
Abstract
Several studies have identified copy number variants (CNVs) as responsible for cardiac diseases associated with sudden cardiac death (SCD), but very few exhaustive analyses in large cohorts of patients have been performed, and they have been generally focused on a specific SCD-related disease. The aim of the present study was to screen for CNVs the most prevalent genes associated with SCD in a large cohort of patients who suffered sudden unexplained death or had an inherited cardiac disease (cardiomyopathy or channelopathy). A total of 1765 European patients were analyzed with a homemade algorithm for the assessment of CNVs using high-throughput sequencing data. Thirty-six CNVs were identified (2%), and most of them appeared to have a pathogenic role. The frequency of CNVs among cases of sudden unexplained death, patients with a cardiomyopathy or a channelopathy was 1.4% (8/587), 2.3% (20/874), and 2.6% (8/304), respectively. Detection rates were particularly high for arrhythmogenic cardiomyopathy (5.1%), long QT syndrome (4.7%), and dilated cardiomyopathy (4.4%). As such large genomic rearrangements underlie a non-neglectable portion of cases, we consider that their analysis should be performed as part of the routine genetic testing of sudden unexpected death cases and patients with SCD-related diseases.
Similar content being viewed by others

Introduction
More than four million people around the world die every year because of sudden cardiac death (SCD). The most common cause of SCD in adults is coronary heart disease, but arrhythmogenic cardiac diseases are the leading cause in population younger than 35 years old. In this latter group, most SCDs are due to cardiomyopathies—mainly hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy (DCM), arrhythmogenic cardiomyopathy (AC), and left ventricular non-compaction (LVNC), or due to electrical abnormalities without structural heart defects, commonly known as channelopathies—predominantly long QT syndrome (LQTS), short QT syndrome (SQTS), catecholaminergic polymorphic ventricular tachycardia (CPVT), and Brugada syndrome (BrS) [1].
Despite the improvements in genetic diagnosis, closely linked to the development of high-throughput sequencing (HTS) technology, the percentage of cases that remain unexplained after genetic screening is still high, ranging from 20% to 80% depending on the disease [2]. Causality in unresolved cases may be explained by variants in as of yet non-associated genes, regulatory regions, splice sites, epigenetic alterations, or structural variants (not detectable by traditional Sanger sequencing). In the last 10 years, scientists have identified abundant and ubiquitous structural variants in the human genome, both in normal population and in disease groups. Among them stand out copy number variants (CNVs), which have been traditionally defined as DNA segments larger than one kilobase (kb) that present variable copy number in comparison with a reference genome. In the last years, several authors consider a CNV any imbalance larger than 50 base pairs [3] (bp) (this latter criterion is the one followed in the present work). Evidence supporting a role of CNVs in SCD-related pathologies has been reported, but robust studies with large cohorts of patients and multiple genes being screened have only been performed for specific SCD-related diseases [4,5,6,7,8,9,10,11,12,13,14,15,16,17,18].
The lack of information on prevalence of CNVs in SCD and related diseases encouraged our group to shed some light on this topic. We have analyzed CNVs in the most prevalent genes associated with SCD in a large cohort of patients diagnosed with a cardiomyopathy or a channelopathy, and patients who died suddenly with a suspected arrhythmogenic cause of death (sudden unexplained death, SUD).
Materials and methods
Patients
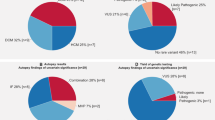
Our cohort includes 1765 unrelated European patients, and is divided in three subgroups: (a) 587 post-mortem blood samples from SUD patients (23 of them were cases of sudden infant death syndrome (SIDS)) (all cases were aged <50 y.o. and had a non-conclusive cause of death after a complete autopsy, including toxicological analyses and macroscopic and microscopic analysis of the heart); (b) 874 patients clinically diagnosed with a cardiomyopathy: 591 HCM, 136 DCM, 118 AC, and 29 LVNC; and (c) 304 patients diagnosed with a channelopathy: 151 BrS, 127 LQTS, 7 SQTS, and 19 CPVT (Fig. 1a). Samples were referred from 11 hospitals from Spain and from Institute of Legal Medicine of Catalonia. The study was approved by the Ethics Committee of Hospital Universitari Dr. Josep Trueta (Girona, Spain) and conforms to the principles outlined in the Declaration of Helsinki.

Cohort distribution a and frequency of CNVs by pathology b. AC arrhythmogenic cardiomyopathy, AF affects function, BrS Brugada syndrome, DCM dilated cardiomyopathy, HCM hypertrophic cardiomyopathy, LQTS long QT syndrome, LVNC left ventricular non-compaction, NAF does not affect function, PAF probably affects function, PNAF probably does not affect function, SIDS sudden infant death syndrome, SQTS short QT syndrome, SUD sudden unexplained death, VUS variant of unknown significance
Custom sequencing panel design and library preparation
Two custom sequencing panels, which included the coding regions and intronic boundaries of 55 or 78 genes associated with SCD, were used. The genes screened in each panel and the reference isoforms analyzed are listed in Table 1. Coordinates of sequence data were based on UCSC human genome version GRCh37/hg19. The 55-gene panel, which included the UTR (Untranslated Region) sequences for some genes, covered 432,512 kb of the human genome and was used for the screening of 701 patients. The 78-gene panel did not include UTR regions, covered 410,308 kb and was used for 1064 patients. A biotinylated complementary RNA probe solution was used to capture the regions of interest (Agilent Technologies, Santa Clara, CA, USA). Probes were designed and optimized by Gendiag.exe SL. Both custom enrichment gene designs are commercialized by Ferrer inCode as SudD inCode®.
For library preparation, genomic DNA was extracted with Chemagic Magnetic Separation Module I (PerkinElmer, Waltham, MA, USA) from post-mortem or fresh whole blood samples. DNA was fragmented with Bioruptor® (Diagenode, Seraing, Belgium) and libraries were prepared following the SureSelect XT Target Enrichment System for Illumina Paired-End Sequencing Library protocol (Agilent Technologies). Indexed libraries were sequenced in 10-sample pools per cartridge. Paired-end sequencing process was performed on a MiSeq platform (Illumina, San Diego, CA, USA), with a read length of 2 × 76 bp.
Detection of genetic variants
For the analysis of single-nucleotide variants (SNVs) and small insertions or deletions (indels), an algorithm developed by Gendiag.exe SL was used. The algorithm follows four analytic steps: (i) adaptors and low quality bases trimming; (ii) mapping with Burrows-Wheeler Aligner - Maximum Exact Match; (iii) SNV and indel detection with SAMtools v.1.2, together with an ad hoc developed script —intronic positions up to 6 bp are interrogated; and (iv) SNVs and indels annotation with dbSNP, Exome Sequencing Project, 1000 Genomes, Exome Aggregation Consortium, Human Gene Mutation Database (HGMD), ClinVar, Ensembl and in home database IDs. Only nonsynonymous variants with a minor allele frequency <1% in general population were reported and confirmed by Sanger sequencing.
The CNV detection was performed with an ad hoc developed algorithm divided in seven major steps: (i) raw coverage extraction from BAM files (using BEDtools v2.23.0); (ii) quality metrics collection and coverage correlation between samples (correlation coefficients >0.97 were expected for comparable samples; those samples displaying a generalized dispersion were removed from analysis and, therefore, not included in the cohort); (iii) raw coverage normalization by library size and GC (guanine-cytosine) content correction per region; (iv) log2-ratio calculation for every sample within each region of interest using a dynamic baseline built with normalized coverages derived from the other samples being analyzed; (v) copy number estimation: if the ratio fell outside a signal-to-noise window (±3 SD) and was greater or lower than the duplication or deletion cutoffs (0.45 and −0.8, respectively), a CNV was inferred (to avoid artifacts caused by abnormally long insert sizes in regions with short nearby exons, the algorithm scanned the surrounding introns for discordant read pairs—if no breakpoints were detected, the signal was discarded due to an anomalous gain/loss of coverage not associated with the presence of a structural variation); (vi) annotation with HGMD, ClinVar, DECIPHER, Database of Genomic Variants, 1000 Genomes, and ClinGen (if the potential CNV was present in global population and healthy individuals with a frequency >1%, it was considered a polymorphism and was not reported); and (vii) CNV quality score generation, which takes into account sample characteristics (total number of reads, enrichment of the regions of interest and differences of insert sizes among samples), signal behavior (robustness through deletion/duplication cutoffs and noise), and region characteristics (anomalous GC percentage, abnormal allelic frequencies for Single Nucleotide Polymorphisms detected in the potentially altered region, and length and distance to nearby regions). Identified CNVs were confirmed by multiplex ligation-dependent probe amplification (MLPA) (MRC-Holland, Amsterdam, The Netherlands) or quantitative polymerase chain reaction (qPCR) with QuantStudio 7 Flex System and Power Up Sybr Green master mix (Thermo Fisher Scientific, Waltham, MA, USA), following manufacturer’s recommendations. Confirmed CNVs were submitted to DECIPHER.
Classification of genetic variants
SNVs and indels were classified as variants that: affect function (AF), probably affect function (PAF), probably does not affect function (PNAF), does not affect function (NAF), or variants of uncertain significance (VUS); according to the recommendations of the Human Genome Variation Society. They are equivalent to the terms used by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology: pathogenic variant, likely pathogenic variant, likely benign variant, benign variant, or variant of uncertain significance, respectively [19].
Regarding CNVs, a CNV was considered a variant that AF if: (a) it had been previously reported as pathogenic for patient’s disease (or for SUD cases, if the reported disease is compatible with a structurally normal heart—this remark applies for all the following classifications); (b) it was a deletion in/of a gene where loss of function is a known mechanism of patient’s disease; (c) it was an intragenic in tandem duplication (not involving the last exon of the gene) in a gene where loss of function is a known mechanism of disease; or (d) it was a whole gene duplication in a gene for which triplosensitivity is known to cause patient’s disease. A CNV was considered a variant that PAF if: (a) it was a deletion in/of a gene associated with the observed disease or an intragenic duplication (not involving the last exon of the gene) in a gene associated with the disease, and the variant was absent from controls; or (b) the CNV cosegregated with the disease in >5 affected family members. A CNV was considered a variant that NAF if previously described as benign, and PNAF if: (a) identified in >10 individuals in general population (for whole gene deletions/duplications, cases of general population involving contiguous genes were considered for comparison), or (b) the CNV did not cosegregate with disease in the family. All remaining scenarios were classified as VUS. Exons involved in CNVs were numbered according to genomic reference sequences (NG_), detailed in Table 2.
Statistical analysis
Comparisons between variables were performed using the chi-square test, with STATA/IC 13.1 for Windows. Two-sided p-values < 0.05 were considered significant.
Results
Study cohort
For the 1765 patients of the cohort, the mean age at clinical diagnosis was of 39.8 ± 19.8 y.o. The majority of patients were males (68%), and their age at diagnosis was not significantly different from that of females.
Genetic screening
An exhaustive screening for SNVs and indels was carried out in the entire cohort. However, since the leading interest of the present work was to explore the frequency of CNVs in SCD and related pathologies, we only reported SNVs and indels present in those samples with CNVs (Table 2).
A total of 79 CNVs were identified in 78 out of the 1765 patients studied. Thirty-six of these variants were confirmed by MLPA or qPCR (in 36 different patients) (Table 2 and Fig. 2). The remaining 43 signals resulted to be false positives of the HTS methodology used (false discovery rate of 54.4%). Thus, the detection rate for large genomic imbalances in our cohort was 2%. The detected CNVs consisted of 18 deletions and 18 duplications, and were identified in the genes described in Table 2 (DECIPHER ID for each case is also specified in Table 2). All variants were heterozygous or hemizygous. According to our classification criteria, 14 CNVs were considered AF, 6 PAF, 14 VUS, and 2 PNAF. The majority of the CNVs identified involved several exons. The smallest CNV detected was a 104-bp deletion including exon 28 of ABCC9 gene (case S21), and the largest one was a 194-kb duplication involving >200 exons of TTN gene (case S28). It is important to specify that as we used targeted gene panels for the screening, no breakpoints could be defined for the detected CNVs, as most of them fell into intronic or intergenic regions excluded from the analysis. Consequently, the size of the detected CNVs could be larger than reported.

Graph of four of the CNVs detected in our cohort (log2 ratios). a Deletion from exons 9 to 24 of DSP in a patient with arrhythmogenic cardiomyopathy (S26, dark blue). b Deletion of exons 8 and 9 of KCNQ1 found in three long QT syndrome patients (S3 shown in the graph, brown). c Duplication from exons 8 to 10 of PKP2 identified in three arrhythmogenic cardiomyopathy patients (S22 shown in the graph, red). d Complex rearrangement involving exons 4, 5, and 8 of TNNI3 in a sudden unexplained death case (S33, light green)
Among SUD cases, the frequency of patients with CNVs was 1.4% (8/587 patients) (Table 3 and Fig. 1b), 0.5% if we only consider variants that AF or PAF (1 and 2 CNVs, respectively). This cohort included 23 cases of SIDS, and 1 of them (4.3%) had a CNV, which was classified as VUS. Regarding cardiomyopathies, we detected a frequency of CNVs in our series of: 1.2% for HCM (7/591 patients; 4 AF and 3 PAF), 4.4% for DCM (6/136 patients; 3 AF, 2 VUS, and 1 PNAF), 5.1% for AC (6/118 patients; 2 AF, 3 PAF, and 1 VUS), and 3.4% for LVNC (1/29 patients; 1 VUS) (Fig. 1b). The HCM subgroup includes 303 patients already published by our group [18]. In relation to channelopathies, the frequency for each disease in our series was: 4.7% for LQTS (6/127 patients; 4 AF, 1 PAF, and 1 VUS), 1.3% for BrS (2/151 patients; 2 VUS), 0% for SQTS (0/7 patients), and 0% for CPVT (0/19 patients) (Fig. 1b).
Algorithm validation
Prior to the realization of the present work, a validation of the algorithm for the detection of CNVs was performed using a cohort of 108 full-screened cardiovascular disease patients. Among these patients, there were 16 carriers of CNVs (the remaining patients were negative for these structural variants). The CNV sizes of the validation set ranged from 1 to 15 exons, with minimal known sizes of 123 bp and 40.1 kb, respectively. The algorithm achieved a sensitivity of 100% and a specificity of 91%. The corresponding positive predicting value was 64% and, therefore, the false discovery rate was 36%. In order to compare our algorithm with other CNV detection softwares, the same validation cohort was analyzed with CONTRA v.2.0.8 [20] and CNVKIT v.0.8.6 [21], for which effectiveness and reliability have been previously widely proved [22]. The best results were those achieved with our algorithm and CNVKIT, both in terms of accuracy (99.9%, whereas for CONTRA it was 99.6%) and sensitivity (100%, whereas for CONTRA it was 87.3%). Moreover, our algorithm gave superior precision in comparison with CNVKIT and CONTRA (85.9% versus 83.3% and 75%, respectively).
HTS panel performance
The average call rate achieved at 30× with the custom enrichment gene designs of 55 and 78 genes was 99.70% and 99.82%, respectively. The median percentage of reads overlapping our target regions was 48% for the first panel and 66% for the second one. The median coverage per sample was 870 and 679, respectively. If a region did not reach a minimum coverage of 30×, conventional Sanger sequencing was performed for that region.
Discussion
Several studies have identified CNVs as causative of cardiac diseases associated with SCD [4,5,6,7,8,9,10,11,12,13,14,15,16,17,18]. However, exhaustive analysis of multiple genes in large cohorts of patients has never been performed for most SCD-related diseases. Motivated by this lack of information, in the present study we screened for CNVs the main genes associated with SCD using HTS technology in a large cohort of SUD cases and patients diagnosed with a cardiomyopathy or a channelopathy.
Among SUD cases, the frequency of patients with CNVs was 1.4%. These results cannot be compared with previously published studies, as the present work is the first screening for CNVs in a large series of SUD cases. There is only one previous publication involving the screening for CNVs in SUD/SIDS cases [4]. The authors performed array-based comparative genomic hybridization in 27 cases of SIDS, and detected 3 CNVs. However, these CNVs were large (all of them >240 kb, involving several genes) and at least one could be associated with a syndromic phenotype.
Regarding cardiomyopathies, we detected a frequency of CNVs in our series of: 1.2% for HCM, 4.4% for DCM, 5.1% for AC, and 3.4% for LVNC. Recently, Ceyhan-Birsoy et al. published a study of similar characteristics and reported the following frequencies: 0.56% for HCM, 0.6% for DCM, 1% for AC, and 1.9% for LVNC [17]. Lopes et al. also published in 2015 a comprehensive screening for CNVs in HCM patients, and reported a frequency of 0.8% [13]. Although detection rates seem to be higher in our cohort, differences are only statistically significant for DCM, which may be due to cohort characteristics. No further studies including a comprehensive screening for CNVs in large series of patients with cardiomyopathy have been performed, but there are several additional reports of CNVs in patients with DCM [5, 6, 9, 12,], AC [15], and HCM [18].
In relation to channelopathies, the frequency of CNVs in our series was: 4.7% for LQTS, 1.3% for BrS, and 0% for SQTS and CPVT. Previously published series involving LQTS patients consist of the study of exclusively 2–5 genes (KCNQ1, KCNH2, KCNE1, KCNE2, and/or SCN5A), with a CNV detection rate of 2–11.5% [8, 10,], which is compatible with our results. In our series, five out of the six CNVs detected in LQTS patients were detected in these five genes, and were considered variants that AF/PAF. Patient S3 was published in 2014 as a case report [10]. For BrS, only the work performed by our group including the screening for SCN5A and for some cases BrS-minor genes in BrS patients has been published (63 cases screened by HTS are included in the present study), and a single duplication in SCN5A was detected [16]. An intragenic deletion in SCN5A was also previously reported in a BrS patient [7]. For CPVT, several CNVs in RYR2 have been identified [11, 14,], but other CPVT-related genes have never been screened for large genomic imbalances up to that time. On the other hand, studies involving the screening for CNVs in SQTS patients have never been previously published.
Accordingly, the CNV detection rate is variable for the different groups studied, ranging from 0% to 5.1% in our cohort. Although the knowledge of the role of CNVs in most SCD-related diseases is scarce, our results are compatible with those already published. As CNVs underlie a non-neglectable portion of cases, we consider that their analysis should be performed as part of the routine genetic testing of SUD cases and patients with SCD-related diseases. This especially applies for patients with cardiomyopathies and LQTS, as the CNV detection rate among these patients is particularly high. Further studies, mainly those focused in the poorly studied cardiac disease groups, will allow to improve the data on the prevalence of CNVs in these populations. Although large genomic imbalances have never been reported for some of the diseases included in the present work, CNVs may be the genetic cause for a portion of these patients. It is noteworthy that the identification of a disease-causing variant in a patient is crucial for diagnosis confirmation in borderline cases, early management of at-risk family members, and avoidance of unnecessary follow-up of non-carriers. Moreover, if genetic testing for SNVs and indels is performed using HTS technologies, the screening for CNVs requires no additional costs (apart from those associated with confirmation tests).
Interestingly, all the CNVs classified as variants that AF or PAF in our series were identified in patients without SNVs or indels considered responsible of the observed phenotype. Eight of these patients were sequenced with the 55-gene panel, whereas 12 with the 78-gene panel (Table 2). This suggests that the non-identification of any variant that AF or PAF is not directly related to the panel used. However, for the patients screened with the 55-gene panel, we cannot exclude the presence of SNVs and/or indels classified as AF or PAF in the not-screened genes.
Regarding the characteristics of the detected CNVs, most of them were novel (Table 2), which may be partly explained by the reduced number of studies focused on the screening of these genes and the low resolution of the techniques used for genotyping structural variants in 1000 Genomes project. On the other hand, seven recurrent CNVs have been identified in our cohort: deletion of exons 8 and 9 of KCNQ1, duplication of exons 8–10 of PKP2, deletion of exon 1 of PKP2, deletion of exons 21–23 of DSP, duplication of KCNE1 and KCNE2, deletion of exon 2 of PLN, and duplication of exons 2–11 of CASQ2 (Table 2). These recurrent CNVs may be the result of a founder effect or due to rearrangement flanking regions particularly rich in interspersed repeats, low complexity DNA sequences, or mobile genetic elements, which are genetic features that tend to generate genomic instability and promote the apparition of DNA rearrangements [23].
Family segregation studies could be performed for patients S3 [10] and S18 [18]. Cosegregation with complete penetrance was observed in both cases (two and one affected family members, respectively). The relative of patient S18 harboring the deletion of MYBPC3 was also carrier of the variants TTN_c.77716C>T and NEBL_c.326T>C, but not the variant TTN_c.6163G>A.
For many diseases, HTS technology has been progressively incorporated into clinical diagnosis, but high quality analysis is required to ensure the reliability of the results. Our CNV detection method offers high reliability for the detection of such genomic rearrangements when using custom sequencing panels (sensitivity of 100%, specificity of 91% and positive predictive value of 64%). Compared with other widely used algorithms for detection of CNVs from HTS data (CONTRA [20] and CNVKIT [21]), our algorithm is the one with higher accuracy, sensitivity, and precision altogether. In part, this is probably due to the capture probe optimization step during the HTS panel design. Our custom gene panel design and the optimized probe distribution (mainly in regions difficult to be properly sequenced) result in samples that exhibit both high coverage homogeneity across all captured regions and high median coverage per sample, despite being short read sequences obtained from a moderate capacity platform. With this sample quality, our CNV detection algorithm shows a high sensitivity with an assumable number of false positives, an imperative fact for its implementation in the routine of a genetic diagnosis laboratory. Moreover, the algorithm is able to detect single and multiple exon deletions/duplications. The detection of small single exon alterations is particularly important, as they tend to be discriminated in exome-sequencing detection assays [13, 24,]. In the present cohort, we had a false discovery rate of 54.4%. The reason for such high frequency of false positives was to avoid as much as possible false negatives, being aware of the limitations of short read sequencing and taking into account that samples were screened for diagnosis purposes. We selected some suspicious signals for validation even though the CNV detection algorithm gave them a low quality score. This resulted in an attempt to validate a set of signals (usually exons with extreme GC content and too far from other exons to detect breakpoints) that turned out to be false positives.
Finally, we believe that both interpretation and clinical translation of genetic data is the current challenge for geneticists as well as cardiologists. Current CNV interpretation guidelines are focused on the interpretation of large genomic rearrangements, generally involving multiple contiguous genes [25, 26,]. Detailed recommendations for the interpretation of intragenic CNVs do not exist, although they need special considerations. For example, duplications are generally considered to be less deleterious than deletions [25], but an intragenic duplication may disrupt a gene in the same way a deletion does. In an attempt to help in the interpretation of such rearrangements, in the present work we describe several criteria for their classification. The mainstay is if loss-of-function of a particular gene is a known mechanism of patient’s disease, which is also the stronger criterion for classifying variants as pathogenic according to the standards and guidelines for the interpretation of sequence variants [19]. If this is the case, any deletion or tandem duplication (not involving the last exon of the gene) within this gene should be considered to affect its normal function, as it will disrupt gene function by leading to a complete absence of the gene product by lack of transcription or nonsense-mediated decay of the altered transcript [19] (if a tandem duplication involves the last exon of a gene, this gene may have normal expression). This statement concerns exclusively confirmed tandem duplications, but it has to be noticed that the 83% of the duplications are tandem duplications in direct orientation [27]. On the other hand, it is important to remember the importance of having family members of patients with CNVs for its proper classification. Cosegregation of a CNV with disease in multiple affected family members supports that the variant affects the normal function of a gene and no cosegregation supports benignity, thus reducing the proportion of VUS. It is also worth mentioning that a portion of the CNVs considered VUS in our cohort because the altered gene had not been previously related to the patient’s disease could be in fact variants that AF or PAF, because it is widely known that the same gene (and even the same genetic variant) can be associated with different SCD-related diseases [28].
In summary, we report the frequency of CNVs in SUD cases and patients diagnosed with a cardiomyopathy or a channelopathy, and interpret the results for its translation into clinics. Although CNVs only explain a small portion of cases for most SCD-related diseases, we support the screening for such rearrangements as part of routine clinical testing with the ultimate aim of identifying the cause of the patient’s disease and providing an appropriate clinical assessment, genetic counseling, and preventive measures for patients and their relatives.
References
Deo R, Albert CM. Epidemiology and genetics of sudden cardiac death. Circulation. 2012;125:620–37.
Ackerman MJ, Priori SG, Willems S, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm. 2011;8:1308–39.
MacDonald JR, Ziman R, Yuen RKC, Feuk L, Scherer SW. The database of genomic variants: a curated collection of structural variation in the human genome. Nucleic Acids Res. 2014;42(Database issue):D986–992.
Toruner GA, Kurvathi R, Sugalski R, et al. Copy number variations in three children with sudden infant death. Clin Genet. 2009;76:63–8.
Norton N, Siegfried JD, Li D, Hershberger RE. Assessment of LMNA copy number variation in 58 probands with dilated cardiomyopathy. Clin Transl Sci. 2011;4:351–2.
Norton N, Li D, Rieder MJ, et al. Genome-wide studies of copy number variation and exome sequencing identify rare variants in BAG3 as a cause of dilated cardiomyopathy. Am J Hum Genet. 2011;88:273–82.
Eastaugh LJ, James PA, Phelan DG, Davis AM. Brugada syndrome caused by a large deletion in SCN5A only detected by multiplex ligation-dependent probe amplification. J Cardiovasc Electrophysiol. 2011;22:1073–6.
Barc J, Briec F, Schmitt S, et al. Screening for copy number variation in genes associated with the long QT syndrome: clinical relevance. J Am Coll Cardiol. 2011;57:40–7.
Herman DS, Lam L, Taylor MRG, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366:619–28.
Campuzano O, Sarquella-Brugada G, Mademont-Soler I, et al. Identification of genetic alterations, as causative genetic defects in long QT syndrome, using next generation sequencingtechnology. PLoS ONE. 2014;9:e114894.
Bhuiyan ZA, van den Berg MP, van Tintelen JP, et al. Expanding spectrum of human RYR2-related disease: new electrocardiographic, structural, and genetic features. Circulation. 2007;116:1569–76.
Truszkowska GT, Bilińska ZT, Kosińska J, et al. A study in Polish patients with cardiomyopathy emphasizes pathogenicity of phospholamban (PLN) mutations at amino acid position 9 and low penetrance of heterozygous null PLN mutations. BMC Med Genet. 2015;16:21.
Lopes LR, Murphy C, Syrris P, et al. Use of high-throughput targeted exome-sequencing to screen for copy number variation in hypertrophic cardiomyopathy. Eur J Med Genet. 2015;58:611–6.
Campbell MJ, Czosek RJ, Hinton RB, Miller EM. Exon 3 deletion of ryanodine receptor causes left ventricular noncompaction, worsening catecholaminergic polymorphic ventricular tachycardia, and sudden cardiac arrest. Am J Med Genet A. 2015;167A:2197–200.
Sonoda K, Ohno S, Otuki S, et al. Quantitative analysis of PKP2 and neighbouring genes in a patient with arrhythmogenic right ventricular cardiomyopathy caused by heterozygous PKP2 deletion. Europace. 2017;19:644–50.
Mademont-Soler I, Pinsach-Abuin ML, Riuró H, et al. Large genomic imbalances in Brugada syndrome. PLoS ONE. 2016;11:e0163514.
Ceyhan-Birsoy O, Pugh TJ, Bowser MJ, et al. Next generation sequencing-based copy number analysis reveals low prevalence of deletions and duplications in 46 genes associated with genetic cardiomyopathies. Mol Genet Genom Med. 2016;4:143–51.
Mademont-Soler I, Mates J, Yotti R, et al. Additional value of screening for minor genes and copy number variants in hypertrophic cardiomyopathy. PLos ONE. 2017;12:e0181465.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med J Am Coll Med Genet. 2015;17:405–24.
Li J, Lupat R, Amarasinghe KC, Thompson ER, et al. CONTRA: copy number analysis for targeted resequencing. Bioinformatics. 2012;28:1307–13.
Talevich E, Shain AH, Botton T, Bastian BC. CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput Biol. 2016;12:e1004873.
Hwang JA, Kim D, Chun SM, et al. Genomic profiles of lung cancer associated with idiopathic pulmonary fibrosis. J Pathol. 2018;244:25–35.
Toffolatti L, Cardazzo B, Nobile C, et al. Investigating the mechanism of chromosomal deletion: characterization of 39 deletion breakpoints in introns 47 and 48 of the human dystrophin gene. Genomics. 2002;80:523–30.
Pfundt R, Del Rosario M, Vissers LELM, et al. Detection of clinically relevant copy-number variants by exome sequencing in a large cohort of genetic disorders. Genet Med J Am Coll Med Genet. 2017;19:667–75.
Miller DT, Adam MP, Aradhya S, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86:749–64.
Kearney HM, Thorland EC, Brown KK, Quintero-Rivera F, South ST, Working Group of the American College of Medical Genetics Laboratory Quality Assurance Committee. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med J Am Coll Med Genet. 2011;13:680–5.
Newman S, Hermetz KE, Weckselblatt B, Rudd MK. Next-generation sequencing of duplication CNVs reveals that most are tandem and some create fusion genes at breakpoints. Am J Hum Genet. 2015;96:208–20.
Bezzina C, Veldkamp MW, van Den Berg MP, et al. A single Na(+) channel mutation causing both long-QT and Brugada syndromes. Circ Res. 1999;85:1206–13.
Kalman L, Leonard J, Gerry N, et al. Quality assurance for Duchenne and Becker muscular dystrophy genetic testing: development of a genomic DNA reference material panel. J Mol Diagn. 2011;13:167–74.
Acknowledgements
We acknowledge the CIBERCV, an initiative of the Instituto de Salud Carlos III/Spanish Ministry of Economy and Competitiveness (Fondos FEDER). The sources of funds supporting the present work are: Fundació “Obra social La Caixa” (IP: Ramon Brugada), Instituto de Salud Carlos III—Fondo Investigación Sanitaria FIS PI14/01773 (IP: Ramon Brugada), Sociedad Española de Cardiología (Proyecto Investigación Básica Cardiología 2015 de los Socios Estratégicos SEC) (IP: Oscar Campuzano). Dr. JM is a beneficiary of the PFIS predoctoral fellowship FIS.09.01.06/13–146, from Instituto de Salud Carlos III.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
RB is consultant of FerrerInCode. Gendiag.exe SL provided support in the form of salaries for authors Dr. CF-C and Dr. PÁ, but did not have any additional role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. The remaining authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Mates, J., Mademont-Soler, I., del Olmo, B. et al. Role of copy number variants in sudden cardiac death and related diseases: genetic analysis and translation into clinical practice. Eur J Hum Genet 26, 1014–1025 (2018). https://doi.org/10.1038/s41431-018-0119-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-018-0119-1
This article is cited by
-
EMQN: Recommendations for genetic testing in inherited cardiomyopathies and arrhythmias
European Journal of Human Genetics (2023)
-
Detection of the Copy Number Variants of Genes in Patients with Familial Cardiac Diseases by Massively Parallel Sequencing
Molecular Diagnosis & Therapy (2023)
-
Molecular Diagnosis of Inherited Cardiac Diseases in the Era of Next-Generation Sequencing: A Single Center’s Experience Over 5 Years
Molecular Diagnosis & Therapy (2021)
-
Re-evaluation of single nucleotide variants and identification of structural variants in a cohort of 45 sudden unexplained death cases
International Journal of Legal Medicine (2021)
-
Epidemiologie des Kreislaufstillstands in Europa
Notfall + Rettungsmedizin (2021)
