
Abstract
Orofacial development is a multifaceted process involving tightly regulated genetic signaling networks, that when perturbed, lead to orofacial abnormalities including cleft lip and/or cleft palate. We and others have shown an association between the cysteine-rich secretory protein LCCL domain containing 2 (CRISPLD2) gene and nonsyndromic cleft lip with or without cleft palate (NSCLP). Further, we demonstrated that knockdown of Crispld2 in zebrafish alters neural crest cell migration patterns resulting in abnormal jaw and palate development. In this study, we performed RNA profiling in zebrafish embryos and identified 249 differentially expressed genes following knockdown of Crispld2. In silico pathway analysis identified a network of seven genes previously implicated in orofacial development for which differential expression was validated in three of the seven genes (CASP8, FOS, and MMP2). Single nucleotide variant (SNV) genotyping of these three genes revealed significant associations between NSCLP and FOS/rs1046117 (GRCh38 chr14:g.75746690 T > C, p = 0.0005) in our nonHispanic white (NHW) families and MMP2/rs243836 (GRCh38 chr16:g.55534236 G > A; p = 0.002) in our Hispanic families. Nominal association was found between NSCLP and CASP8/rs3769825 (GRCh38 chr2:g.202111380 C > A; p < 0.007). Overtransmission of MMP2 haplotypes were identified in the Hispanic families (p < 0.002). Significant gene–gene interactions were identified for FOS-MMP2 in the NHW families and for CASP8-FOS in the NHW simplex family subgroup (p < 0.004). Additional in silico analysis revealed a novel gene regulatory network including five of these newly identified and 23 previously reported NSCLP genes. Our results demonstrate that animal models of orofacial clefting can be powerful tools to identify novel candidate genes and gene regulatory networks underlying NSCLP.
Similar content being viewed by others
Introduction
Normal craniofacial development results from the convergence and fusion of the facial and palatal processes and involves interactions between genes that regulate cell growth, proliferation, differentiation, epithelial-to-mesenchymal transition, and apoptosis [1,2,3]. The cranial processes are derived from neural crest cells that subsequently undergo migration and differentiation to form the maxillary, lateral nasal, and medial nasal processes, which fuse to form a continuous upper lip and palate [4,5,6]. Characterizing the normal biological functions of the genes that control the craniofacial complex, including their potential interactions, has led to important insights about the biologic mechanisms contributing to craniofacial anomalies, with the most frequent being nonsyndromic cleft lip and palate (NSCLP).
NSCLP is a common birth defect affecting one out of every 700 newborns and resulting in ~4000 new cases in the US and ~135,000 worldwide each year [7]. NSCLP requires surgical, dental, and speech interventions, which place significant financial and psychological burdens on these families. Therefore, identifying the genetic liability underlying this common birth defect is critical towards the development of prevention and better prediction strategies [1, 8]. The causes of NSCLP are multifactorial with both genetic and environmental factors contributing to the etiology [9,10,11,12]. Approximately 40 genes with moderate evidence for involvement in NSCLP have been identified by chromosomal, genome-wide association (GWAS), candidate gene, and animal model approaches [12,13,14,15].
Gene regulatory networks such as fibroblast growth factor (FGF) signaling, transforming growth factor beta (TGF-β) signaling, and wingless-type MMTV integration site (WNT) family genes, are known to be important in craniofacial development and genes within these networks have shown association with NSCLP. Two other gene networks have also recently been implicated in NSCLP, the first network composed by Pbx1, Wnt3, Wnt9b, Fgf8, and p63, which function to form the murine midface, and second network represented by the Irf6 krüppel-like factor (Klf) gene network that is expressed in the oral periderm of mice and zebrafish and regulates keratin expression [16, 17]. Variation in many of these network genes have previously been associated with orofacial clefting, thus supporting the use of a network-based approach to identify gene contributions to NSCLP [18,19,20,21,22,23,24].
We and others have shown an association between cysteine-rich secretory protein Limulus factor C-related region domain containing 2 (CRISPLD2) and NSCLP in ethnically diverse populations [25,26,27,28]. CRISPLD2, also known as late gestation lung 1 (Lgl1) in mice, is a member of the Cysteine-rich secretory proteins, Antigen 5 and Pathogenesis-related 1 (CAP) superfamily [25, 28, 29]. In mice, Crispld2/Lgl1 plays a critical role in fibroblast and epithelial cell migration and epithelial-to-mesenchymal transition and Crispld2/Lgl1 null mice are embryonic lethals, demonstrating that it is necessary for embryonic development [30]. In previous studies, we showed that CRISPLD2 is expressed in the developing murine craniofacies and zebrafish embryo heads, and that knockdown of Crispld2 in zebrafish alters neural crest cell migration and increases apoptosis resulting in jaw and palatal anomalies [28, 31, 32]. While these observations support a role for CRISPLD2 in craniofacial development, there is little information about interacting genes and gene networks that would further elucidate its role in NSCLP. In this study, we applied RNA-seq and in silico network approaches to identify genes that were (1) differentially expressed between wild type and Crispld2 morphant zebrafish and (2) previously known to play a role in craniofacial development. We also tested the association of single nucleotide variants (SNVs) in the identified genes with NSCLP in our nonHispanic white (NHW) and Hispanic multiplex and simplex families. We then applied in silico network approaches to our RNA-seq derived network and previously implicated NSCLP genes to identify a novel candidate NSCLP gene network.
Materials and methods
RNA-seq analysis
Zebrafish (Danio rerio) were raised and housed following standard techniques [33] and fertilized eggs were obtained through in-tank breeding. One-cell stage embryos were injected with 0.2 ng of crispld2 antisense morpholino (MO) targeting the ATG start site (TTGATGATTTCAGGCCC GGACTCTA), as previously described [31]. RT-PCR validation studies also utilized mismatch MO (TTcATcATTTgAGcCCCcGACTCTA). Fifty MO-injected, mismatch MO-injected, and uninjected embryos were collected at the 15-somite stage (16 h post fertilization, hpf) and the yolk was removed. The anterior third of each embryo was isolated, pooled into injected and uninjected samples, and total RNA was isolated. For RNA-seq analysis, four micrograms of total RNA from MO-injected and uninjected samples were used for comparative RNA-seq analysis using Cuffdiff (LC Sciences, Houston, TX, USA). The abundance, or fragments per kilobase of exon per million (FPKM) reads, and the dispersion were estimated using the negative binomial model, and differences between uninjected and MO-injected embryos were tested using a Student’s t-test. Transcripts were sorted by the q-value (False Discovery Rate adjusted p-value) with a cutoff of 0.05. RNA-seq data have been deposited in the ArrayExpress database at EMBL-EBI (www.ebi.ac.uk/arrayexpress) under accession number E-MTAB-6705.
In silico pathway analyses
Differentially expressed genes identified in RNA-seq analysis were subjected to pathway primary analysis using Ingenuity (Qiagen, San Francisco, CA, USA) [34]. Pubmatrix (NIH, Bethesda, MD, USA) was used to determine the craniofacial relevance and previous species-specific publications of the gene network using MESH words: “oral”, “cleft”, “lip”, “palate”, “facial”, “tooth”, “zebrafish”, “mouse”, “CLP”, and “RNA-seq” [35].
Validation
The seven newly identified differentially expressed pathway genes, bag3, casp8, fgfr1, fos, hoxb1b, kif1b, and mmp2, were subjected to RT-PCR validation using beta actin (actb1) as an endogenous control gene [32]. Gene expression assays were obtained from either Life Technologies (Foster City, CA, USA) or Qiagen (Valencia, CA, USA) and total RNA was run on a ViiA7 Automatic Sequence Detection System (Foster City, CA, USA). All samples were run in triplicate following manufacturer’s protocol. The comparative cycle threshold (CT) and the 2-ddCt method were used to assess change in mRNA levels between the target genes and the endogenous control gene [36]. Student’s t-test was used to compare the mRNA expression levels between wild type, morpholino-injected, and mismatch morpholino-injected embryos; p < 0.05 was considered significant.
Genotyping and association analysis
As previously described, probands and their families were ascertained at Boston Children’s Hospital, Texas Children’s Hospital, Houston, the Texas Cleft-Craniofacial Clinic at the University of Texas McGovern Medical School, and the Cleft Clinic of Shriners Hospital, Houston [23, 28, 37, 38]. Briefly, all individuals were evaluated for the presence/absence of NSCLP and to exclude syndromic forms of clefting. The dataset was composed of 241 multiplex families (152 NHW and 89 Hispanic) and 589 simplex parent-child trios (367 NHW and 222 Hispanic) with ethnicity based on self-report (Supplemental Table 1). After obtaining informed consent, saliva and/or blood samples were collected and DNA was extracted using either Oragene Purifier for saliva (DNA Genotek, Inc., Ontario, Canada) or Roche DNA Isolation Kit for Mammalian Blood (Roche, Switzerland) following the manufacturers’ protocols.
Nine SNVs in/nearby the validated RNA-seq genes were selected based on: (1) HapMap reported minor allele frequencies greater than 0.2 in the CEPH population, (2) Haploview linkage disequilibrium (LD) plots, and (3) location within the gene, with preference given for SNVs in potential regulatory regions (Table 1) [28]. SNVs were genotyped using TaqMan Genotyping Assays (Life Technologies, Foster City, CA, USA) following manufacturer’s protocol. Allele calls were detected on a ViiA7 Automatic Sequence Detection System (Life Technologies, Foster City, CA, USA). Genotyping data from SNVs with call rate of 90% or higher were entered into Progeny Laboratory (South Bend, IN, USA) and assessed for Mendelian errors using PedCheck [39].
Genotype data were stratified by ethnicity and presence/absence of family history of NSCLP in each population. Single-SNV analysis was performed using Family Based Association Test (FBAT), with the “-e” option to correct for complex pedigree structures [40, 41] and Association in the Presence of Linkage (APL) test [42]. Haplotype analysis was performed using the “HBAT” function in FBAT. Gene–gene interaction analysis was performed using APL [42]. Genotyping data for CRISPLD2 SNVs previously associated with NSCLP was used in the gene–gene analysis [28]. To correct for multiple testing, a p-value, corrected for the number of SNVs, of 0.0056 (0.05/9 SNVs) was considered significant and a p-value, corrected for the number of genes, of 0.017 (0.05/3 genes) was considered nominal. For the gene–gene interaction analysis, a p-value of 0.0042 (0.05/3!/((3–2)!2!)) was considered significant.
Gene network In silico analyses
Pathway analysis using Ingenuity (Qiagen, San Francisco) was applied to the validated RNA-seq genes as well as 41 previously confirmed, likely, or intensely studied NSCLP genes (Supplemental Table 2) [12, 13].
Results
RNA-seq, in silico analysis and validation
RNA-seq analysis identified 249 unique differentially expressed genes between wild type and Crispld2 knockdown zebrafish (Supplemental Table 3). In silico pathway analysis of these genes identified five gene networks, including one 52- and three 2-gene pathways (Supplemental Fig. 1A-D). Using PubMatrix, the 52-gene pathway was collapsed and yielded a smaller 7-gene network containing BAG3, CASP8, FGFR1, FOS, KIF1B, MMP2, and HOXA1/hox1b1 (Fig. 1a) [35]. All seven genes are plausible candidates for involvement in craniofacial development because of their expression patterns, role in animal craniofacial development, and human syndromes and/or human association studies (Supplemental Table 4).

RNA-seq network analysis. a Network of seven craniofacial-relevant genes generated by Ingenuity and Pubmatrix. These genes showed differential expression following Crispld2 morpholino knockdown in zebrafish compared to uninjected controls. b RNA-seq differential expression results for seven craniofacial-relevant genes (*p < 0.05). c Differential expression of casp8, fos, and mmp2 (*p < 0.05) was validated by RT-PCR. d Ingenuity-derived network of three validated RNA-seq genes. Dotted lines represent gene–gene interactions identified in this study
RT-PCR analysis of MO-injected and mismatch MO-injected zebrafish embryos confirmed the expression of bag3, casp8, fgfr1, fos, hoxb1b, kif1b, and mmp2 in both wild type and Crispld2 morphant zebrafish. In the RNA-seq results, all genes, with the exception of kif1b, showed increased expression in the crispld2 morpholino-injected embryos compared to uninjected controls (UIC) (Fig. 1b). However, RT-PCR validated the differential expression patterns from the RNA-seq data for only three genes: casp8, fos, and mmp2 (Fig. 1c; Supplemental Table 5). Each of these genes showed differential expression compared to both UIC and mismatch MO-controls. While the RNA-seq results showed upregulation of bag3 and hoxb1b (HOXA1 in humans) in the morphant, RT-PCR results showed no difference in bag3 expression between morphant and UIC embryos and downregulation of hoxb1b in the morphant embryos. Additionally, fgfr1 and kif1b expression in MO-injected embryos were not different from mismatch MO-injected embryos. Therefore, only validated genes were further subjected to in silico network analysis, resulting in a three-gene network (Fig. 1d).
Association analysis in NSCLP families
Nine intra- and intergenic SNVs for CASP8, FOS, and MMP2 were tested for association with NSLCP in 241 multiplex and 589 simplex families (Table 1). Genotypes had a >90% allele call rate and all SNVs were in Hardy–Weinberg equilibrium (data not shown). Genotyping data is available in supplemental table 6.
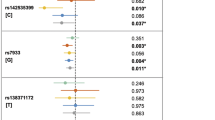
As shown in Table 2, the strongest association was found between FOS/rs1046117:T > C and NSCLP in the NHW simplex families (p = 0.0007), and when all NHW families were analyzed together (p = 0.0005; Table 2). There was only suggestive association with FOS in the Hispanic multiplex subset (p = 0.02). In the Hispanic families, MMP2/rs243836:G > A) was associated with NSCLP in only the multiplex group (p = 0.002); while no association was found for MMP2 in the NHW families (Table 2; Supplemental Table 7). There was suggestive evidence for association between NSCLP and several SNVs in CASP8 in both the NHW and Hispanic families (0.017 ≥ p ≥ 0.0056; Table 2).
Haplotype analysis identified both significant and suggestive associations to the same genes identified in the single-SNV analysis (Table 3). MMP2 haplotypes were associated in the Hispanic multiplex subset (p < 0.0056) and one haplotype was nominal in the NHW families (p = 0.01). In each case, the haplotype contained the associated allele from the single-SNV analyses. Haplotype analysis was not performed on FOS because only one SNV was genotyped.
Gene–gene interaction analysis was performed between the three RNA-seq-confirmed genes; CRISPLD2 was included in this analysis using previously genotyped and reported SNVs [28]. In the NHW multiplex families, there was evidence for an interaction between the FOS/rs1046117:T > C and two CASP8 SNVs (p = 0.002) and suggestive evidence for an interaction between FOS/rs1046117:T > C and MMP2/rs7201:A > C; p = 0.004) (Table 4; Fig. 1d).
In silico craniofacial development gene network
In silico network analysis including the five validated RNA-seq genes and strong candidate NSCLP genes (from published GWAS, linkage, and animal model studies) resulted in a 26-gene network potentially involved in craniofacial development (Fig. 2). In this 26-gene network, eleven known craniofacial genes were directly connected to our three validated RNA-seq genes; of note, FOS and CASP8 were each linked to five or more previously identified NSCLP genes, while MMP2 was linked to one (Fig. 2).

Craniofacial Gene Network. Ingenuity-derived network including previously identified NSCLP genes that have been confirmed, very likely, and intensively studied (shown in white) and RNA-seq genes from this study (shown in gray) identifies a 26-gene network that connect to our three genes of interest, either directly (solid line) or indirectly through another gene (dotted lines)
Discussion
In this study, we utilized RNA-seq findings from our Crispld2 morphant zebrafish to identify new candidate genes for NSCLP. We identified a three-gene network active in craniofacial development that when perturbed may contribute to NSCLP. This was accomplished by identifying differentially expressed genes in craniofacial development in our crispld2 morphant zebrafish [31, 32] and then testing for association in our multiethnic multiplex and simplex NSCLP families (Fig. 3). This unique network has not previously been reported to contribute to NSCLP, although some of the individual genes have been implicated in craniofacial development or NSCLP (Supplemental Table 4). This approach provides a new network of genes to be interrogated.

Paradigm used to identify novel NSCLP genes
FOS was connected to both CASP8 and MMP2 in the RNA-seq derived gene network and showed the strongest association with NSCLP in the NHW families (Table 2). FOS is an oncogene that promotes epithelial-to-mesenchymal transition, a critical process during craniofacial development [43]. Neural crest cells undergo EMT before migrating into the craniofacies and form the precursors to the processes that will develop into critical facial structures including the lip and palate [44, 45]. Based on these observations, FOS should be considered a plausible candidate gene for NSCLP. FOS is located on chromosome 14q24.3, spans 3456 basepairs, and is located in a single linkage disequilibrium block; therefore, only one synonymous SNV (rs1046117:T > C) was genotyped. While additional studies of the role of FOS in NSCLP are needed, our findings implicate it as a potential candidate gene for NSCLP.
MMP2 showed connections to three of the network genes (Fig. 1d) and showed the strongest association with NSCLP in the Hispanic families (Table 2). MMP2 belongs to the matrix metalloproteinase (MMP) gene family, which collectively are responsible for extracellular matrix (ECM) remodeling and subsequent fusion of the palatal shelves have previously been implicated in orofacial clefting [46, 47]. Several MMPs and their tissue inhibitors (TIMPs) are expressed in the medial edge epithelium during palatogenesis and MMP2 expression was found in a sample of palatal soft tissue adjacent to a cleft site that was excised during a child’s palatal repair [48]. Moreover, Tgfß3 null mice, which have cleft palate, show decreased MMP2 and MMP13 expression at the palatal midline, further supporting the role of this gene network in palatal fusion [46, 47]. Further, genetic studies in humans have reported an association between NSCLP and variants in MMP3, MMP25, TIMP2, and TIMP3, with functional promoter activity changes shown for SNVs in MMP3 and TIMP2 [49,50,51,52,53,54]. Our findings provide additional support for the role of MMPs in orofacial clefting.
Although only nominal associations were found for CASP8/rs3769825:C > A) in NHW simplex families and CASP8/rs6754084:C > T) in Hispanic simplex families, our results support previous findings suggesting a role for these genes in NSCLP. For example, CASP8 plays a role in apoptosis, which is critical to maintaining tissue homeostasis during embryonic development [55] and has been shown to contribute to cancer pathogenesis [56]. Recent studies have suggested a link between NSCLP genes and oral, gastric, and colorectal cancers [57,58,59,60,61]. Interestingly, the CASP8/rs3769825:C > A) “C” allele has been reported as a potentially protective allele for cancer risk in an Asian population [62]. Future research should focus on the potential interplay between NSCLP and cancer genes with emphasis on genes involved in the apoptotic pathway.
Our gene–gene interaction analyses revealed significant or nominal gene–gene interactions between MMP2, CASP8, and FOS, confirming the expression network analysis and thereby suggesting a biological interaction between these genes (Fig. 1d). MMP2 and FOS have previously been shown to interact in studies that demonstrate that the inhibition of MMP2 suppresses the induction of c-FOS and both of these genes are activated in metastatic gastric adenocarcinoma [13, 63]. FOS, which has been shown to play a role in cellular functions including apoptosis, is a target for CASP8 [64]. While neither of these connections are specific to craniofacial development; however, our gene–gene interaction analyses suggest that they might also have a role together for normal development of the lip and palate (Fig. 1d). Remarkably, when utilizing 41 known or suspected NSCLP genes, network analysis found direct connections between the three validated RNA-seq genes and 11 previously identified NSCLP genes, whereas indirect connections were also found with an additional twenty-two NSCLP genes (Fig. 2). It is particularly interesting that FOS, a novel finding in this study, has already been biologically connected to seven additional NSCLP genes as shown in Fig. 2. The biological implications of the RNA-seq identified gene connections and other craniofacially relevant genes opens new avenues of investigation.
Intriguingly, although the RNA-seq data was generated using zebrafish crispld2 morphant embryos, we did not detect any evidence of interactions between variations in CRISPLD2 and other genes identified in the network analysis. There are a number of possible explanations for this observation. The most likely is that the in silico analyses are based on curated data from published studies, and most of the studies on CRISPLD2 have focused on cancer and lung and not on craniofacial development [34, 35]. Alternatively, it is possible that the potential interactions involve rare variants in one or more genes, or that the effect size may be too small to be identified in our dataset. Interactions involving more than two genes or the presence of extrinsic unidentified factors that are critical for proper development are also possible. Functional studies will clarify if biological interactions between these genes indeed exist.
The power of this study lies in our multi-pronged approach (Fig. 3), taking discoveries in humans to an animal model and then back to humans to identify novel and biologically plausible NSCLP candidate gene networks. Functional studies of the identified gene network in animal models may reveal whether the individual genes interact during craniofacial development and will potentially improve our understanding of the role of CRISPLD2 in craniofacial development. Finally, these results support the integration of gene network analysis as an unbiased approach to discover the genetic etiology of NSCLP and other complex birth defects.
References
Gorlin RJ, Cohen MM, Hennekam RCM. Syndromes of the head and neck. Fourth Edition. New York: Oxford University Press; 2001.
Johnston MC, Hassell JR, Brown KS. The embryology of cleft lip and cleft palate. Clin Plast Surg. 1975;2:195–203.
Trasler DG, Fraser FC. Handbook of teratology. New York: Plenum Press; 1977.
Jiang R, Bush JO, Lidral AC. Development of the upper lip: morphogenetic and molecular mechanisms. Dev Dyn. 2006;235:spc1.
Nanci A. Ten cate’s oral histology—development, structure, and function. 6th edn. Los Angelos: Mosby; 2003.
Senders CW, Peterson EC, Hendrickx AG, Cukierski MA. Development of the upper lip. Arch Facial Plast Surg. 2003;5:16–25.
Parker SE, Mai CT, Canfield MA, et al. Updated national birth prevalence estimates for selected birth defects in the United States, 2004–2006. Birth Defects Res A Clin Mol Teratol. 2010;88:1008–16.
Hashmi SS, Waller DK, Langlois P, Canfield M, Hecht JT. Prevalence of nonsyndromic oral clefts in Texas: 1995-1999. Am J Med Genet A. 2005;134:368–72.
Artama M, Auvinen A, Raudaskoski T, Isojarvi I, Isojarvi J. Antiepileptic drug use of women with epilepsy and congenital malformations in offspring. Neurology. 2005;64:1874–8.
Azarbayjani F, Danielsson BR. Phenytoin-induced cleft palate: evidence for embryonic cardiac bradyarrhythmia due to inhibition of delayed rectifier K+ channels resulting in hypoxia-reoxygenation damage. Teratology. 2001;63:152–60.
Lidral AC, Moreno LM. Progress toward discerning the genetics of cleft lip. Curr Opin Pediatr. 2005;17:731–9.
Dixon MJ, Marazita ML, Beaty TH, Murray JC. Cleft lip and palate: understanding genetic and environmental influences. Nat Rev Genet. 2011;12:167–78.
Beaty TH, Marazita ML, Leslie EJ. Genetic factors influencing risk to orofacial clefts: today’s challenges and tomorrow’s opportunities. F1000Res. 2016;5:2800.
Khandelwal KD, van Bokhoven H, Roscioli T, Carels CE, Zhou H. Genomic approaches for studying craniofacial disorders. Am J Med Genet C Semin Med Genet. 2013;163C:218–31.
Ludwig KU, Mangold E, Herms S, et al. Genome-wide meta-analyses of nonsyndromic cleft lip with or without cleft palate identify six new risk loci. Nat Genet. 2012;44:968–71.
Ferretti E, Li B, Zewdu R, et al. A conserved Pbx-Wnt-p63-Irf6 regulatory module controls face morphogenesis by promoting epithelial apoptosis. Dev Cell. 2011;21:627–41.
Liu H, Leslie EJ, Jia Z, et al. Irf6 directly regulates Klf17 in zebrafish periderm and Klf4 in murine oral epithelium, and dominant-negative KLF4 variants are present in patients with cleft lip and palate. Hum Mol Genet. 2015;25:766–76.
Blanton SH, Cortez A, Stal S, Mulliken JB, Finnell RH, Hecht JT. Variation in IRF6 contributes to nonsyndromic cleft lip and palate. Am J Med Genet A. 2005;137:259–62.
Ghassibe M, Bayet B, Revencu N, et al. Interferon regulatory factor-6: a gene predisposing to isolated cleft lip with or without cleft palate in the Belgian population. Eur J Hum Genet. 2005;13:1239–42.
Scapoli L, Palmieri A, Martinelli M, et al. Strong evidence of linkage disequilibrium between polymorphisms at the IRF6 locus and nonsyndromic cleft lip with or without cleft palate, in an Italian population. Am J Hum Genet. 2005;76:180–3.
Srichomthong C, Siriwan P, Shotelersuk V. Significant association between IRF6 820G- A and non-syndromic cleft lip with or without cleft palate in the Thai population. J Med Genet. 2005;42:e46.
Zucchero TM, Cooper ME, Maher BS, et al. Interferon regulatory factor 6 (IRF6) gene variants and the risk of isolated cleft lip or palate. N Engl J Med. 2004;351:769–80.
Chiquet BT, Blanton SH, Burt A, et al. Variation in WNT genes is associated with non-syndromic cleft lip with or without cleft palate. Hum Mol Genet. 2008;17:2212–8.
Leslie EJ, Murray JC. Evaluating rare coding variants as contributing causes to non-syndromic cleft lip and palate. Clin Genet. 2013;84:496–500.
Letra A, Menezes R, Cooper ME, et al. CRISPLD2 variants including a C471T silent mutation may contribute to nonsyndromic cleft lip with or without cleft palate. Cleft Palate-Craniofacial J. 2011;48:363–70.
Shen X, Liu RM, Yang L, et al. The CRISPLD2 gene is involved in cleft lip and/or cleft palate in a Chinese population. Birth Defects Res A Clin Mol Teratol. 2011;91:918–24.
Shi J, Jiao X, Song T, Zhang B, Qin C, Cao F. CRISPLD2 polymorphisms are associated with non-syndromic cleft lip with or without cleft palate in a northern Chinese population. Eur J Oral Sci. 2010;118:430–3.
Chiquet BT, Lidral AC, Stal S, et al. CRISPLD2: a novel NSCLP candidate gene. Hum Mol Genet. 2007;16:2241–8.
Gibbs GM, Roelants K, O’Bryan MK. The CAP superfamily: cysteine-rich secretory proteins, antigen 5, and pathogenesis-related 1 proteins--roles in reproduction, cancer, and immune defense. Endocr Rev. 2008;29:865–97.
Zhang H, Sweezey NB, Kaplan F. LGL1 modulates proliferation, apoptosis, and migration of human fetal lung fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2015;308:L391–402.
Yuan Q, Chiquet BT, Devault L, et al. Craniofacial abnormalities result from knock down of nonsyndromic clefting gene, crispld2, in zebrafish. Genesis. 2012;50:871–81.
Swindell EC, Yuan Q, Maili LE, Tandon B, Wagner DS, Hecht JT. Crispld2 is required for neural crest cell migration and cell viability during zebrafish craniofacial development. Genesis. 2015;53:660–7.
Westerfield M: The zebrafish book: a guide for the laboratory use of zebrafish (danio rerio). Eugene: University of Oregon Press; 1995.
Calvano SE, Xiao W, Richards DR, et al. A network-based analysis of systemic inflammation in humans. Nature. 2005;437:1032–7.
Becker KG, Hosack DA, Dennis G Jr, et al. PubMatrix: a tool for multiplex literature mining. BMC Bioinforma. 2003;4:61.
Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45.
Chiquet BT, Hashmi SS, Henry R, et al. Genomic screening identifies novel linkages and provides further evidence for a role of MYH9 in nonsyndromic cleft lip and palate. Eur J Human Genet. 2009;17:195–204.
Chiquet BT, Henry R, Burt A, et al. Nonsyndromic cleft lip and palate: CRISPLD genes and the folate gene pathway connection. Birth Defects Res A Clin Mol Teratol. 2011;91:44–9.
O’Connell JR, Weeks DE. PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet. 1998;63:259–66.
Laird NM, Horvath S, Xu X. Implementing a unified approach to family-based tests of association. Genet Epidemiol. 2000;19(Suppl 1):S36–42.
Laird NM, Lange C. Family-based designs in the age of large-scale gene-association studies. Nat Rev Genet. 2006;7:385–94.
Chung RH, Hauser ER, Martin ER. The APL test: extension to general nuclear families and haplotypes and examination of its robustness. Human Hered. 2006;61:189–99.
Reichmann E, Schwarz H, Deiner EM, et al. Activation of an inducible c-FosER fusion protein causes loss of epithelial polarity and triggers epithelial-fibroblastoid cell conversion. Cell. 1992;71:1103–16.
Berndt JD, Clay MR, Langenberg T, Halloran MC. Rho-kinase and myosin II affect dynamic neural crest cell behaviors during epithelial to mesenchymal transition in vivo. Dev Biol. 2008;324:236–44.
Kang P, Svoboda KK. Epithelial-mesenchymal transformation during craniofacial development. J Dent Res. 2005;84:678–90.
Blavier L, Lazaryev A, Groffen J, Heisterkamp N, DeClerck YA, Kaartinen V. TGF-beta3-induced palatogenesis requires matrix metalloproteinases. Mol Biol Cell. 2001;12:1457–66.
Brown NL, Yarram SJ, Mansell JP, Sandy JR. Matrix metalloproteinases have a role in palatogenesis. J Dent Res. 2002;81:826–30.
Smane L, Pilmane M, Akota I. Apoptosis and MMP-2, TIMP-2 expression in cleft lip and palate. Stomatologija. 2013;15:129–34.
Blanton SH, Bertin T, Serna ME, Stal S, Mulliken JB, Hecht JT. Association of chromosomal regions 3p21.2, 10p13, and 16p13.3 with nonsyndromic cleft lip and palate. Am J Med Genet A. 2004;125:23–27.
Letra A, da Silva RA, Menezes R, et al. Studies with MMP9 gene promoter polymorphism and nonsyndromic cleft lip and palate. Am J Med Genet A. 2007;143:89–91.
Letra A, Silva RA, Menezes R, et al. MMP gene polymorphisms as contributors for cleft lip/palate: association with MMP3 but not MMP1. Arch Oral Biol. 2007;52:954–60.
Letra A, Zhao M, Silva RM, Vieira AR, Hecht JT. Functional significance of MMP3 and TIMP2 polymorphisms in cleft lip/palate. J Dent Res. 2014;93:651–6.
Nikopensius T, Kempa I, Ambrozaityte L, et al. Variation in FGF1, FOXE1, and TIMP2 genes is associated with nonsyndromic cleft lip with or without cleft palate. Birth Defects Res A Clin Mol Teratol. 2011;91:218–25.
Machado RA, Messetti AC, de Aquino SN, et al. Association between genes involved in craniofacial development and nonsyndromic cleft lip and/or palate in the Brazilian population. Cleft Palate Craniofac J. 2015;53:550–6.
Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–62.
Ghavami S, Hashemi M, Ande SR, et al. Apoptosis and cancer: mutations within caspase genes. J Med Genet. 2009;46:497–510.
Dunkhase E, Ludwig KU, Knapp M, et al. Nonsyndromic cleft lip with or without cleft palate and cancer: evaluation of a possible common genetic background through the analysis of GWAS data. Genom Data. 2016;10:22–29.
Menezes R, Marazita ML, Goldstein McHenry T, et al. AXIS inhibition protein 2, orofacial clefts and a family history of cancer. J Am Dent Assoc. 2009;140:80–84.
Letra A, Menezes R, Granjeiro JM, Vieira AR. AXIN2 and CDH1 polymorphisms, tooth agenesis, and oral clefts. Birth Defects Res A Clin Mol Teratol. 2009;85:169–73.
Andrade Filho PA, Letra A, Cramer A, et al. Insights from studies with oral cleft genes suggest associations between WNT-pathway genes and risk of oral cancer. J Dent Res. 2011;90:740–6.
Frebourg T, Oliveira C, Hochain P, et al. Cleft lip/palate and CDH1/E-cadherin mutations in families with hereditary diffuse gastric cancer. J Med Genet. 2006;43:138–42.
Zhang YJ, Zhong XP, Chen Y, Liu SR, Wu G, Liu YF. Association between CASP-8 gene polymorphisms and cancer risk in some Asian population based on a HuGE review and meta-analysis. Genet Mol Res. 2013;12:6466–76.
Cvjetkovic N, Maili L, Weymouth KS, et al. Regulatory variant in FZD6 gene contributes to nonsyndromic cleft lip and palate in an African-American family. Mol Genet Genom Med. 2015;3:440–51.
Westerman K, L. Maili, R. Plant, A. Letra J.T. Hecht, and B.T. Chiquet. “Association of β-catenin-mediated Wnt Pathway Genes with Non-syndromic Cleft Lip with or without Cleft Palate.” American Association of Dental Research. Los Angeles, CA. March 18, 2016.
Acknowledgements
We thank Maria Elena Serna and Rosa Martinez for expert clinical coordination and for managing the databases. We also thank all of the NSCLP families who kindly participated in this study. This study was approved by the Committee for the Protection of Human Subjects at UTHealth (HSC-MS-03-090) and the Animal Welfare Committee at UTHealth (AWC-14-0032). This work was funded by NIH grant R01-DE011931 to JTH.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Chiquet, B.T., Yuan, Q., Swindell, E.C. et al. Knockdown of Crispld2 in zebrafish identifies a novel network for nonsyndromic cleft lip with or without cleft palate candidate genes. Eur J Hum Genet 26, 1441–1450 (2018). https://doi.org/10.1038/s41431-018-0192-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-018-0192-5
This article is cited by
-
The genetic factors contributing to the risk of cleft lip-cleft palate and their clinical utility
Oral and Maxillofacial Surgery (2022)
-
The functional variant of NTN1 contributes to the risk of nonsyndromic cleft lip with or without cleft palate
European Journal of Human Genetics (2020)
