
Abstract
Height is a complex quantitative trait with a high heritability. Short stature is diagnosed when height is significantly below the average of the general population for that person’s age and sex. We have recently found that the retinoic acid degrading enzyme CYP26C1 modifies SHOX deficiency phenotypes toward more severe clinical manifestations. Here, we asked whether damaging variants in CYP26C1 alone could lead to short stature. We performed exome and Sanger sequencing to analyze 856 individuals with short stature where SHOX deficiency was previously excluded. Three different damaging missense variants and one splicing variant were identified in six independent individuals; the functional significance of the identified variants was tested in vitro or in vivo using zebrafish as a model. The genetic and functional data reported here indicate that CYP26C1 represents a novel gene underlying growth disorders and that damaging variants in the absence of SHOX variants can lead to short stature.
Similar content being viewed by others
Introduction
Short stature is diagnosed when the height of an individual is 2 SD below the mean height of a population of the same ethnic group, age, and gender [1]. The height of an individual is the result of the interactions of different genetic and environmental factors. Hormones, extracellular matrix proteins, membrane and nuclear receptors, enzymes and transcription factors, together with environmental cues such as nutrient availability establish a complex network that leads the growth of the long bones to their final length, hereby deciding on a person’s final height. Genetic variants negatively affecting the nodes of such a delicate system may result in readjustments that lead from height within the normal range to diseases with various skeletal defects, altered bone growth, and stature below the mean. More than 150 genes are known to be involved in the etiology of short stature [2, 3].
In a recent study, we identified CYP26C1 as a genetic modifier driving SHOX deficiency toward a more severe phenotype [4]. All the combined SHOX (MIM:312865) and CYP26C1 (MIM: 608428) variants were found so far in patients with Léri-Weill dyschondrosteosis (MIM:127300), the severe form of SHOX deficiency (MIM: 300582). CYP26C1 is an enzyme belonging to the cytochrome P450 superfamily and is involved in the degradation of intracellular retinoic acid (RA). RA has been shown to play an important role during skeletogenesis and alterations of its physiological levels can lead to dramatic effects on bone development [5, 6]. Excess of RA hinders longitudinal bone growth by inhibiting proliferation and hypertrophy of growth plate chondrocytes and matrix synthesis in postnatal rats [7]. CYP26B1, another member of the CYP26 family, has been shown to be involved in skeleton development and, variants in this gene, lead to craniosynostosis and multiple skeletal anomalies including shorter limbs in humans [8]. Loss of cyp26c1 in zebrafish embryos leads to accumulation of RA resulting in reduction of SHOX expression and smaller limbs [4].
In this study, we asked whether damaging variants in CYP26C1 could account for short stature in individuals where SHOX deficiency was excluded. A cohort of 856 individuals with short stature and an intact SHOX gene were therefore analyzed for variants within the CYP26C1-coding sequence. Functional studies on the detected variants were performed to test their effect in cell culture or zebrafish embryos.
Materials and methods
Cohort of short-stature patients and controls
The sample comprised 856 unrelated individuals with idiopathic short stature (834 with proportionate short stature and 22 with disproportionate/mesomelic short stature). The 834 individuals with proportional short stature comprised 546 Germans, 265 Japanese, 13 Belgians, and 10 Americans. The 22 individuals with disproportionate short stature were of German origin.
Sequencing results were compared to publicly available databases (gnomAD, TGP, and EVS) and to 350 control individuals with normal stature (240 Germans, 110 Japanese).
Sequencing
The DNA samples from the cohort of short-stature patients were sequenced as follows: 230 individuals with idiopathic short stature were sequenced by whole-exome sequencing as described below; 234 individuals with idiopathic short stature were sequenced with the GS Roche Junior System (Roche), as recommended by the manufacturers; 392 individuals with short stature were sequenced by Sanger sequencing. Index patient of family 1 was identified in the cohort of short-stature individuals analyzed by Sanger sequencing. Index patient of family 2 was identified in the cohort of short-stature individuals analyzed with the GS Roche Junior System. Families 3 and 4 were identified in the cohort of short-stature individuals analyzed by whole-exome sequencing. Individuals 5 and 6 were identified in the cohort of short-stature individuals analyzed by Sanger sequencing. Variants identified by the GS Roche Junior System or whole-exome sequencing were validated by Sanger sequencing. CGH arrays were also performed on families 3 and 4 to exclude copy-number variants.
CYP26C1 accession (NM_183374.2; NG_007958.1; NP_899230.2; ENST 00000285949.5). Exons are numbered as in NG_007958.1. Data on identified variants has been submitted to the gene variant database www.LOVD.nl/CYP26C1 (patient IDs 144626–144629).
Whole-exome sequencing
Whole-exome sequencing was performed on 230 individuals with short stature as previously described [9]. Briefly, DNA from the patients was enriched using the Agilent’s SureSelect Human All Exon Kit V5 (Agilent Technologies), and sequenced in paired-end reads on a HiSeq2500 system (Illumina). Sequenced reads were mapped with BWA to the reference human genome assembly hg19 (GRCh37). Genotypes were additionally called with GATKHap, GATKUG [10]. An in-house database consisting of 1200 sequenced exomes was also used to exclude sequence errors and frequent variants.
Zebrafish experiments
Experiments were carried out in wild-type Danio rerio strains (AB, TL, Tübingen). Sense-capped RNA of human CYP26C1 wild type and mutants was synthesized using the mMESSAGE mMACHINE system (Ambion) from pCS2 [11].
Whole-mount in situ hybridization was performed as described previously [12]. Probes were designed and synthesized as previously described [4]. Images were taken with the microscope SZX16, CellD Imaging Software (Olympus). Pectoral fin area was measured with Fiji ImageJ [13].
Minigene splicing assay
The minigene splicing assay was performed as previously described [14]. PCR fragments of CYP26C1 intron3-exon4-intron4 genomic region were cloned into the splicing vector pSPL3 using specific primers linking the EcoRI and BamHI restriction enzyme sites (forward primer 5′-GTACAGGGAAAGGGCAATGG-3′; reverse primer 5′-GAGTTTGATCCTGAGCCCCT-3′). Wild-type (Wt) and mutant constructs were verified by Sanger sequencing.
U2OS cells (human osteosarcoma cells, ATCC; mycoplasma-free) were cultured in DMEM containing 10% FBS and penicillin/streptomycin at 37 °C, 5% CO2, 95% humidity. Minigene splicing assays were performed as follows: cells were seeded in six-well plates at 2 × 106 cells each well. After 24 h, cells were transfected with Lipofectamine 2000 (Invitrogen) according to standard protocols. For each well, cells were transfected with 2.5 µg of pSPL3 empty vector, pSPL3-CYP26C1 intron3-exon4-intron4 wt, or pSPL3-CYP26C1 intron3-exon4-intron4 c.706-2A>C mutant. After 24 h, RNA was extracted with Trizol (Invitrogen) following standard protocols. Complementary DNA synthesis was performed with SuperScript™ II according to the manufacturer’s protocol. To evaluate splicing, vector-specific primers were used for PCR (forward primer SD6 5′-TCTGAGTCACCTGGACAACC-3′; reverse primer SA2 5′-ATCTCAGTGGTATTTGTGAGC-3′). PCR was performed using Q5® high-fidelity DNA polymerase (New England Biolabs) according to the manufacturer´s protocols. The PCR products were separated by electrophoresis on a 1.5% agarose gel. All transcripts were verified by Sanger sequencing.
Statistics
Samples for zebrafish embryos experiments were randomly assigned to experimental groups, to processing order, and positioned in multi-well devices. No statistical method was used to predetermine the sample size. Group sample sizes for experiments were chosen based on previous studies. Zebrafish embryos that died before analysis were excluded. Pectoral fin pictures were taken and analyzed randomized blindly. Statistical analyses were performed using GraphPad Prism version 5 for Windows (GraphPad Software). Data were tested for normality using the D’Agostino and Pearson omnibus normality test. Differences were analyzed by analysis of variance test. For all experiments, data are expressed as the mean ± SD. P-values <0.05 were considered significant. Pairwise comparison between wild type and mutants resulted in a P-value for the mutants (c.148C>T, p.(Pro50Ser), c.356A>C, p.(Gln119Pro), and c.910G>A, p.(Ala304Thr)) that withstand correction for multiple testing using the Bonferroni threshold (P < 0.0125; n = 4 tests).
Bioinformatics resources
Primers for sequencing were designed using Primer3 [15]. Variants were tested with PolyPhen2 [16], MutationTaster [17], SIFT [18], PROVEAN [19], and CADD [20].
Ethical approval
This study was approved by the Institutional Review Board Committee at the Heidelberg University, Hamamatsu University, and National Center for Child Health and Development, and performed after obtaining written informed consent from the participants.
Results
To evaluate whether damaging CYP26C1 variants could lead to short stature, we performed exome and Sanger sequencing on a cohort of 856 individuals with short stature and 350 individuals with normal height (Supplementary Table S1). In all patients, defects in the SHOX gene or its enhancers were previously ruled out. We identified three different rare missense variants, c.148C>T, p.(Pro50Ser), c.356A>C, p.(Gln119Pro), and c.910G>A, p.(Ala304Thr), and a splice variant change, c.706-2A>C, in CYP26C1 in four unrelated individuals with short stature (Fig. 1). The variant c.356A>C, p.(Gln119Pro) was identified in two additional unrelated patients. The identified variants were either not reported or described as rare in the major public variant databases, genome Aggregation database (gnomAD), Exome Variant Server (EVS), and 1000 Genomes Project (TGP), and were not found in 350 control individuals with normal height (Supplementary Table S1). All identified missense variants in the patient cohort were predicted as damaging by three or four of the four prediction tools used. The splice variant was predicted as damaging by MutationTaster (Supplementary Table S1). In the control group with normal height, two rare missense variants c.479G>A, p.(Arg160His) and c.610G>C, p.(Glu204Gln) and the common missense variant c.734G>A, p.(Arg245Gln) were identified (Supplementary Table S2).

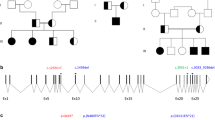
Family pedigree charts of the screened short-stature individuals and CYP26C1 variants identified. a Pedigree of the families studied. Families 3 and 4 were screened by exome sequencing. b Schematic representation of the genomic structure of the CYP26C1 gene. Variants identified are indicated below. Filled symbol individual with short stature, N.A. DNA not available
In family 1, the affected German individual with a height SD of −2.9 carried the missense variant c.148C>T, p.(Pro50Ser) (Fig. 1). This variant was reported as rare by the gnomAD and TGP databases (Supplementary Table S1). Neither clinical data nor DNA from both parents was available to test association and inheritance. To assess the functional significance of p.(Pro50Ser), we performed in vivo experiments in zebrafish. One-cell stage zebrafish embryos were injected with sense-capped RNA encoding for hCYP26C1 wild type or mutant. We included the common CYP26C1 missense variant, c.734G>A, p.(Arg245Gln), found in individuals with normal height as an internal negative control. As expected, injection of hCYP26C1 wild type or hCYP26C1 p.(Arg245Gln) resulted in the complete loss of the pectoral fins as a result of its enzymatic activity (Fig. 2). In contrast, embryos injected with RNA encoding for the variant p.Pro50Ser developed normal pectoral fins, suggesting that this variant affects CYP26C1 activity (Fig. 2).

Functional significance of the CYP26C1 missense variants identified in zebrafish embryos. a Wild-type embryos injected with sense-capped RNA coding for human CYP26C1 wild type or the variants identified in families 1–3, p.(Pro50Ser) (family 1), p.(Ala304Thr) (family 2), p.(Gln119Pro) (family 3). The common missense variant p.(Arg245Gln) was used as control. a (left) Dorsal views of the embryos at 55 h post fertilization (hpf). Embryos injected with CYP26C1 wild-type RNA displayed absent pectoral fins (non-injected = 118; WT = 123; p.(Pro50Ser) = 90; p.(Ala304Thr) = 90; p.(Gln119Pro) = 57; p.(Arg245Gln) = 37). Arrows indicate pectoral fin. a (right) Magnification on the pectoral fins of col2a1 expression at 55 hpf. b Pectoral fin area was measured by ImageJ. The box represents the interquartile range. The whiskers represent Min to Max. ****P-value < 0.0001; two-way ANOVA. Dotted line pectoral fin
In family 2, the Japanese index patient with a height SD of −4.35 (Fig. 1, II:1) carrying the CYP26C1 variant c.356A>C, p.(Gln119Pro) presented with short stature. This variant was also carried by the mother with borderline height (SD −1.85) and found in two further unrelated Japanese individuals with short stature in our cohort (SD −2.8; SD −3.9; Supplementary Table S3). Functional analysis of Gln119Pro showed a highly significant impact on CYP26C1 in zebrafish embryos when compared to wild type or the p.(Arg245Gln) control (Fig. 2). Based on the fact that this variant was inherited from the unaffected/borderline mother in family 2, we cannot reliably conclude that this variant is causative for short stature in this family, however, incomplete penetrance cannot be ruled out.
In family 3, the affected girl of German origin presented with a height of −2.9 SD and intellectual disability (ID). The patient was born at term to non-consanguineous parents after normal pregnancy with a weight of 3200 g (~50th centile and a birth length of 49 cm, 50th centile). After birth, a congenital hip dysplasia and an umbilical hernia were noticed and treated. Her development was in the lower normal range and she received special training for her learning disability. Puberty was normal. Her growth was reported to constantly be below the 3rd centile. Other than myopia and her growth deficit, no further medical issues were present. At the age of 14.5 years, her height was 2.9 SD below the average height. Her facial gestalt was not specific. Weight and head circumference were below the 3rd centile. The patient carried a de novo CYP26C1 variant, c.910G>A, p.(Ala304Thr) (Fig. 1). This variant is not reported in the gnomAD, TGP, and EVS databases (Supplementary Table S1). No further variant explaining the patient phenotype could be found in the whole-exome sequencing data. Functional analysis in zebrafish embryos displayed a strong and significant effect of p.(Ala304Thr) on the development of the pectoral fin (Fig. 2).
In family 4, we identified the splice variant c.706-2A>C. The affected boy of German origin presented with short stature (−4.3 SD) and autism spectrum disorder (ASD) at the age of 8 years (Fig. 1, family 4). The child was born from non-consanguineous parents. He was born after a prenatal infection at 25th week of gestation with a weight of 765 g and a length of 30 cm. The variant was inherited from the mother, who also had short stature (−2.5 SD), but did not display ASD. Height of the father was within the normal range (−1.2 SD). The identified variant c.706-2A>C altered the AG acceptor splice site of intron 3 in CYP26C1. No further variant explaining the patient phenotype could be found in the whole-exome sequencing data. In order to test the functional significance of c.706-2A>C, we performed a minigene splicing assay with the pSPL3 vector [14] (Fig. 3a). This variant strongly affected splicing, leading to the retention of intron 3, as shown by the minigene splicing assay (Fig. 3b). Intron 3 contains a stop codon and its retention would lead to an insertion of 67 amino acids and a stop codon after position p.(Lys235), resulting in a truncated protein missing the P450 domain. Although we cannot exclude that during development and growth of the bone alternative or cryptic splicing events occur leading to the synthesis of functional CYP26C1 protein, our data suggest that c.796-2A>C compromises CYP26C1 splicing. RNA from this family was not available to assess this possibility.

Minigene splicing assay for the variant c.706-2A>C. a The pSLP3 vector contains a functional intron and a multiple cloning site within the two exons SD and SA. PCR fragments containing the CYP26C1 genomic sequence intron3-exon4-intron4 wt or c.706-2A>C were cloned within the multiple cloning site. The expected splicing events are indicated in the scheme. b Agarose gel electrophoresis of the RT-PCR amplicons. Forward SD6 and reverse SA2 primers were used for RT-PCR of cDNA generated from U2OS cells transfected with pSPL3 empty vector, pSPL3-CYP26C1 intron3-exon3-intron4 wt, or pSPL3-CYP26C1 intron3-exon4-intron4 c.706-2A>C. MCS multiple cloning site
Discussion
We have recently reported that CYP26C1 acts as a genetic modifier for SHOX deficiency [4]. Patients bearing damaging variants in both SHOX and CYP26C1 presented with more severe phenotypes when compared to family members carrying only SHOX variants. We also demonstrated that damaging CYP26C1 variants lead to high RA levels which, in turn, repressed SHOX expression, thereby exacerbating the SHOX deficiency phenotype, as seen in patients with Léri-Weill dyschondrosteosis [21].
In this study, we investigated whether variants in CYP26C1 alone could lead to short stature. We screened 856 patients with short stature, where SHOX deficiency had previously been ruled out and identified four variants, three missense variants, and one splicing variant, in six independent individuals (6/856; ~0.7%). All four identified variants displayed a significant effect on CYP26C1 activity or splicing compared to wild type, whereas this was not the case for the three missense variants found in individuals with normal height (this study; and ref. 4).
The c.910G>A, p.(Ala304Thr) variant and the splice variant c.706-2A>C were not present in gnomAD, TGP, or the EVS databases. The c.148C>T, p.(Pro50Ser) variant was not found in the TGP and EVS databases, but in 7/75893 individuals from the gnomAD. GnomAD provides a dataset of exome and whole-genome sequencing from unrelated individuals sequenced as part of disease-specific and population genetic studies (e.g., the Cancer Genome Atlas, 1000 Genomes, Myocardial Infarction Genetics Consortium). It is therefore likely that the height of most individuals was not reported as a phenotype during the process of data collection; therefore, it cannot be excluded that some of these individuals are also short.
Variant c.356A>C, p.(Gln119Pro) was not found in (non-Finnish) Europeans but in two individuals (Han Chinese) of a total 2501 of individuals in the TGP, suggesting that this variant is more frequent in the East Asian population. It was also found in 27 individuals of East Asian origin in the gnomAD (27/81,453 individuals; allele frequency 0.00017). Just considering the East Asian population in this database (27/11,628), the allele frequency is 0.0023. This variant was also rather frequent in the Finnish population (246/81,453, allele frequency 0.0030; 246/11,134, Finnish individuals allele frequency 0.022), clearly not a rare variant in this population. These data show that c.356A>C, p.(Gln119Pro) is a variant with very different population frequencies. Our data also argues for reduced penetrance. It would be interesting to analyze this variant specifically in short individuals from Finland, China, or Japan to get further insight into its penetrance.
A previous study has associated CYP26C1 homozygous and compound heterozygous 7-bp duplications (predicting a c.844_851dupCCATGCA, p.(Glu284fs128) frameshift) with focal facial dysplasia type IV [22]. The 7-bp duplication was also present in 0.3% of healthy control individuals and 0.3% of patients with other birth defects. In contrast to our results, short stature or a neurodevelopmental phenotype were not reported in the affected homozygous/compound heterozygous and non-affected heterozygous individuals. We would have expected that loss of both CYP26C1 alleles leads to a short-stature phenotype. The observed phenotypic variability may be explained by allelic variation, modifier genes, and complex genetic and environmental interactions [23]. A recent screen in 589,306 genomes identified 13 healthy adult individuals bearing known variants for eight severe Mendelian diseases, indicating that there are genetic and environmental factors that can buffer the phenotypic effect of highly penetrant deleterious variants and that phenotypic variability is more common than anticipated [24]. Excess of RA affects multiple organs during development and in adulthood including the limb, eye, face, and central nervous system [25] resulting in a broad spectrum of phenotypes. It is therefore possible that variants in CYP26C1 leading to increased levels of RA are buffered and/or compensated by variants in other genes within and without the RA pathway resulting in low penetrance. Finally, neither height nor IQ scores were analyzed in the focal facial dysplasia type IV study and, therefore, it cannot be excluded that those individuals presented with reduced height and/or lower IQ scores compared to other family members. Two of the six cases in our study presented with short-stature and a neurodevelopmental phenotype. In family 3, the affected individual presented with short-stature and mild ID. In family 4, the affected son displayed short stature as well as ASD associated with ID, which did not show in his mother. A recent study reported an individual with a de novo 8.3 Mb microdeletion on chromosome 10q23.2–23.33 associated with accelerated skeletal and dental development, craniosynostosis, retinal scarring, and ASD [26]. The patient displayed elevated levels of total RA metabolites. The deleted region contains CYP26C1 and other genes that play a role in vitamin A metabolism including CYP26A1, CYP2C8, CYP2C9, and RBP4. Our patients did not show craniosynostosis, eye, and dental phenotypes, likely because other RA metabolism genes, e.g. CYP26A1, compensated for the loss of CYP26C1. However, as RA plays a role in the development and maintenance of different tissues and organs including the brain [25] and since Cyp26c1 as well has been proposed to play a role in brain development [27], compromised CYP26C1 function alone could be the cause of the ID found in these two patients. Alternatively, another so far unknown gene(s) may contribute to the neurodevelopmental aspects of the present phenotype. This may be more likely, considering the fact that the mother of family 4 with the same variant shares short stature with her son but not the ID/ASD phenotype, although other scenarios including variable penetrance cannot fully be ruled out.
Our analysis in zebrafish gave a straightforward answer on whether a variant proved to affect function or not. Limb development can be dramatically affected by manipulating RA levels; both excess and deficiency of this signaling molecule can alter limb formation [25]. Loss of RA in the early stages of development of zebrafish embryos causes forelimb initiation defects [27]. Injection of wild-type CYP26C1-capped RNA in zebrafish embryos results in ectopic overexpression of this enzyme leading to depletion of RA in the embryo and consequently to defects in limb development. The CYP26C1 missense variants identified in our cohort of short-stature patients, c.148C>T, p.(Pro50Ser), c.356A>C, p.(Gln119Pro) and c.910G>A, p.(Ala304Thr), alter CYP26C1 enzymatic activity. Mutant CYP26C1 protein, even if expressed ectopically, cannot deplete RA. We hypothesize that individuals bearing CYP26C1 damaging variants have a defective catabolism of RA resulting in an excess of RA, which then can lead to altered limb patterning and culminate in short stature.
Together, our compiled genetic and functional data support the conclusion that rare damaging variants in CYP26C1 are causative for short stature with incomplete penetrance. The zebrafish model has become an important tool for modeling genetic diseases in the context of basic research [28, 29] and will continue to be of use for high-throughput screening of small molecules to accelerate drug discovery [30].
References
Ranke MB. The KIGS aetiology classification system. In: Ranke MB, Gunnarson R, editors. Progress in growth hormone therapy-5 years of KIGS. Mannheim: J&J VerlagGmBH; 1994; p. 51–61.
Durand C, Rappold GA. Height matters – from monogenic disorders to normal variation. Nat Rev Endocrinol. 2013;9:171–7.
Baron J, Sävendahl L, De Luca F, et al. Short and tall stature: a new paradigm emerges. Nat Rev Endocrinol. 2015;11:735–46.
Montalbano A, Juergensen L, Roeth R, et al. Retinoic acid catabolizing enzyme CYP26C1 is a genetic modifier in SHOX deficiency. EMBO Mol Med. 2016;8:1455–69.
Weston AD, Hoffman LM, Underhill TM. Revisiting the role of retinoid signaling in skeletal development. Birth Defects Res C Embryo Today. 2003;69:156–73.
Kuss P, Villavicencio-Lorini P, Witte F, et al. Mutant Hoxd13 induces extra digits in a mouse model of synpolydactyly directly and by decreasing retinoic acid synthesis. J Clin Invest. 2009;119:146–56.
De Luca F, Uyeda JA, Mericq V, et al. Retinoic acid is a potent regulator of growth plate chondrogenesis. Endocrinology. 2000;141:346–53.
Laue K, Pogoda HM, Daniel PB, et al. Craniosynostosis and multiple skeletal anomalies in humans and zebrafish result from a defect in the localized degradation of retinoic acid. Am J Hum Genet. 2011;89:595–606.
Kessler K, Wunderlich I, Uebe S, et al. DYNC2LI1 mutations broaden the clinical spectrum of dynein-2 defects. Sci Rep. 2015;15:11649 https://doi.org/10.1038/srep11649
McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303.
Hassel D, Dahme T, Erdmann J, et al. Nexilin mutations destabilize cardiac Z‐disks and lead to dilated cardiomyopathy. Nat Med. 2009;15:1281–8.
Jowett T, Lettice L. Whole-mount in situ hybridizations on zebrafish embryos using a mixture of digoxigenin- and fluorescein-labelled probes. Trends Genet. 1994;10:73–4.
Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: an open-source platform for biological image analysis. Nat Methods. 2012;9:676–82.
Zhao X, Cui L, Lang Y, et al. A recurrent deletion in the SCL5A2 gene including the intron 7 branch site responsible for familiar renal glucosuria. Sci Rep. 2016;6. https://doi.org/10.1038/srep33920.
Untergasser A, Cutcutache I, Koressaar T, et al. Primer3‐new capabilities and interfaces. Nucleic Acids Res. 2012;40:e115.
Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen‐2. Curr Protoc Hum Genet. 2013;7:Unit7.20 doi: 10.1002/0471142905.hg0720s76.
Schwarz JM, Roedelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease‐causing potential of sequence alterations. Nat Methods. 2010;7:575–6.
Vaser R, Adusumalli S, Leng SN, Sikic M, Ng PC. SIFT missense predictions for genomes. Nat Protoc. 2016;11:1–9.
Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31:2745–7.
Kircher M, Witten DM, Jain P, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–5.
Marchini A, Ogata T, Rappold GA. A track record on SHOX: from basic research to complex model and therapy. Endocr Rev. 2016;37:417–48.
Slavotinek AM, Mehrotra F, Nazarenko I, et al. Focal facial dermal dysplasia, type IV, is caused by mutations in CYP26C1. Hum Mol Genet. 2013;22:696–703.
Nadeau JH. Modifier genes in mice and humans. Nat Rev Genet. 2001;2:165–74.
Chen R, Shi L, Hakenberg J, et al. Analysis of 589,306 genomes identifies individuals resilient to severe Mendelian childhood diseases. Nat Biotech. 2016;34:531–8.
Cunningham TJ, Duester G. Mechanisms of retinoic acid signalling and its roles in organ and limb development. Nat Rev Mol Cell Biol. 2015;16:110–23.
Nilsson O, Isoherranen N, Guo MH, et al. Accelerated skeletal maturation in disorders of retinoic acid metabolism: a case report and focused review of the literature. Horm Metab Res. 2016;48:737–44.
Uehara M, Yashiro K, Mamiya S, Nishino J, et al. CYP26A1 and CYP26C1 cooperatively regulate anterior‐posterior patterning of the developing brain and the production of migratory cranial neural crest cells in the mouse. Dev Biol. 2006;302:399–411.
Grandel H, Lun K, Rauch GJ, et al. Retinoic acid signalling in the zebrafish embryo is necessary during pre-segmentation stages to pattern the anterior-posterior axis of the CNS and to induce a pectoral fin bud. Development. 2002;129:2851–65.
MacRae CA, Peterson RT. Zebrafish as tools for drug discovery. Nat Rev Drug Discov. 2015;14:721–31.
White DT, Froglu AU, Wang G, et al. ARQiv-HTS, a versatile whole-organism screening platform enabling in vivo drug discovery at high-throughput rates. Nat Protoc. 2016;11:2432–53.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Montalbano, A., Juergensen, L., Fukami, M. et al. Functional missense and splicing variants in the retinoic acid catabolizing enzyme CYP26C1 in idiopathic short stature. Eur J Hum Genet 26, 1113–1120 (2018). https://doi.org/10.1038/s41431-018-0148-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-018-0148-9
