: characterization of the ‘set’ gene. Mol Cell Biol. 1992;12:3346–55.
Wang D, Kon N, Lasso G, et al. Acetylation-regulated interaction between p53 and SET reveals a widespread regulatory mode. Nature 2016;538:118–22.
Kim DW, KK, Kim JY, Lee KS, Seo SB. Negative regulation of neuronal cell differentiation by INHAT subunit SET/TAF-Iβ. Biochem Biophys Res Commun. 2010;3:419–25.
Madeira A, Pommet JM, Prochiantz A, Allinquant B. SET protein (TAF1beta, I2PP2A) is involved in neuronal apoptosis induced by an amyloid precursor protein cytoplasmic subdomain. FASEB J. 2005;19:1905–7.
Leung JW, Leitch A, Wood JL, et al. SET nuclear oncogene associates with microcephalin/MCPH1 and regulates chromosome condensation. J Biol Chem. 2011;286:21393–400.
Hoischen A, van Bon BW, Gilissen C, et al. De novo mutations of SETBP1 cause Schinzel-Giedion syndrome. Nat Genet. 2010;42:483–5.
Filges I, Shimojima K, Okamoto N, et al. Reduced expression by SETBP1 haploinsufficiency causes developmental and expressive language delay indicating a phenotype distinct from Schinzel-Giedion syndrome. J Med Genet. 2011;48:117–22.
Wang S, Wang Y, Lu Q, et al. The expression and distributions of ANP32A in the developing brain. Biomed Res Int. 2015;2015:8.
Chai G-S, Feng Q, Wang Z-H, et al. Downregulating ANP32A rescues synapse and memory loss via chromatin remodeling in Alzheimer model. Mol Neurodegener. 2017;12:34.
Acknowledgements
The authors thank the families for their participation. The DDD study presents independent research commissioned by the Health Innovation Challenge Fund (grant number HICF-1009-003), a parallel funding partnership between the Wellcome Trust and the Department of Health, and the Wellcome Trust Sanger Institute (grant numberWT098051). The views expressed in this publication are those of the author(s) and not necessarily those of the Wellcome Trust or the Department of Health. The study has UK Research Ethics Committee approval (10/H0305/83, granted by the Cambridge South REC, and GEN/284/12 granted by the Republic of Ireland REC). The research team acknowledges the support of the National Institute for Health Research, through the Comprehensive Clinical Research Network.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Richardson, R., Splitt, M., Newbury-Ecob, R. et al. SET de novo frameshift variants associated with developmental delay and intellectual disabilities. Eur J Hum Genet 26, 1306–1311 (2018). https://doi.org/10.1038/s41431-018-0199-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-018-0199-y
This article is cited by
-
Whole exome sequencing and transcriptome analysis in two unrelated patients with novel SET mutations
Journal of Human Genetics (2023)
-
Balanced SET levels favor the correct enhancer repertoire during cell fate acquisition
Nature Communications (2023)
