
Abstract
Familial hypercholesterolemia (FH) is caused by mutations in LDLR (low-density lipoprotein receptor), APOB (apolipoprotein B), PCSK9 (proprotein convertase subtilisin/kexin type 9), or APOE (apolipoprotein E) genes in approximately 80% of the cases. Polygenic forms of hypercholesterolemia may be present among patients clinically diagnosed with FH but with no identified mutation (FH mutation-negative (FH/M−)). To address whether polygenic forms may explain phenocopies in FH families, we calculated a 6-single-nucleotide polymorphism (SNP) genetic risk score (GRS) in all members from five French FH families where a mutation was identified (FH/M+) as well as some phenocopies (FH/M−). In two families, three FH/M− patients present a high GRS suggesting a polygenic hypercholesterolemia for these phenocopies. However, a high GRS is also observed in nine FH/M+ patients and in four unaffected relatives from three families. These observations indicate that the GRS does not seem to be a good diagnostic tool at the individual level. Nevertheless, the GRS seems to be a contributor of the severity of hypercholesterolemia since patients who cumulate a mutation and a high GRS exhibit higher low-density lipoprotein cholesterol levels when compared to patients with only FH (p = 0.054) or only polygenic hypercholesterolemia (p = 0.0039). In conclusion, the GRS can be used as a marker of the severity of hypercholesterolemia but does not seem to be a reliable tool to distinguish phenocopies within FH families.
Similar content being viewed by others


Introduction
Familial hypercholesterolemia (FH) (OMIM #143890) is an inherited disorder of lipoprotein metabolism characterized by severe elevation of low-density lipoprotein cholesterol (LDL-C) levels responsible of premature atherosclerotic plaque deposition in the coronary arteries leading to higher risk of coronary heart disease (CHD) [1, 2]. In addition to CHD, individuals with heterozygous hypercholesterolemia (HeFH) can develop tendinous xanthomas, xanthelasma, and corneal arcus.
The prevalence of HeFH is estimated to be 1/300–1/200, suggesting that there are between 14 and 34 million individuals with FH worldwide. These data can vary in certain populations with founder effect where higher prevalence are observed in Afrikaners, French Canadians, Lebanese, and Finns [1, 3].
FH is caused by mutations in four genes: those encoding the LDL receptor (LDLR at 19p13.3; OMIM #606945) [4, 5], its ligands apolipoprotein B (APOB at 2p23–p24; OMIM #107730) [6] and apolipoprotein E (APOE at 19q13.32; OMIM #107741) [7], and the proprotein convertase subtilisin/kexin type 9 gene (PCSK9 at 1p32.3; OMIM #607786) [8]. The discovery of the LDLR and then its defective function led to a great advance in the understanding of the pathophysiology of FH [4]. Few years later, in 1987, the contribution of APOB-causing variants to the FH phenotype was shown [6]. In 2003, after the exclusion of LDLR and APOB genes and through a positional cloning strategy in two large French FH families, our team identified PCSK9, the third FH-causing gene which is actually a major therapeutic target in the treatment of FH [8]. Lately, in 2013, we identified a causative variant in the APOE gene in a large French FH family and showed that screening of APOE, along with LDLR, APOB, and PCSK9, is warranted in the molecular diagnosis of FH genes [7]. We then proposed to rename this genetically heterogeneous hypercholesterolemia ADH for “Autosomal Dominant Hypercholesterolemia” including FH initially defined for LDLR defects, FDB (familial ligand-defective apoB-100 caused by variants in APOB), and hypercholesterolemia due to mutations in PCSK9, APOE, and other genes. However, since a large majority of the literature refers to FH for all types of molecular defects, we will use the term FH in this article to encompass all monogenic forms of hypercholesterolemia.
FH-causing variants in LDLR, APOB, PCSK9, and APOE cover around 80% of FH cases [9]. Several reasons can explain mutation-negative cases: current methods fail to detect all DNA changes, the defects are present in uncovered genetic regions, there is a possible existence of other genes which are yet to be discovered and might become new targets for lipid-lowering therapy [10,11,12]. However, an FH-causing variant in a novel gene would be very rare [10, 13]. The Global Lipid Genetic Consortium (GLGC) meta-analysis of genome-wide association studies identified several loci where common variants (single-nucleotide polymorphisms (SNPs)) affect LDL-C concentrations [14]. In 2013, Talmud et al. [15] genotyped participants for 12 of these SNPs and constructed a weighted LDL-C raising gene score (genetic risk score or GRS). They showed that this GRS may explain the high LDL-C level observed in the 772 FH mutation-negative (FH/M−) patients due to the combined impact of several LDL-C raising SNPs. They also demonstrated that in polygenic hypercholesterolemia, the proportion of relatives who are likely to also have raised LDL-C is <50% predicted for monogenic hypercholesterolemia [15]. Futema et al. [11] in 2015, by using statistical methods, showed that a score of 6 SNPs performed as well as the 12-SNP score in discriminating between FH/M− patients and control individuals [11]. It should be noted that the GRS was associated with hypercholesterolemia in patients with and without a defect in the four known FH genes. However, Sjouke et al. [12], in their study of 2016, showed that FH/M− patients do not have a higher GRS than unaffected individuals or patients with known mutation. They concluded that the GRS could not be used as a diagnostic tool to define clinically diagnosed FH patients with polygenic hypercholesterolemia [12].
In this study, we selected five families with causative variant in one of the FH genes and calculated the GRS, corresponding to the weighted sum of 6 LDL-C raising SNPs, in all family members. Our aim is to address whether polygenic forms may explain phenocopies for FH/M− relatives, and to see if the GRS can be considered a good diagnostic tool at the individual level, and thus could help to phenotype family members for linkage analysis in the search of new FH genes.
Materials/subjects and methods
Patients
Through the French Research Network for FH, which includes 14 clinical centers in France, more than 3000 hypercholesterolemic probands were recruited. Patients were eligible if they had a total LDL-C values above the 95th percentile when compared to a sex- and age-matched French population (STANISLAS cohort) [16] (B Herbeth, G Siest, and S Visvikis-Siest, personal communication 2009), triglycerides below 1.5 mmol/L (133 mg/dL), normal HDL-C levels, personal and/or familial xanthomas, and/or early coronary artery disease. Exclusion criteria comprised all diseases-causing secondary hypercholesterolemia.
Molecular genetic analysis
Blood samples were obtained from all recruited subjects in accordance with French bioethics regulations and written, informed consent was obtained for all subjects. Genomic DNA was isolated using standard methods. Direct Sanger sequencing was performed to search for variants in all 18 exons of the LDLR gene, all 12 exons of the PCSK9 gene, all 4 exons of the APOE gene, and a part of APOB gene encoding the binding domain (from amino acids 3157 to 3657 in exon 26 and from amino acids 4181 to 4540 in exon 29). Multiplex ligation-dependent probe amplification (MLPA) was also performed to detect gross deletions and insertions in the LDLR gene. This allows us to identify variants in these four FH-causing genes in more than 80% of the probands. The families of some of these FH/M+ probands were enlarged to study the segregation of the identified variant and to confirm the presence of a monogenic form of the disease. The p.(Leu167del) in APOE was submitted to LOVD database (http://www.lovd.nl/) (patient’s ID: CAD966), whereas the four remaining variants were submitted to ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/) under the accession numbers: SUB3073981 for the p.(Ala3396Thr) in APOB, SCV000503221 for the p.(Glu228Lys) in LDLR, SUB3141089 for the c.(1586 + 1_1587−1)_(1845 + 1_1846−1)del in LDLR, and SCV000503500 for the p.(Ser127Arg) in PCSK9.
Genotyping
DNA samples of 70 individuals from five different families with an FH-causing variant were genotyped using TaqMan® SNP Genotyping Assays for discriminating between two alleles of a specific SNP. A set of 6 SNPs based on the GLGC LDL-C gene score was used to genotype 32 FH/M+ patients, 5 FH/M− patients, and 33 unaffected family members. These 6 SNPs include 4 LDL-C-associated SNPs and 2 SNPs (rs429358:C>T and rs7412:T>C) that determine APOE haplotypes (E2/E3/E4) and result in different isoforms of the apoE protein which is a major determinant of LDL-C levels [15].
GRS calculation
For each individual, the GRS was calculated using the weighted sum of the risk allele for the 4 selected SNPs plus the 2 APOE SNPs. The weights used correspond to the beta coefficient reported by the GLGC as previously described [15] (Table 1). GRS were then compared to those of 3020 normocholesterolemic men and women of European ancestry from the UK Whitehall II (WHII) cohort study (SE Humphries and M Futema, personal communication). Scores in quartile I have a strong probability of monogenic FH while scores in quartile IV have strong probability of polygenic hypercholesterolemia.
MoM calculation
The MoM (multiple of median) was calculated for each individual. The MoM is a measure of how far an individual test result deviates from the median of a reference population. It allows the comparison of LDL-C levels adjusted for age and gender using a French population (STANISLAS cohort) [16]. It is a ratio determined by the following:
Statistical analysis
Statistical analyses were performed using GraphPad Prism 7.03. Unpaired t test was used to assess differences between conditions. A value of p < 0.05 was considered statistically significant for all experiments.
Results
Patients characteristics and mutation spectrum
Sanger sequencing, MLPA, and whole-exome sequencing allowed us to identify five families with FH-causing variant which were included in this study (Table 2). A total of 70 individuals were genotyped: 5 index cases and 65 relatives. Pedigree of studied families, LDL-C levels and GRS are shown in Fig. 1. Mean LDL-C levels (before treatment) are 6.62 ± 2.20 mmol/L in FH/M+ patients, 5.01 ± 0.89 mmol/L in FH/M− patients, and 3.22 ± 0.68 mmol/L in unaffected relatives. A variant in one FH-causing gene segregates in each family and few phenocopies are present in some of these families.

a–e Pedigree of studied families. The characteristics of each individual are represented as following: individual number, age (in years), LDL-C levels (in mmol/L), GRS, quartile, and the segregation of the mutation (+/− indicates the presence of the mutation in its heterozygous state and −/− indicates the absence of the mutation). Individuals with clinically diagnosed FH are indicated by black symbols; gray symbols are for individuals with borderline LDL-C levels; normocholesterolemic relatives are shown as unfilled symbols. Circles represent females; squares represent males. Symbols with a slash through them indicate deceased subjects. *LDL-C values under therapy
Genetic risk score
In each family, the calculated GRS for FH/M− patients was compared to GRS identified in unaffected or FH/M+ relatives in order to determine if the hypercholesterolemia in these phenocopies could be explained by a polygenic origin.
The recruitment of HC126 family (Fig. 1a) was initiated through the proband II-2 and expanded to 34 individuals over 4 generations. We have previously identified a deletion, p.(Leu167del) (c.500_502delTCC; rs746494694), in the APOE gene [7]. This variant was found in all affected patients of this family, except in patients I-3, II-15, III-16, and III-17. Two of these four phenocopies, I-3 and III-16, have a high GRS (0.931 and 0.831, respectively) in the top quartile which is in favor of a polygenic hypercholesterolemia. The two remaining phenocopies, II-15 and III-17, have a GRS of 0.681 and 0.781, respectively, in the quartile III and thus we cannot confirm the presence of a polygenic hypercholesterolemia. Note that individuals II-16 and III-10, two unaffected relatives with normal LDL-C, also have a high GRS in quartile IV. Interestingly, all affected mutation carriers have a GRS in quartile I or II, except for individuals II-3 and III-7 whose GRS is in quartile III (0.681 for both). In these two patients, LDL-C levels are significantly higher (mean MoM: 2.80 ± 0.14) than in the other mutation carriers (N = 9; mean MoM: 1.77 ± 0.52; p = 0.0282).
The HC138 family (Fig. 1b) comprises five FH patients with a missense variant in the APOB gene, p.(Ala3396Thr) (c.10186G>A) [17]. Three of them (II-1, III-2, and III-4) also have a high GRS in quartile IV, whereas in the two others (I-1 and II-4) the GRS are lower (quartiles I and III, respectively). The two children (III-2 and III-4) with the APOB variant have different LDL-C levels (5.0 and 6.7 mmol/L, respectively) which can be explained by the difference in their GRS (0.831 and 0.931, respectively).
In family HC218 (Fig. 1c), all affected individuals have high GRS in quartile IV, which is in favor of a polygenic hypercholesterolemia in the FH/M− patient III-2. However, in the four remaining FH patients with the variant in the LDLR gene, p.(Glu228Lys) (c.682G>A; rs121908029), the GRS is also high in quartile IV. In addition, the unaffected individual III-1 also has a high GRS in quartile IV like individuals II-6 and III-10 from the HC126 family (Fig. 1a).
The eight FH patients of family HC436 (Fig. 1d) have a major rearrangement in the LDLR gene, c.(1586 + 1_1587−1)_(1845 + 1_1846−1)del, where exons 11 and 12 are deleted. No phenocopies are found in this family. The GRS of four patients (III-2, III-4, III-5, and III-6) is low, in quartile I or II, which is in favor of a monogenic form of hypercholesterolemia in this family. In some patients of this family, the GRS can be considered as a contributor of the severity of the hypercholesterolemia. Indeed, patient III-5 with a GRS of 0.580 in quartile II has a higher LDL-C levels than III-6 with a GRS of 0.480 in quartile I (4.6 and 4.2 mmol/L, respectively).
In family HC806 (Fig. 1e), the missense mutation p.(Ser127Arg) (c.381 T > A; rs28942111) in PCSK9 gene is responsible of the hypercholesterolemic phenotype in the affected family members. No phenocopies are identified in this family. Two affected members, II-2 and II-4, have high GRS of 0.831 and 0.931, respectively, in quartile IV. Their LDL-C levels are also higher than III-2 (same age and sex) who has a GRS in quartile III. Concerning the four unaffected individuals, their GRS are in quartile II and III, except the individual II-3 whose GRS of 0.860 is in the top quartile. This 56-year-old woman has higher MoM (1.29) than the three other unaffected individuals (mean MoM = 0.93 ± 0.14).
The 70 participants of this study were then divided into seven groups: unaffected subjects with a GRS in quartile I, II, III, or IV (1–4), hypercholesterolemic patients with strong probability of polygenic hypercholesterolemia (FH/M− and GRS in quartile IV) (5), hypercholesterolemic patients with strong probability of monogenic FH (FH/M+ and GRS in quartile I) (6), and patients who cumulate the familial variation and a high GRS (FH/M+ and GRS in quartile IV) (7) (Table 3). The LDL-C levels were not different between the four groups of unaffected subjects who have significantly lower levels (mean MoM: 0.96 ± 0.16) than patients with either a strong probability of polygenic hypercholesterolemia (mean MoM 1.38 ± 0.02; p = 0.0005), a monogenic FH (mean MoM 1.5 ± 0.52; p < 0.0001), or the cumulative effect of the familial variation and a GRS in the top quartile (mean MoM: 2.23 ± 0.37; p = < 0.0001). This latter group of patients exhibit the most severe phenotype with significantly higher LDL-C levels than patients with only polygenic hypercholesterolemia (p = 0.0039). They also present higher LDL-C levels than monogenic FH patients, but the difference is not significant.
Discussion
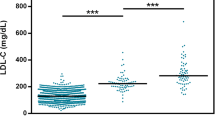
FH is characterized by a high genetic heterogeneity [18] with a least four causal genes, the presence of many others loci (16q22.1, 8q24.22, 3q25.1-26.1, 21q22.3) and an allelic heterogeneity among mutations in the LDLR gene. In a French population, our team reported that carriers of mutations in the LDLR or PCSK9 gene had the highest LDL-C levels, whereas mutations in the APOB gene were associated with a less severe phenotype [19]. We conducted this study in five families with a clear monogenic inheritance of FH due to mutations in the four FH-causing genes. Despite the genetic heterogeneity observed in these families, there is no significant difference in LDL-C levels between the five molecular groups (Fig. 2).

Mean MoM in the five families. Mean MoM of FH/M+ patients is calculated in each family. N represents the number of FH/M+ patients
The weighted 6-SNP GRS was calculated in all recruited family members. Our first observation is that we cannot use the GRS to distinguish phenotypically affected and unaffected individuals from the same family because FH patients did not exhibit higher GRS than controls. In family HC126, the GRS of patients I-3 who has a high LDL-C levels (5.7 mmol/L) is in quartile IV like the GRS of individuals II-16 and III-10, two unaffected individuals in the family. In this family, which is the only family large enough to allow the formation of homogeneous groups of patients, no significant difference (p = 0.18) is observed between the mean GRS of FH patients (0.60 ± 0.15, n = 15) and the mean GRS of unaffected relatives (0.51 ± 0.24, n = 17). The same result was obtained by Sjouke et al. [12] who conclude that phenotypically affected and unaffected individuals could not be distinguished based on the risk scores in their study population.
The second conclusion is that the GRS cannot always explain phenocopies in families with an identified causative mutation. Unlike the results obtained in cohorts, at the individual’s level, the GRS is not always higher in FH/M− compared to FH/M+ and unaffected relatives. This is observed in this study even with a small number of phenocopies. In family HC218, for example, FH/M+ and FH/M− patients have their GRS in the top quartile. It is possible that FH/M− patients with a low GRS have another unknown cause such as an unidentified gene since only 20% of FH/M− patients are likely to have a polygenic explanation for their high LDL-C levels [20]. Concerning unaffected individuals with high GRS in quartile IV (II-16 and III-10 in family HC126, III-1 in family HC218, and II-3 in family HC806), the presence of an unknown factor can reduce the effect of the high GRS and explain the normal LDL-C levels. It can be: (1) an LDL-C-lowering variant in the APOB gene which abolish the hypercholesterolemia phenotype since we have not sequenced the whole gene [21], (2) the combined impact of several LDL-C-lowering SNPs such as rs6511720 and rs57217136 located in intron 1 of the LDLR [22] and rs11591147 (p.(Arg46Leu)) in PCSK9 [23] that have been reported to be associated with lower levels of LDL-C, (3) loss-of-function mutations in ANGPTL3 [21], or (4) environmental factors.
Nevertheless, in this study, we can conclude that the GRS can be a contributor of the severity of hypercholesterolemia in affected patients and may explain the variability of the FH phenotype observed among patients in the same family. In family HC126, two affected mutation carriers have a relatively high GRS (in quartile III) and higher LDL-C levels than other mutation carriers with a low GRS (in quartile I and II): patients II-3 and III-7 (mean MoM: 2.8 ± 0.14) have higher LDL-C than other mutation carriers (N = 9, mean MoM: 1.78 ± 0.53; p = 0.0282). Also, in family HC218, patient II-1 has higher LDL-C levels than II-4 (MoM = 2.40 and 2.31, respectively) and its GRS is also higher (1.031 and 0.860, respectively). However, two exceptions should be noted. In family HC218, the twin sisters III-4 and III-5 have the same GRS (0.831) in quartile IV, but their LDL-C levels are very different (9.25 and 6.70 mmol/L, respectively). In these patients with the same GRS, non-genetic factors can explain the difference in the LDL-C levels. This association between the GRS and lipid traits was also demonstrated in a study conducted by Paquette et al. [24] in 725 individuals who carried an FH-causing mutation. They divided their score into tertile and showed that patients in the highest GRSLDL tertile presented higher LDL-C levels than subjects in the lowest tertile (p = 0.01). They also showed that the GRSLDL is not associated with risk of coronary artery disease or cardiovascular disease [24]. Furthermore, when individuals were divided into seven groups based on their mutational status and their GRS (Table 3), carriers of a variation in one FH-causing gene with a high GRS (quartile IV) have the highest MoM (2.23 ± 0.37) compared to carriers of the mutation only (1.5 ± 0.52, p = 0.054) or affected individual with high GRS only (1.38 ± 0.02, p = 0.0039). Therefore, patients who combine a high GRS and the presence of the FH-causing variants exhibit a higher LDL-C level compared to patients with monogenic FH or polygenic hypercholesterolemia alone. Moreover, patients with monogenic FH have higher LDL-C levels (mean MoM: 1.5 ± 0.52) than those with polygenic hypercholesterolemia (mean MoM: 1.38 ± 0.02), but the difference is not significant probably due to the small sample size. However, other teams suggested that the accumulation of “LDL-C burden” was higher in FH patients since they have had genetically determined lifelong high LDL-C levels. While patients with polygenic hypercholesterolemia reach the LDL-C treshold of FH patients later in life [20].
Previous studies, especially studies that defined and calculated the GRS, did not include a family history of hypercholesterolemia. These studies were conducted on independent cohorts of patients diagnosed with FH and controls. Our study, however, includes probands and their families where a mutation in an FH-causing gene was identified. Our aim was to explain phenocopies reported in these FH families using the GRS. Unlike cohorts, the GRS analysis on individual samples is not very straightforward and does not give us a clear answer. Thus, the GRS cannot be considered as a reliable tool to diagnose clinically defined hypercholesterolemic patients with polygenic hypercholesterolemia as stated by Sjouke et al. [12]
Some limitations must be taken into consideration. First is the small number of families included in the study (5 FH families) and the small number of individuals genotyped in each family. Except the family HC126 which can be considered as a large family with 15 FH patients and 19 unaffected individuals, the 4 remaining families have a small number of participants, especially FH/M− patients. Therefore, we might miss the presence of effective phenotype/genotype correlation. A second limitation is the genetic score itself which is based on a few polymorphisms. We calculated the GRS according to cohort studies showing that a 6-SNP score performed as well as the 12-SNP score and that increasing the number of SNPs to 33 did not improve the ability of the score to discriminate between FH/M− and controls [11]. We hypothesized in this study that this is also true for family studies. However, the six SNPs included in the GRS have a relatively high frequency in the general population, and with a small number of combinations, it is very likely to lose sensitivity. A score based on a greater number of less frequent SNPs with a high effect on plasma LDL-C levels might be more sensitive.
In conclusion, the 6-SNP score used in this study does not seem to be a reliable diagnostic tool to identify polygenic hypercholesterolemia at the individual level and to explain phenocopies within FH families.
References
Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013;34:3478–3490a.
Cartier JL, Goldberg AC. Familial hypercholesterolemia: advances in recognition and therapy. Prog Cardiovasc Dis. 2016;59:125–34.
de Ferranti SD, Rodday AM, Mendelson MM, Wong JB, Leslie LK, Sheldrick RC. Prevalence of familial hypercholesterolemia in the 1999 to 2012 United States National health and nutrition examination surveys (NHANES). Circulation. 2016;133:1067–72.
Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986;232:34–47.
Goldstein JL, Schrott HG, Hazzard WR, Bierman EL, Motulsky AG. Hyperlipidemia in coronary heart disease. II. Genetic analysis of lipid levels in 176 families and delineation of a new inherited disorder, combined hyperlipidemia. J Clin Invest. 1973;52:1544–68.
Innerarity TL, Mahley RW, Weisgraber KH, et al. Familial defective apolipoprotein B-100: a mutation of apolipoprotein B that causes hypercholesterolemia. J Lipid Res. 1990;31:1337–49.
Marduel M, Ouguerram K, Serre V, Bonnefont-Rousselot D, Marques-Pinheiro A, Berge KE, et al. Description of a large family with autosomal dominant hypercholesterolemia associated with the APOE p.Leu167del mutation. Hum Mutat. 2013;34:83–87.
Abifadel M, Varret M, Rabès J-P, Allard D, Ouguerram K, Devillers M, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34:154–6.
Wang J, Dron JS, Ban MR, Robinson JF, McIntyre AD, Alazzam M, et al. Polygenic versus monogenic causes of hypercholesterolemia ascertained clinically. Arterioscler Thromb Vasc Biol. 2016;36:2439–45.
Futema M, Plagnol V, Li K, Whittall RA, Andrew H, Neil W, et al. Whole exome sequencing of familial hypercholesterolaemia patients negative for LDLR/APOB/PCSK9 mutations. J Med Genet. 2014;51:537–44.
Futema M, Shah S, Cooper JA, Li KW, Whittall RA, Sharifi M, et al. Refinement of variant selection for the LDL cholesterol genetic risk score in the diagnosis of the polygenic form of clinical familial hypercholesterolemia and replication in samples from 6 countries. Clin Chem. 2015;61:231–8.
Sjouke B, Tanck MWT, Fouchier SW, Defesche JC, Hutten BA, Wiegman A, et al. Children with hypercholesterolemia of unknown cause: Value of genetic risk scores. J Clin Lipidol. 2016;10:851–9.
Kwon M, Han SM, Kim D-I, Rhee MY, Lee BK, Ahn YK, et al. Evaluation of polygenic cause in Korean patients with familial hypercholesterolemia—a study supported by Korean Society of Lipidology and Atherosclerosis. Atherosclerosis. 2015;242:8–12.
Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–13.
Talmud PJ, Shah S, Whittall R, Futema M, Howard P, Cooper JA, et al. Use of low-density lipoprotein cholesterol gene score to distinguish patients with polygenic and monogenic familial hypercholesterolaemia: a case-control study. Lancet Lond Engl. 2013;381:1293–301.
Siest G, Visvikis S, Herbeth B, Gueguen R, Vincent-Viry M, Sass C, et al. Objectives, design and recruitment of a familial and longitudinal cohort for studying gene-environment interactions in the field of cardiovascular risk: the Stanislas cohort. Clin Chem Lab Med. 1998;36:35–42.
Stitziel NO, Peloso GM, Abifadel M, Cefalu AB, Fouchier S, Motazacker MM, et al. Exome sequencing in suspected monogenic dyslipidemias. Circ Cardiovasc Genet. 2015;8:343–50.
Varret M, Abifadel M, Rabès J-P, Boileau C. Genetic heterogeneity of autosomal dominant hypercholesterolemia. Clin Genet. 2008;73:1–13.
Marduel M, Carrié A, Sassolas A, Devillers M, Carreau V, Di Filippo M, et al. Molecular spectrum of autosomal dominant hypercholesterolemia in France. Hum Mutat. 2010;31:E1811
Sharifi M, Higginson E, Bos S, Gallivan A, Harvey D, Li KW, et al. Greater preclinical atherosclerosis in treated monogenic familial hypercholesterolemia vs. polygenic hypercholesterolemia. Atherosclerosis. 2017;263:405–11.
Huijgen R, Sjouke B, Vis K, de Randamie JS, Defesche JC, Kastelein JJ, et al. Genetic variation in APOB, PCSK9, and ANGPTL3 in carriers of pathogenic autosomal dominant hypercholesterolemic mutations with unexpected low LDL-Cl Levels. Hum Mutat. 2012;33:448–55.
Fairoozy RH, White J, Palmen J, Kalea AZ, Humphries SE. Identification of the functional variant(s) that explain the low-density lipoprotein receptor (LDLR) GWAS SNP rs6511720 association with lower LDL-C and risk of CHD. PLoS ONE. 2016;11:e0167676.
Cohen JC, Boerwinkle E, Mosley TH, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354:1264–72.
Paquette M, Chong M, Thériault S, Dufour R, Paré G, Baass A, Polygenic risk score predicts prevalence of cardiovascular disease in patients with familial hypercholesterolemia. J Clin Lipidol. 2017;11:725–732.e5.
Acknowledgements
We thank all patients and their relatives for their participation in this study. We would like to thank Dr. Marta Futema and Pr. Steve Humphries for constructive discussions and for providing us some of their data. This work was supported by a grant from Leducq Foundation (FLQ # 13 CVD 03) through the Transatlantic Networks of Excellence in Cardiovascular Research program (“The function and regulation of PCSK9: a novel modulator of LDLR activity”); and Institut National de la Santé et de la Recherche Médicale (INSERM); and Conseil de la Recherche de l’Université Saint-Joseph, Beirut, Lebanon; and Lebanese National Council for Scientific Research (CNRS-L). This work was also supported by the national project CHOPIN (CHolesterol Personalized Innovation) granted by the Agence Nationale de la Recherche (ANR-16-RHUS-0007) and coordinated by the Centre Hospitalo-Universitaire (CHU) de Nantes. YG and SE are supported by grants from Lebanese National Council for Scientific Research (CNRS-L); and Council of Research of Saint-Joseph University of Beirut, Lebanon and Institut Français du Liban.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ghaleb, Y., Elbitar, S., El Khoury, P. et al. Usefulness of the genetic risk score to identify phenocopies in families with familial hypercholesterolemia?. Eur J Hum Genet 26, 570–578 (2018). https://doi.org/10.1038/s41431-017-0078-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-017-0078-y
This article is cited by
-
Age and sex specific effects of APOE genotypes on ischemic heart disease and its risk factors in the UK Biobank
Scientific Reports (2021)
-
The complex molecular genetics of familial hypercholesterolaemia
Nature Reviews Cardiology (2019)
-
The Present and the Future of Genetic Testing in Familial Hypercholesterolemia: Opportunities and Caveats
Current Atherosclerosis Reports (2018)
