
Abstract
Communesins, isolated from the mycelium of a strain of Penicillium sp., are cytotoxic heptacyclic indole alkaloids bearing a bis-aminal structure and two contiguous quaternary carbon centers. Toward a total synthesis of communesin F, we synthesized a pentacyclic ABCDG ring skeleton via carboborylation of 1,3-diene and a Friedel–Crafts-type cyclization, resulting in the formation of an azepine ring through a Bi(OTf)3-catalyzed SN2’ reaction.
Similar content being viewed by others
Introduction
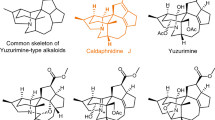
Communesins A and B, which were originally isolated by Numata et al. from the mycelium of a strain of Penicillium sp. attached to the marine alga Enteromorpha intestinalis, are heptacyclic indole alkaloids (Fig. 1) [1]. Spectroscopic analyses, including nuclear magnetic resonance (NMR) spectroscopy (1H NMR, 13C NMR including 2D NMR) and high-resolution mass spectrometry, have revealed that their structures are quite unique. They are characterized by a heptacyclic skeleton bearing two aminals and two contiguous quaternary carbon centers. To date, nine congeners have been reported [2,3,4,5,6], and perophoramidine is also known as a structurally related bis-amidine indole alkaloid [7]. Recently, Tang et al. confirmed that communesins can be biosynthetically produced through the coupling of aurantioclavine and tryptamine based on genetic inactivation studies [8]. Communesins show cytotoxicity against P388 lymphocytic leukemia cells (ED50 A: 3.5 µg/mL, B: 0.45 µg/mL) and potent insecticidal activity toward silkworms (LD50 D: 300 µg/g, E: 80 µg/g). Because of their unique structure and biological activity, many research groups have conducted synthetic studies of communesins, in which various synthetic methods were developed [9,10,11,12,13,14]. The first racemic total synthesis of communesin F was achieved by Qin et al. based on an intramolecular cyclopropanation strategy [15]. Weinreb and Funk also reported total synthesis of communesin F, independently [16, 17]. The first asymmetric total syntheses of communesins A, B, and F were accomplished by Ma et al. [18, 19]. Asymmetric total syntheses were also reported by Stoltz, Movassaghi, Yang, and Chen, independently [20,21,22,23]. We have also engaged in the development of synthetic strategies for this class of alkaloids, including communesins, perophoramidine, and aurantioclavin [24,25,26,27,28,29,30,31].

Communesins and related alkaloids
Results and discussion
Recently, we have developed palladium(Pd)-catalyzed carbosilylation of 1,3-diene with carbamoyl chloride for the synthesis of several spirooxindoles [32]. Extending this reaction, a Pd-catalyzed carboborylation of 1,3-diene was developed for a synthesis of iminoindoline [30]. Considering our developed method, it was envisioned that communesin F would be accessed from a pentacyclic skeleton II through intermediate I by the introduction of an aminoethyl unit and the formation of amidine. The pentacyclic skeleton II would be constructed from a tetracyclic compound IV via III by the introduction of an allyl alcohol unit, resulting in an SN2’ reaction for the formation of an azepine ring and a reduction of amidine. The tetracyclic compound IV can be synthesized by a carboborylation of 1,3-diene VI and an intramolecular Friedel–Crafts-type reaction of a resultant iminoindoline V [30]. Following this retrosynthetic analysis, we have recently succeeded in the construction of tetracyclic skeleton IV (R = OMe) from diene VI (R = OMe) through iminoindoline V (R = OMe). However, compound 1 could not be converted to compound 2 through removal of the methyl group, although we tried various conditions, including BBr3, BCl3, AlCl3, LiCl, Ph2PLi, and pMeC6H4SLi (Scheme 1b, also see Supplementary Information, Figure S1). These reaction conditions resulted in the removal of a Cbz group or the decomposition of compound 1. Therefore, we needed to revise our initial synthetic route and planned to employ a 1,3-diene-containing triflate (R = OTf) to avoid a protecting group manipulation. The use of a substrate bearing a triflate group for Pd-catalyzed carboborylation would extend its reaction scope, and it might react itself under the reaction conditions. In this paper, we report the construction of a pentacyclic skeleton of communesin F by extending our strategy based on carboborylation of 1,3-diene.

a Retrosynthesis of communesin F and b failed attempt at removing a methyl group from compound 1
The synthesis started with a removal of a methyl group on a phenolic hydroxy group. A methoxy aniline derivative 3, which was prepared from tert-butyl(3-methoxyphenyl)carbamate in four steps [30], was treated with BBr3 to give a phenol (Scheme 2). The resultant phenolic hydroxy group was silylated with tert-butyldimethylsilyl (TBS) chloride and imidazole to give compound 4. A half reduction of a lactone with diisobutylaluminum hydride (DIBAL-H) was followed by Wittig olefination, which gave diene 5 through internal transfer of a TBS group. After the formation of urea by a treatment with phenyl isocyanate, a phenolic hydroxy group was protected as a triflate. A removal of a TBS group was followed by a Mitsunobu reaction with pNsNHBoc [33] to give compound 8, which was converted to carbodiimide 9 through dehydration with CBr4, PPh3, and Et3N. Because compound 9 was unstable, it was necessary to use it immediately for the next reaction.

Synthesis of 3,3-disubstituted iminoindoline 10 based on the Pd-catalyzed carboborylation of 1,3-diene and its derivatization
With carbodiimide 9 containing a diene moiety, we investigated whether the triflate is intact under the reaction conditions of Pd-catalyzed carboborylation of 1,3-diene. In the previous literature, there is no report concerning Pd(II)-catalyzed Miyaura borylation of triflates and diborone without a ligand, but reactions using diphenylphosphinoferrocene [34] or the reaction of arylbromide have been reported [35]. Therefore, it was expected that a triflate group would be intact during the carboborylation of 1,3-diene. As expected, the reaction of 9 proceeded smoothly under the established conditions (Pd(OAc)2, (pinB)2, xylene, 50 °C) to give an allyl borane, which was treated with NaBO3·4H2O to give allyl alcohol 10. After silylation of allyl alcohol 10, a tert-butoxycarbonyl (Boc) group was introduced to an amidine nitrogen for further transformation. The treatment of compound 11 with tetrabutylammonium fluoride gave an allyl alcohol along with the removal of the trifluoromethanesulfonyl (Tf) group, which was converted to allyl bromide 12 under standard conditions. Unfortunately, the resultant allyl bromide 12 could not be converted to compound 13 through a treatment with Tf2O and pyridine. On the other hand, when HF·pyridine was used, a triethylsilyl group was selectively removed with the triflate group intact. The resultant allyl alcohol was also converted to allyl bromide 13 containing a triflate group, while a small amount of compound 14 was also obtained through the removal of a Boc group.
Next, we investigated Friedel–Crafts-type cyclization of allyl bromides 12 and 13 to construct a tetracyclic ABCD ring skeleton. Previously, we have reported the cyclization of compound 15 containing a methoxy group using 10 mol% of Bi(OTf)3 and 3.5 equivalents of AgOTf (Table 1, entry 1) [30, 36,37,38]. The reaction gave compound 18a in 49% yield along with 18b in 30% yield. We initially applied these conditions to a cyclization of compound 13 containing a triflate group. However, the reaction gave a complex mixture instead of any cyclized products 17a and 17b (entry 2). On the other hand, the cyclization of compound 12 containing a phenolic hydroxy group proceeded under the same conditions to give compounds 16a and 16b in 63 and 30% yields with excellent stereochemistry, respectively (entry 3). The stereochemistry was determined by a comparison with our previous results [30] and a NOESY experiment of a derivatized compound 28 (vide infra). When 3.5 equivalents of AgOTf was reduced to 1.2 equivalents, the formation of byproduct 16b was suppressed to 17% yield (entry 4). Finally, the yield of the desired product 16a was improved to 80% yield using 1.05 equivalents of AgOTf (entry 5). AgOTf was essential for this Friedel–Crafts-type reaction (entry 6).
After the construction of a tetracyclic ABCD ring skeleton containing an amidine, we turned our attention to the formation of an azepine ring (G ring). A treatment of compound 16a with Tf2O and pyridine gave compound 17a in 91% yield (Scheme 3). To introduce an allyl alcohol unit, Suzuki–Miyaura coupling with vinyl boronic ester 19 was examined. When compound 17a and vinyl boronic ester 19 were treated with a catalytic amount of Pd(dba)2, SPhos and K3PO4, or Pd(PPh3)4 and Na2CO3 in N,N-dimethylformamide (DMF) at 100 °C, respectively, these reactions gave the desired product 20 in low yield (Table 2, entries 1 and 2). However, conditions involving Pd(PPh3)4 and Na2CO3 in toluene and ethanol at 100 °C improved the yield to 56% (entry 3). The removal of the pNs and trimethylsilyl (TMS) group gave allyl alcohol 21 in 66% yield over two steps. To construct the azepine ring, mesylation of a tertiary alcohol was initially attempted through a treatment with methanesulfonyl chloride (MsCl) and Et3N [18]. However, a dehydration occurred to give diene 23 instead of the desired cyclized product 22. Interestingly, when compound 21 was treated with pyridinium p-toluenesulfonate (PPTS) [15], ortho-amide 24 was observed (as assessed using 1H NMR analysis). A related structure was observed in synthetic studies of dehaloperophoramidine reported by Somfai et al. [13, 14]. We considered the thermodynamic stability of possible equilibrium products such as simplified compounds 25, 26, and 27 through density functional theory (DFT) calculations (Fig. 2). These calculations revealed that ortho-amide 26 was the most stable isomer among these compounds. These results indicate that the formation of the ortho-amide through acid activation using PPTS from amidine would be a competitive process with the formation of the azepine ring via the SN2’ reaction of the tertiary alcohol, and the equilibrium tends to be biased toward the ortho-amides, such as compounds 24 and 26. Therefore, we expected that it would be difficult to achieve the formation of azepine 22 from compound 21 containing the amidine moiety.

Failed attempt at the formation of an azepine ring

Comparison of the thermodynamic stability of formable compounds 25, 26, and 27, calculated using Gaussian ‘09 at the B3LYP/6-31G(d) level of theory (DFT)
Therefore, a reduction of amidine 20 was investigated prior to the formation of the azepine ring to avoid the formation of the ortho-amide (Scheme 4). When compound 20 was treated with NaBH4, the desired product was not obtained. In the case of DIBAL-H, the removal of a Boc group occurred instead of the reduction of the amidine. However, in sharp contrast, treatment with catechol borane [39] gave the desired product 28 in 65% yield as a 3.3:1 mixture of diastereomers. A NOESY experiment indicated that the stereochemistry of the major isomer was a trans-fused structure, which would be epimerized to a cis-fused structure later. Because a reducing reagent (catechol borane) would approach from the opposite side of the sterically hindered substituent (−CH2CH2N(Boc)(pNs)) of the angular position of the BC ring, the trans isomer was obtained as a major product in this reaction. After the removal of pNs and the TMS groups, the formation of an azepine ring was investigated again. When compound 29 was treated with MsCl and Et3N [18], the reaction gave diene 31 in 48% yield and the desired cyclized product 30 was not detected at all (Table 3, entry 1). When Bi(OTf)3 was employed at −15 °C as a Lewis acid, the reaction proceeded to give the desired product 30 as a major product albeit in low yield (entry 2) [40, 41]. The reaction using Bi(OTf)3 at −40 °C gave the desired product 30 in 17% yield with recovery of the starting material (entry 3). However, under room temperature reaction conditions, the starting material 29 was consumed completely to give the desired azepine 30 in 55% yield, while diene 31 was obtained in 34% yield (entry 4). The newly generated stereochemistry of compound 30 was confirmed to have the desired stereochemistry using NOESY experiments (Fig. 3a). In this cyclization, it was supposed that transition state B would not be favored than transition state A because of the presence of the steric repulsion between the allyl alcohol and vinyl group (Fig. 3b). Thus, compound 30 was obtained as a single diastereomer through transition state A. The obtained pentacyclic compound 30 would be useful for further derivatization, and now we are investigating further transformations to achieve a total synthesis of communesin F.

Synthesis of the ABCDG ring skeleton 30

a NOESY experiment of compounds 30, b proposed transition state A and B
In summary, we have investigated the synthesis of a pentacyclic ABCDG ring skeleton of communesin F based on carboborylation of 1,3-diene, a Bi(OTf)3-catalyzed Friedel–Crafts-type reaction and azepine ring formation. It is interesting that a triflate group was intact under the conditions required for Pd-catalyzed carboborylation of 1,3-diene. Additionally, it was essential that the resultant amidine was reduced prior to the formation of the azepine ring through Bi(OTf)3-catalyzed cyclization to avoid an undesired formation of ortho-amide. We are currently investigating further transformation of the pentacyclic compound to complete the synthesis of communesin F.
Experimental procedure
General
All non-aqueous reactions were carried out under a positive pressure of argon in oven-dried glassware. Analytical thin-layer chromatography was performed using Silica gel 60 plates (Merck, Darmstadt, Germany). Silica gel column chromatography was performed using Kanto silica gel 60 (particle size of 63–210 μm, Kanto, Tokyo, Japan) and Chromatorex BW-300 (Fuji silysia, Aichi, Japan). Proton nuclear magnetic resonance (1H NMR) spectra were recorded using a JNM-ECA 500 (JEOL, Tokyo, Japan) at 500 MHz or a JNM-AL 400 (JEOL) at 400 MHz. Chemical shifts were reported relative to Me4Si (δ 0.00) in CDCl3 or the residual solvent peak in C6D6 (δ 7.16). Multiplicity was indicated by one or more of the following: s (singlet); d (doublet); t (triplet); q (quartet); m (multiplet); br (broad). Carbon nuclear magnetic resonance (13C NMR) spectra were recorded using a JNM-ECA 500 at 126 MHz or a JNM-AL 400 at 100 MHz. Chemical shifts were reported relative to CDCl3 (δ 77.0) or C6D6 (δ 128.0). Infrared spectra were recorded using a FT/IR-4100 Fourier-transform infrared spectrometer (JASCO, Tokyo, Japan) with ATR (attenuated total reflectance). Low- and high-resolution mass spectra were recorded using a JMS-700 mass spectrometer (JEOL) for FAB-MS and a LCMS-IT-TOF (Shimadzu, Kyoto, Japan) for ESI-MS.
Experimental procedures and spectroscopic data

Silylether 4: To a solution of aniline 3 (2.06 g, 9.40 mmol) in CH2Cl2 (94.0 mL) was added a solution of BBr3 (25.0 g, 94.0 mmol) in CH2Cl2 (94.0 mL) at −78 °C. The mixture was stirred at −78 °C for 20 min, and then warmed to room temperature. After 2 h, saturated aqueous NaHCO3 and 1 M aqueous NaOH were added to the reaction mixture until the mixture became basic. The mixture was extracted with EtOAc. The combined organic layers were washed with brine, and dried over Na2SO4. Concentration under reduced pressure gave a crude demethylated lactone.
To a solution of the above crude lactone in anhydrous DMF (20.0 mL) were added TBSCl (2.80 g, 18.8 mmol) and imidazole (1.90 g, 28.2 mmol) at 0 °C. The mixture was stirred at room temperature for 3 h. After addition of water, the mixture was extracted with extracted with Et2O. The combined organic layers were washed with brine, and dried over Na2SO4. After concentration under reduced pressure, purification by flash column chromatography on silica gel (5–40% EtOAc/hexane) gave silylether 4 (1.88 g, 63% in two steps) as a pale yellow solid: 1H NMR (500 MHz, CDCl3) δ 6.99 (dd, 1 H, J = 8.0, 8.0 Hz), 6.35 (dd, 1 H, J = 8.0, 1.1 Hz), 6.27 (dd, 1 H, J = 8.0, 1.1 Hz), 6.06 (dd, 1 H, J = 1.7, 1.2 Hz), 4.53 (dd, 2 H, J = 6.3, 5.8 Hz), 3.76 (br, 2 H), 2.72 (dd, 2 H, J = 6.3, 5.7 Hz), 0.94 (s, 9 H), 0.21 (s, 6 H); 13C NMR (126 MHz, CDCl3) δ 164.4, 156.1, 153.1, 144.0, 129.8, 120.3, 115.5, 108.7, 108.6, 66.5, 28.2, 25.6, 18.1, −4.1; IR (ATR, cm−1) 3369, 2954, 2891, 2857, 1716, 1625, 1580, 1462, 1398, 1302, 1257, 1219, 1081, 1020; MS (FAB) m/z 320 [M + H]+; HRMS calcd for C17H26NO3Si [M + H]+ 320.1682; found: m/z 320.1685. 
(E)-Dienylaniline 5: To a solution of silylether 4 (1.25 g, 3.91 mmol) in CH2Cl2 (40.0 mL) was added DIBAL-H (1 M in toluene, 7.80 mL, 7.80 mmol) at −78 °C. After the mixture was stirred at −78 °C for 2 h, saturated aqueous Na/K tartrate was added to the reaction solution. The resultant mixture was stirred vigorously at room temperature for 2 h, and extracted with EtOAc. The combined organic layers were washed with brine, and dried over Na2SO4. Concentration under reduced pressure gave a crude acetal.
To a suspension of MePPh3Br (4.89 g, 13.7 mmol) in anhydrous THF (25.0 mL) was added KHMDS (1 M solution in THF; 12.0 mL, 11.7 mmol) at 0 °C, and the mixture was stirred at 0 °C for 30 min. To the yellow mixture was then added a solution of the above crude acetal in anhydrous THF (15 mL) via cannula. The reaction mixture was stirred at room temperature for 2 h. After addition of water, the resultant mixture was extracted with EtOAc. The combined organic layers were washed with brine, and dried over Na2SO4. After concentration under reduced pressure, purification by flash column chromatography on silica gel (5–40% EtOAc/hexane) gave (E)-dienylaniline 5 (963.1 mg, 77% in two steps) as a yellow oil: 1H NMR (500 MHz, CDCl3) δ 6.95 (dd, 1 H, J = 8.0, 8.0 Hz), 6.77 (ddd, 1 H, J = 16.9, 10.9, 10.3 Hz), 6.36 (dd, 1 H, J = 8.0, 0.8 Hz), 6.29 (d, 1 H, J = 11.1 Hz), 6.25 (dd, 1 H, J = 10.3 Hz), 5.27 (dd, 1 H, J = 16.9, 1.2 Hz), 5.24 (d, 1 H, J = 10.3 Hz), 3.75 (br, 1 H), 3.63 (br, 1 H), 3.60 (br, 2 H), 2.98 (br, 1 H), 2.38 (br, 1 H), 0.88 (s, 9 H), 0.08 (s, 6 H); 13C NMR (126 MHz, CDCl3) δ 154.9, 144.7, 136.6, 132.4, 132.1, 128.7, 119.3, 115.8, 106.9, 106.0, 60.6, 33.3, 25.7, 18.1, −5.5; IR (ATR, cm−1) 3375, 2955, 2924, 2857, 1618, 1581, 1464, 1234, 1088; MS (FAB) m/z 320 [M + H]+; HRMS calcd for C18H30NO2Si [M + H]+ 320.2046; found: m/z 320.2045. 
(E)-Dienylurea 6: To a solution of (E)-dienylaniline 5 (847.9 mg, 2.65 mmol) in CH2Cl2 (26.0 mL) was added phenyl isocyanate (317.0 µL, 2.92 mmol) at 0 °C. The mixture was stirred at 0 °C for 13 h. After addition of water, the reaction mixture was extracted with EtOAc. The combined organic layers were washed with brine, and dried over Na2SO4. After concentration under reduced pressure, purification by short-column chromatography on silica gel (10–20% EtOAc/hexane) gave a crude urea as a white solid.
To a solution of the above crude urea in CH2Cl2 (50.0 mL) were added Et3N (2.10 mL, 15.1 mmol) and PhNTf2 (6.15 g, 17.2 mmol) in some portions. The resultant solution was refluxed at 55 °C for 3 days. The reaction mixture was then cooled to room temperature. After addition of saturated aqueous NH4Cl, the mixture was extracted with EtOAc. The combined organic layers were washed with brine, and dried over Na2SO4. After concentration under reduced pressure, purification by flash column chromatography on silica gel (5–20% EtOAc/hexane) gave (E)-dienylurea 6 (1.37 g, 90% in 2 steps) as a pale yellow solid: 1H NMR (500 MHz, CDCl3) δ 8.21 (dd, 1 H, J = 8.3, 0.9 Hz), 7.39 (br, 1 H), 7.34–7.30 (m, 5 H), 7.13–7.09 (m, 1 H), 9.97 (dd, 1 H, J = 8.3, 0.9 Hz), 6.69 (ddd, 1 H, J = 16.6, 10.9, 10.3 Hz), 6.65 (br, 1 H), 6.21 (d, 1 H, J = 11.1 Hz), 5.38–5.34 (m, 2 H), 3.71–3.69 (m, 1 H), 3.55–3.51 (m, 1 H), 2.97–2.95 (m, 1 H), 2.39–2.36 (m, 1 H), 0.83 (s, 9 H), 0.02 (s, 6 H); 13C NMR (126 MHz, CDCl3) δ 152.6, 147.2, 138.8, 137.6, 137.1, 131.4, 129.7, 129.3, 128.9, 126.8, 124.4, 121.4, 121.1, 120.6, 119.7, 118.4 (q, J = 321 Hz), 115.1, 61.6, 35.0, 25.9, 18.5, −5.5; IR (ATR, cm−1) 3332, 2954, 2857, 1659, 1550, 1524, 1446, 1420, 1296, 1250, 1207, 1139, 1054, 962; MS (FAB) m/z 571 [M + H]+; HRMS calcd for C26H34F3N2O5SSi [M + H]+ 571.1910; found: m/z 571.1910.
(E)-Dienylalcohol 7: To a solution of (E)-dienylurea 6 (42.7 mg, 0.0748 mmol) in THF (1.0 mL) was added TBAF (1 M in THF, 83.0 µL, 0.083 mmol) at 0 °C. The mixture was stirred at 0 °C for 1 h. After addition of saturated aqueous NH4Cl, the mixture was extracted with EtOAc. The combined organic layers were washed with brine, and dried over Na2SO4. After concentration under reduced pressure, purification by flash column chromatography on silica gel (10–40% EtOAc/hexane) gave (E)-dienylalcohol 7 (35.3 mg, quant.) as a pale yellow solid: 1H NMR (500 MHz, CDCl3) δ 8.25 (d, 1 H, J = 8.6 Hz), 7.77 (br, 1 H), 7.32–7.18 (m, 5 H), 7.18 (br, 1 H), 7.07 (dd, 1 H, J = 7.1, 6.9 Hz), 6.94 (d, 1 H, J = 8.3 Hz), 6.72 (ddd, 1 H, J = 16.9, 10.9, 10.3 Hz), 6.24 (d, 1 H, J = 10.9 Hz), 5.39–5.33 (m, 2 H), 3.81 (br, 1 H), 3.46 (br, 1 H), 3.04 (br, 1 H), 2.35 (d, 1 H, J = 14.6 Hz), 2.23 (br, 1 H); 13C NMR (126 MHz, CDCl3) δ 153.2, 147.3, 139.3, 137.9, 137.8, 131.3, 129.2, 129.1, 129.1, 126.1, 124.1, 121.4, 120.9, 119.6, 118.4 (q, J = 321 Hz), 114.8, 60.2, 34.2; IR (ATR, cm−1) 3337, 3010, 2926, 1670, 1579, 1550, 1446, 1420, 1297, 1210, 1138, 1051, 963; MS (FAB) m/z 457 [M + H]+; HRMS calcd for C20H20F3N2O5S [M + H]+ 457.1045; found: m/z 457.1042.

(E)-Dienylurea 8: To a solution of (E)-dienylalcohol 7 (992.0 mg, 2.17 mmol), pNsNHBoc (786.0 mg, 2.60 mmol) and PPh3 (682.0 mg, 2.60 mmol) in THF (12.0 mL) was added a solution of di-tert-butyl azodicarboxylate (598.7 mg, 2.60 mmol) in THF (10.0 mL). The mixture was stirred at room temperature for 13.5 h. After addition of saturated aqueous NH4Cl, the mixture was extracted with EtOAc. The combined organic layers were washed with brine, and dried over Na2SO4. After concentration under reduced pressure, purification by flash column chromatography on silica gel (10–40% EtOAc/hexane) gave the mixture of (E)-dienylurea 8 and pNsNHBoc. The mixture was dissolved in CHCl3, washed with 1 M aqueous NaOH and brine, and dried over Na2SO4. Concentration under reduced pressure gave (E)-dienylurea 8 (1.43 g, 89%) as a pale yellow solid: 1H NMR (500 MHz, CDCl3) δ 8.35–8.32 (m, 3 H), 8.04 (d, 2 H, J = 9.1 Hz), 7.47 (br, 1 H), 7.37–7.29 (m, 5 H), 7.17 (br, 1 H), 7.13–7.09 (m, 1 H), 6.98 (d, 1 H, J = 8.3 Hz), 6.79 (ddd, 1 H, J = 16.3, 10.9, 10.6 Hz), 6.24 (d, 1 H, J = 10.9 Hz), 5.42–5.38 (m, 2 H), 3.88–3.78 (m, 2 H), 3.15–3.09 (m, 1 H), 2.76–2.70 (m, 1 H), 1.32 (s, 9 H); 13C NMR (126 MHz, CDCl3) δ 152.6, 150.9, 150.5, 147.3, 145.1, 138.5, 138.4, 137.7, 131.3, 129.4, 129.4, 129.1, 128.1, 125.9, 124.6, 124.1, 122.6, 121.3, 120.0, 118.4 (q, J = 321 Hz), 115.1, 86.5, 46.2, 33.4, 27.9; IR (ATR, cm-1) 3349, 2929, 2854, 1732, 1668, 1534, 1446, 1420, 1368, 1351, 1291, 1249, 1212, 1139, 1055, 961; MS (FAB) m/z 741 [M + H]+; HRMS calcd for C31H32F3N4O10S2 [M + H]+ 741.1512; found: m/z 741.1512. 
(E)-Dienylcarbodiimide 9: To a solution of (E)-dienylurea 8 (62.5 mg, 0.0844 mmol) and PPh3 (73.5 mg, 0.270 mmol) in CH2Cl2 (2.0 mL) were added Et3N (47.0 µL, 0.338 mmol) and CBr4 (83.9 mg, 0.253 mmol) at 0 °C. The mixture was stirred at 0 °C for 2.5 h. After concentration of the mixture under reduced pressure, purification of the residue by flash column chromatography on neutral silica gel (5–20% EtOAc/hexane) gave (E)-dienylcarbodiimide 9 (56.4 mg, 92%) as a pale yellow oil. The product was not stable, thus it was used for the next reaction immediately: 1H NMR (500 MHz, CDCl3) δ 8.33 (d, 2 H, J = 8.9 Hz), 8.07 (d, 2 H, J = 8.8 Hz), 7.37–7.27 (m, 4 H), 7.19 (dd, 1 H, J = 7.5, 7.4 Hz), 7.16–7.13 (m, 3 H), 6.80 (ddd, 1 H, J = 16.6, 10.6, 10.6 Hz), 6.25 (d, 1 H, J = 11.2 Hz), 5.37–5.32 (m, 2 H), 3.89 (dd, 2 H, J = 7.2, 7.2 Hz), 2.99 (br, 2 H), 1.31 (s, 9 H); 13C NMR (126 MHz, CDCl3) δ 150.3, 150.2, 147.6, 145.4, 139.3, 137.3, 137.2, 132.4, 132.1, 131.1, 129.5, 129.3, 129.1, 127.9, 125.9, 124.9, 124.4, 123.9, 121.6, 118.4 (q, J = 321 Hz), 118.2, 85.2, 45.9, 33.4, 27.7; IR (ATR, cm−1) 3105, 2938, 2857, 2141, 1731, 1591, 1563, 1533, 1476, 1452, 1421, 1366, 1351, 1285, 1250, 1213, 1137, 909 (Compound 9 was too unstable to measure HRMS). 
2-Iminoindoline 10: To a solution of carbodiimide 9 (56.4 mg, 0.0780 mmol) in anhydrous xylene (1.0 mL) were added bis(pinacolato)diboron (39.6 mg, 0.156 mmol) and Pd(OAc)2 (3.5 mg, 0.0156 mmol) and the reaction atmosphere was replaced by the Ar atmosphere. The mixture was stirred at 50 °C for 1 h, and then cooled to 0 °C. After addition of water (1.0 mL) and sodium perborate tetrahydrate (72.0 mg, 0.468 mmol), the mixture was stirred vigorously at room temperature for 1 h. The mixture was then extracted with EtOAc. The combined organic layers were washed with brine, and dried over Na2SO4. After concentration under reduced pressure, purification by flash column chromatography on silica gel (20–60% EtOAc/hexane) gave 2-iminoindoline 10 (42.4 mg, 73%) as a yellow oil: 1H NMR (500 MHz, CDCl3) δ 8.26 (d, 2 H, J = 8.9 Hz), 7.94 (d, 2 H, J = 8.9 Hz), 7.79 (d, 2 H, J = 8.0 Hz), 7.40–7.34 (m, 4 H), 7.13 (dd, 1 H, J = 7.5, 7.4 Hz), 6.98 (br, 1 H), 6.93–6.90 (m, 1 H), 6.07 (ddd, 1 H, J = 15.8, 5.2, 4.8 Hz), 5.78 (d, 1 H, J = 16.0 Hz), 4.23 (d, 2 H, J = 4.9 Hz), 3.45 (ddd, 1 H, J = 14.0, 11.8, 4.0 Hz), 3.22 (ddd, 1 H, J = 14.3, 12.0, 4.3 Hz), 2.89 (ddd, 1 H, J = 12.6, 12.6, 4.3 Hz), 2.54 (ddd, 1 H, J = 12.6, 12.4, 4.0 Hz), 1.94 (br, 1 H), 1.31 (s, 9 H); 13C NMR (126 MHz, CDCl3) δ 170.5, 158.9, 150.3, 150.0, 145.1, 144.9, 138.6, 133.2, 131.1, 129.2, 129.1, 127.6, 127.5, 124.1, 123.9, 120.0, 118.5 (q, J = 320 Hz), 117.9, 114.4, 85.7, 62.8, 59.0, 43.4, 33.0, 27.7; IR (ATR, cm−1) 3380, 3106, 2936, 2877, 1732, 1561, 1534, 1439, 1420, 1349, 1247, 1213, 1138, 1083, 1014, 907; MS (FAB) m/z 741 [M + H]+; HRMS calcd for C31H32F3N4O10S2 [M + H]+ 741.1512; found: m/z 741.1508. 
N-Boc-iminoindoline 11: To a solution of 2-iminoindoline 10 (39.4 mg, 0.0532 mmol) in CH2Cl2 (1.0 mL) were added Et3N (23.0 µL, 0.160 mmol) and TESCl (16.0 µL, 0.106 mmol) at 0 °C. The mixture was stirred at room temperature for 2 h. After addition of water, the mixture was extracted with EtOAc. The combined organic layers were washed with brine, and dried over Na2SO4. After concentration under reduced pressure, purification by short-column chromatography on neutral silica gel (10–30% EtOAc/hexane) gave a crude TES-protected iminoindoline.
To a solution of the above crude iminoindoline in CH2Cl2 (1.0 mL) were added Pr2NEt (37.0 µL, 0.213 mmol), Boc2O (34.9 mg, 0.160 mmol), and DMAP (6.5 mg, 0.0532 mmol) at 0 °C. The mixture was stirred at room temperature for 1.5 h. After concentration of the resultant mixture under reduced pressure, purification of the residue by flash column chromatography on silica gel (10–30% EtOAc/hexane) gave N-Boc-iminoindoline 11 (33.7 mg, 84% in two steps) as a colorless oil: 1H NMR (500 MHz, CDCl3) δ 8.26 (d, 2 H, J = 8.9 Hz), 8.05 (d, 2 H, J = 8.8Hz), 7.73 (d, 1 H, J = 8.0 Hz), 7.41 (dd, 1 H, J = 8.6, 8.3 Hz), 7.31 (dd, 2 H, J = 7.8, 7.7 Hz) 7.12 (d, 1 H, J = 8.3 Hz), 7.06–7.02 (m, 3 H), 5.93 (d, 1 H, J = 15.5 Hz), 5.66 (d, 1 H, J = 15.5 Hz), 4.17 (d, 2 H, J = 4.3 Hz), 3.85–3.79 (m, 1 H), 3.65–3.62 (m, 1 H), 2.70–2.63 (m, 2 H), 1.27 (s, 9 H), 1.18 (s, 9 H), 0.92 (dd, 9 H, J = 8.0, 7.8Hz), 0.57 (q, 6 H, J = 7.8 Hz); 13C NMR (126 MHz, CDCl3) δ 153.2, 150.2, 150.1, 149.0, 147.9, 145.9, 145.6, 143.5, 131.5, 130.5, 129.4, 129.1, 128.7, 123.9, 123.8, 122.1, 120.5, 118.3 (q, J = 321 Hz), 115.8, 114.1, 85.3, 84.8, 62.6, 54.0, 43.2, 34.7, 27.7, 27.4, 6.7, 4.3; IR (ATR, cm−1) 2955, 2876, 1731, 1698, 1617, 1594, 1535, 1456, 1421, 1370, 1348, 1287, 1251, 1218, 1141, 1046, 1014, 917, 822; MS (FAB) m/z 955 [M + H]+; HRMS calcd for C42H54F3N4O12S2Si [M + H]+ 955.2901; found: m/z 955.2900. 
Allyl bromide 12: To a solution of N-Boc-iminoindoline 11 (21.9 mg, 0.0229 mmol) in THF (0.5 mL) was added TBAF (48.1 µL, 0.0481 mmol) at 0 °C. The mixture was stirred at room temperature for 30 min. After addition of saturated aqueous NH4Cl, the mixture was extracted with EtOAc. The combined organic layers were washed with brine, and dried over Na2SO4. After concentration under reduced pressure, purification by flash column chromatography on silica gel (20–60% EtOAc/hexane) gave an allyl alcohol (15.1 mg, 93%) as a pale yellow oil: 1H NMR (500 MHz, CDCl3) δ 8.24 (d, 2 H, J = 8.8 Hz), 8.07 (d, 2 H, J = 8.8 Hz), 7.29 (dd, 2 H, J = 7.5, 7.2 Hz), 7.19 (d, 1 H, J = 7.7 Hz), 7.13–7.09 (m, 1 H) 7.04–6.99 (m, 3 H), 6.59 (d, 1 H, J = 8.0 Hz), 6.03 (d, 1 H, J = 15.1 Hz), 5.77 (d, 1 H, J = 15.4 Hz), 4.09 (br, 2 H), 3.86–3.80 (m, 1 H), 3.69–3.63 (m, 1 H), 2.78 (ddd, 1 H, J = 12.3, 12.1, 4.6 Hz), 2.58 (ddd, 1 H, J = 12.1, 12.0, 4.3 Hz), 1.27 (s, 9 H), 1.21 (s, 9 H); 13C NMR (126 MHz, CDCl3) δ 156.0, 152.7, 150.3, 150.2, 149.2, 148.4, 145.5, 142.2, 131.3, 129.8, 129.6, 129.4, 129.1, 123.8, 123.7, 120.4, 115.2, 112.7, 106.6, 85.2, 84.2, 62.9, 53.5, 43.9, 35.2, 27.7, 27.5; IR (ATR, cm−1) 3445, 2980, 1729, 1695, 1535, 1450, 1360, 1352, 1270, 1085, 910, 730; MS (FAB) m/z 709 [M + H]+; HRMS calcd for C35H41N4O10S [M + H]+ 709.2543; found: m/z 709.2543.
To a solution of the above allyl alcohol (169.6 mg, 0.239 mmol) and PPh3 (157.4 mg, 0.598 mmol) in CH2Cl2 (2.5 mL) was added CBr4 (158.5 mg, 0.478 mmol) at 0 °C. The mixture was stirred at 0 °C for 30 min. After addition of water, the mixture was extracted with EtOAc. The combined organic layers were washed with brine, and dried over Na2SO4. After concentration under reduced pressure, purification by flash column chromatography on silica gel (10–40% EtOAc/hexane) gave allyl bromide 12 (180.3 mg, 98%) as a white solid: 1H NMR (500 MHz, CDCl3) δ 8.26 (d, 2 H, J = 8.6 Hz), 8.08 (d, 2 H, J = 8.8 Hz), 7.32 (dd, 2 H, J = 8.0, 7.7 Hz), 7.32–7.27 (m, 1 H), 7.21–7.17 (m, 1 H) 7.07–7.02 (m, 3 H), 6.63 (d, 1 H, J = 8.0 Hz), 6.05 (d, 1 H, J = 15.2 Hz), 5.87–5.81 (m, 1 H), 3.96–3.89 (m, 2 H), 3.82 (ddd, 1 H, J = 14.7, 11.1, 4.6 Hz), 3.67 (dd, 1 H, J = 12.0, 11.2 Hz), 2.77 (dd, 1 H, J = 12.3, 10.9 Hz), 2.58 (ddd, 1 H, J = 12.3, 12.0, 4.3 Hz), 1.29 (s, 9 H), 1.20 (s, 9 H); 13C NMR (126 MHz, CDCl3) δ 154.8, 152.2, 150.3, 150.2, 149.2, 148.3, 145.5, 142.4, 134.6, 130.1, 129.4, 129.1, 126.8, 123.8, 120.5, 114.8, 112.8, 107.4, 107.3, 85.3, 84.2, 53.5, 43.8, 34.8, 32.1, 27.8, 27.4; IR (ATR, cm−1) 3445, 2980, 1729, 1695, 1599, 1532, 1460, 1366, 1348, 1277, 1250, 1143, 1085, 1061, 968, 909, 852, 730, 605, 578; MS (FAB) m/z 771 [M + H]+; HRMS calcd for C35H40BrN4O9S [M + H]+ 771.1699; found: m/z 771.1696. 
Tetracyclic compound 16a: To a suspension of allyl bromide 12 (300.0 mg, 0.389 mmol), Bi(OTf)3 (25.5 mg, 0.0389 mmol), AgOTf (104.8 mg, 0.408 mmol), MS4Å (300 mg), and K2CO3 (161.7 mg, 1.17 mmol) in CH2Cl2 (40.0 mL) was stirred at room temperature for 15 min. After addition of water, the mixture was then filtered through Celite pad. The filtrate was extracted with EtOAc. The combined organic layers were dried over Na2SO4. After concentration under reduced pressure, purification by flash column chromatography on silica gel (10–60% EtOAc/hexane) gave a tetracyclic compound 16a (215.8 mg, 80%) as a yellow oil: 1H NMR (500 MHz, CDCl3) δ 8.21 (d, 2 H, J = 8.8 Hz), 7.89 (d, 2 H, J = 8.9 Hz), 7.52 (d, 1 H, J = 8.3 Hz), 7.41 (dd, 1 H, J = 7.8, 1.1 Hz), 7.36 (dd, 1 H, J = 7.5, 7.4 Hz), 7.24 (ddd, 1 H, J = 8.3, 8.3, 0.8 Hz), 7.18 (dd, 1 H, J = 7.5, 7.4 Hz), 7.13 (d, 1 H, J = 7.4 Hz), 6.69 (d, 1 H, J = 8.0 Hz), 6.48 (ddd, 1 H, J = 17.4, 10.0, 9.1 Hz), 5.81 (d, 1 H, J = 9.1 Hz), 5.67 (d, 1 H, J = 17.5 Hz), 4.00 (d, 1 H, J = 9.7 Hz), 3.57–3.50 (m, 1 H), 3.26–3.20 (m, 1 H), 2.19–2.11 (m, 2 H), 1.70 (s, 9 H), 1.26 (s, 9 H); 13C NMR (126 MHz, CDCl3) δ 163.0, 152.9, 150.2, 149.9, 149.3, 145.0, 143.6, 142.9, 136.2, 130.7, 129.5, 128.6, 126.5, 126.0, 125.8, 125.1, 124.7, 123.8, 114.5, 114.2, 108.0, 85.1, 84.2, 50.1, 47.9, 43.8, 28.2, 27.7, 27.3; IR (ATR, cm−1) 3449, 2979, 2919, 1731, 1654, 1599, 1533, 1460, 1368, 1348, 1282, 1236, 1148, 1088, 889,; MS (FAB) m/z 691 [M + H]+; HRMS calcd for C35H39N4O9S [M + H]+ 691.2438; found: m/z 691.2439. 
Triflate 17a: To a solution of tetracyclic compound 16a (213.0 mg, 0.308 mmol) in CH2Cl2 (5.0 mL) were added pyridine (87.3 µL, 1.08 mmol) and Tf2O (103.5 µL, 0.616 mmol) at 0 °C. The mixture was stirred at 0 °C for 2.5 h. After addition of water, the mixture was extracted with EtOAc. The combined organic layers were washed with brine, and dried over Na2SO4. After concentration under reduced pressure, purification by flash column chromatography on silica gel (5–20% EtOAc/hexane) gave triflate 17a (230.3 mg, 91%) as a yellow oil: 1H NMR (500 MHz, CDCl3) δ 8.24 (d, 2 H, J = 8.6 Hz), 7.97 (d, 1 H, J = 8.3 Hz), 7.91 (d, 2 H, J = 8.9 Hz), 7.45 (dd, 1 H, J = 8.6, 8.3 Hz), 7.37–7.32 (m, 2 H), 7.23-7.14 (m, 3 H), 6.28 (ddd, 1 H, J = 16.9, 10.0, 9.8 Hz), 5.55 (d, 1 H, J = 10.0 Hz), 5.30 (d, 1 H, J = 16.9 Hz), 3.88 (d, 1 H, J = 9.7 Hz), 3.53-3.49 (m, 1 H), 3.38-3.32 (m, 1 H), 2.26-2.22 (m, 2 H), 1.70 (s, 9 H), 1.27 (s, 9 H); 13C NMR (126 MHz, CDCl3) δ 162.3, 150.2, 149.7, 149.1, 147.1, 145.2, 144.5, 143.4, 133.6, 131.0, 129.5, 128.4, 126.8, 126.3, 125.7, 125.5, 123.8, 121.5, 119.5, 118.2 (q, J = 318 Hz), 114.9, 114.3, 85.3, 84.8, 51.0, 48.1, 43.2, 28.1, 27.8, 27.6; IR (ATR, cm−1) 2982, 2933, 1729, 1661, 1613, 1534, 1455, 1423, 1369, 13647, 1291, 1217, 1143, 1086, 1033, 922; MS (FAB) m/z 823 [M + H]+; HRMS calcd for C36H38F3N4O11S2 [M + H]+ 823.1931; found: m/z 823.1929.

Coupling product 20: To a solution of triflate 17a (30.0 mg, 0.0365 mmol) and vinyl boronate 19 (20.8 mg, 0.0730 mmol) in toluene (1.0 mL) and EtOH (0.1 mL) were added 0.5 M aqueous Na2CO3 (220.0 µL, 0.110 mmol) and Pd(PPh3)4 (4.2 mg, 3.65 × 10−3 mmol). The reaction atmosphere was replaced by the Ar atmosphere, and the mixture was stirred at 100 °C for 7 h. After the reaction mixture was then cooled to room temperature, the resultant mixture was extracted with EtOAc. The combined organic layers were washed with brine, and dried over Na2SO4. After concentration under reduced pressure, purification by flash column chromatography on silica gel (5–20% EtOAc/hexane) gave coupling product 20 (16.9 mg, 56%) as a yellow oil: 1H NMR (500 MHz, CDCl3) δ 8.21 (d, 2 H, J = 8.9 Hz), 7.86 (d, 2 H, J = 9.2 Hz), 7.35–7.31 (m, 5 H), 7.18–7.17 (m, 2 H), 6.90 (d, 1 H, J = 15.5 Hz), 6.30 (ddd, 1 H, J = 16.9, 10.1, 10.0 Hz), 6.13 (d, 1 H, J = 15.7 Hz), 5.48 (dd, 1 H, J = 10.0, 1.5 Hz), 5.27 (d, 1 H, J = 16.9 Hz), 3.85 (d, 1 H, J = 10.0 Hz), 3.41 (ddd, 1 H, J = 14.3, 13.7, 4.0 Hz), 3.26–3.19 (m, 1 H), 2.29 (ddd, 1 H, J = 12.9, 12.8, 5.5 Hz), 2.16 (ddd, 1 H, J = 12.9, 12.0, 4.0 Hz), 1.67 (s, 9 H), 1.68 (s, 3 H), 1.30 (s, 3 H), 1.29 (s, 9 H), 0.15 (s, 9 H); 13C NMR (126 MHz, CDCl3) δ 164.3, 149.8, 149.6, 149.7, 145.2, 144.1, 142.6, 139.0, 136.1, 134.6, 129.4, 129.0, 128.2, 126.7, 126.3, 125.7, 125.3, 125.3, 125.0, 123.8, 123.7, 122.5, 122.4, 113.5, 85.1, 84.0, 74.0, 51.5, 48.1, 44.3, 30.1, 30.1, 28.2, 27.9, 2.6; IR (ATR, cm−1) 2978, 1727, 1655, 1597, 1575, 1533, 1474, 1452, 1368, 1347, 1291, 1249, 1150, 1087, 1034, 840, 748, 713, 685, 628, 602; MS (FAB) m/z 831 [M + H]+; HRMS calcd for C43H55N4O9SSi [M + H]+ 831.3459; found: m/z 831.3448.

Aminal 28: To a solution of coupling product 20 (50.0 mg, 0.0602 mmol) in THF (6.0 mL) was added catechol borane solution (1 M in THF, 75.3 µL, 0.0753 mmol) at 0 °C. The mixture was stirred at 0 °C for 2 h. After addition of water, the resultant mixture was extracted with EtOAc. The combined organic layers were washed with brine, and dried over Na2SO4. After concentration under reduced pressure, purification by flash column chromatography on silica gel (5–20% EtOAc/hexane) gave aminal 28 (32.6 mg, 65%, dr = 3.3:1) as a yellow oil: (major diastereomer) 1H NMR (500 MHz, CDCl3) δ 8.14 (d, 2 H, J = 8.9 Hz), 7.93 (d, 2 H, J = 8.9 Hz), 7.75 (br, 1 H), 7.30 (d, 1 H, J = 8.0 Hz), 7.27–7.24 (m, 1 H), 7.20–7.14 (m, 2 H), 7.06 (d, 1 H, J = 15.8 Hz), 6.89 (dd, 1 H, J = 7.8, 7.4 Hz), 6.83 (d, 1 H, J = 8.0 Hz), 6.07 (d, 1 H, J = 15.8 Hz), 6.05–5.98 (m, 2 H), 5.61 (dd, 1 H, J = 10.0, 1.7 Hz), 5.35 (dd, 1 H, J = 16.9, 1.5 Hz), 4.89 (s, 1 H), 4.15 (d, 1 H, J = 10.3 Hz), 4.13–4.08 (m, 1 H), 3.29 (ddd, 1 H, J = 14.1, 14.1, 4.0 Hz), 2.08 (ddd, 1 H, J = 12.6, 12.6, 4.3 Hz), 1.86 (ddd, 1 H, J = 12.9, 12.9, 4.3 Hz), 1.65 (s, 9 H), 1.64 (s, 3 H), 1.28 (s, 3 H), 1.25 (s, 9 H), 0.15 (s, 9 H); 13C NMR (126 MHz, CDCl3) δ 150.4, 150.2, 145.7, 144.7, 140.6, 137.8, 137.1, 131.5, 129.5, 129.2, 128.9, 127.8, 127.7, 127.0, 125.4, 123.8, 123.7, 123.5, 121.9, 120.1, 116.9, 113.7, 84.6, 83.3, 78.3, 74.1, 54.8, 50.6, 44.8, 30.6, 30.4, 28.6, 27.9, 2.8; IR (ATR, cm−1) 2997, 2918, 1731, 1696, 1534, 1467, 1370, 1347, 1235, 1089, 887, 627; MS (FAB) m/z 833 [M + H]+; HRMS calcd for C43H57N4O9SSi [M + H]+ 833.3616; found: m/z 833.3616.
Aminal 29: To a solution of aminal 28 (10.8 mg, 0.0130 mmol) in THF (1.3 mL) was added TBAF (1 M in THF, 15.6 µL, 0.0156 mmol) at 0 °C. The mixture was stirred at 0 °C for 4 h. After addition of saturated aqueous NH4Cl, the resultant mixture was extracted with EtOAc. The combined organic layers were washed with brine, and dried over Na2SO4. After concentration under reduced pressure, purification by flash column chromatography on silica gel (5–30% EtOAc/hexane) gave an alcohol (7.6 mg, 77%) as a yellow oil.
To a solution of the above alcohol (7.6 mg, 9.99 × 10−3 mmol) in MeCN (1.0 mL) were added K2CO3 (9.6 mg, 0.0695 mmol) and PhSH (6.3 µL, 0.0614 mmol). The mixture was stirred at room temperature for 12 h, and then diluted with EtOAc. The organic layer was washed with water and brine, and then dried over Na2SO4. After concentration under reduced pressure, purification by flash column chromatography on silica gel (5–30% EtOAc/hexane) gave aminal 29 (4.0 mg, 70%) as a yellow oil: 1H NMR (500 MHz, CDCl3) δ 7.70 (br, 1 H), 7.33-7.26 (m, 2 H), 7.21 (dd, 1 H, J = 8.0, 8.0 Hz), 7.14–7.10 (m, 2 H), 6.84 (ddd, 1 H, J = 7.8, 7.8, 1.2 Hz), 6.78 (d, 1 H, J = 7.7 Hz), 6.14–6.07 (m, 2 H), 5.97 (br, 1 H), 5.61 (dd, 1 H, J = 10.0, 1.7 Hz), 5.40 (dd, 1 H, J = 17.2, 1.8 Hz), 4.85 (s, 1 H), 4.40 (br, 1 H), 4.15 (d, 1 H, J = 9.7 Hz), 2.94 (br, 1 H), 2.73 (br, 1 H), 1.93 (br, 1 H), 1.86 (br, 1 H), 1.63 (s, 9 H), 1.44 (s, 3 H), 1.40 (s, 3 H), 1.32 (s, 9 H); 13C NMR (126 MHz, CDCl3) δ 155.6, 144.7, 142.9, 140.7, 139.3, 137.2, 131.3, 129.9, 129.1, 128.8, 127.6, 127.3, 124.0, 122.9, 120.9, 120.1, 117.2, 113.9, 83.2, 78.9, 78.4, 71.2, 55.5, 50.8, 37.1, 30.2, 29.3, 28.53, 28.49; IR (ATR, cm−1) 2978, 2916, 1469, 1384, 1283, 1234, 1089, 888, 628; MS (FAB) m/z 576 [M + H]+; HRMS calcd for C34H45N3O5 [M]+ 575.3359; found: m/z 575.3359. 
Pentacyclic compound 30: To a mixture of aminal 29 (6.9 mg, 0.0120 mmol) and MS4Å (7.0 mg) in CH2Cl2 (1.2 mL) was added Bi(OTf)3 (0.8 mg, 1.2 × 10−3 mmol) at 0 °C. The mixture was stirred at 0 °C for 2 h, and then warmed to room temperature and stirred for 1 h. After addition of saturated aqueous NaHCO3, and the mixture was diluted with EtOAc. The organic layer was washed with brine and dried over Na2SO4. After concentration under reduced pressure, purification by flash column chromatography on silica gel (5–20% EtOAc/hexane) gave a pentacyclic compound 30 (3.7 mg, 55%) as a yellow oil: 1H NMR (500 MHz, CDCl3) δ 7.74 (br, 1 H), 7.23-7.18 (m, 2 H), 7.11 (dd, 1 H, J = 7.2, 7.1 Hz), 7.00 (d, 1 H, J = 7.8 Hz), 6.81 (dd, 1 H, J = 8.3, 7.9 Hz), 6.75 (d, 1 H, J = 8.0 Hz), 5.94 (d, 1 H, J = 9.2 Hz), 5.92–5.88 (m, 1 H), 5.84 (br, 1 H), 5.42 (dd, 1 H, J = 16.6, 2.3 Hz), 5.38 (dd, 1 H, J = 9.5, 2.3 Hz), 5.05 (s, 1 H), 5.00 (d, 1 H, J = 8.3 Hz), 4.10 (d, 1 H, J = 10.0 Hz), 3.90 (dd, 1 H, J = 14.0, 4.0 Hz), 2.10 (ddd, 1 H, J = 14.6, 11.4, 5.4 Hz), 2.03-1.96 (m, 2 H), 1.85 (s, 3 H), 1.65 (s, 3 H), 1.63 (s, 9 H), 1.46 (s, 9 H); 13C NMR (126 MHz, CDCl3) δ 155.4, 153.8, 144.7, 142.8, 138.5, 137.7, 132.7, 131.2, 130.7, 128.2, 127.6, 126.0, 124.7, 122.8, 120.0, 118.2, 116.7, 114.6, 82.8, 79.2, 78.7, 58.7, 58.2, 50.7, 41.0, 28.5, 28.4, 25.2, 23.5, 18.4; IR (ATR, cm−1) 2977, 2919, 1691, 1466, 1391, 1341, 1279, 1235, 1089, 889, 756, 628, 523; MS (FAB) m/z 558 [M + H]+; HRMS calcd for C34H42N3O4 [M − H] − 556.3175; Found: m/z 556.3177. (ESI) HRMS calcd for C34H44N3O4 [M + H]+ 558.3332; found: m/z 558.3311.
References
Numata A, et al. Communesins, cytotoxic metabolites of a fungus isolated from a marine alga. Tetrahedron Lett. 1993;34:2355–8.
Jadulco R, et al. New communesin derivatives from the fungus Penicillium sp. derived from the Mediterranean Sponge Axinella verrucosa. J Nat Prod. 2004;67:78–81.
Hayashi H, Matsumoto H, Akiyama K. New insecticidal compounds, communesins C, D and E, from Penicillium expansum Link MK-57. Biosci Biotechnol Biochem. 2004;68:753–6.
Dalsgaard PW, Blunt JW, Munro MHG, Frisvad JC, Christophersen C, Communesins G, New H. Alkaloids from the Psychrotolerant Fungus Penicillium rivulum. J Nat Prod. 2005;68:258–61.
Kerzaon I, et al. Structural investigation and elucidation of new communesins from a marine‐derived Penicillium expansum Link by liquid chromatography/electrospray ionization mass spectrometry. Rapid Commun Mass Spectrom. 2009;23:3928–38.
Fan YQ, et al. Alkaloids with cardiovascular effects from the marine-derived fungus Penicillium expansum Y32. Mar Drugs. 2015;13:6489–504.
Verbitski SM, Mayne CL, Davis RA, Concepcion GP, Ireland CMIsolation. Structure determination, and biological activity of a novel alkaloid, Perophoramidine, from the Philippine Ascidian Perophora namei. J Org Chem. 2002;67:7124–6.
Lin HC, et al. Elucidation of the concise biosynthetic pathway of the communesin indole alkaloids. Angew Chem Int Ed. 2015;54:3004–7.
Trost BM, Osipov M. Recent advances on the total syntheses of communesin alkaloids and perophoramidine. Chem Eur J. 2015;21:16318–43.
Siengalewicz P, Gaich T, Mulzer J. It all began with an error: the nomofungin/communesin story. Angew Chem Int Ed. 2008;47:8170–276.
Johnston CA, et al. Polycyclic ethers and an unexpected dearomatisation reaction during studies towards the bioactive alkaloid, perophoramidine. Tetrahedron. 2018;74:3339–47.
Shao W, You SL. Highly diastereo- and enantioselective synthesis of tetrahydro-5H-Indolo[2,3-b]quinolines through copper-catalyzed propargylic dearomatization of indoles. Chem Eur J. 2017;23:12489–93.
Hoang A, Popov K, Somfai P. An efficient synthesis of (±)-dehaloperophoramidine. J Org Chem. 2017;82:2171–6.
Popov K, Hoang A, Somfai P. Concise total synthesis of dehaloperophoramidine. Angew Chem Int Ed. 2016;55:1801–4.
Yang J, Wu H, Shen L, Qin Y. Total synthesis of (±)-communesin F. J Am Chem Soc. 2007;129:13794–5.
Liu P, Seo JH, Weinreb SM. Total synthesis of the polycyclic fungal metabolite (±)-communesin F. Angew Chem Int Ed. 2010;49:2000–3.
Belmar J, Funk RL. Total synthesis of (±)-communesin f via a cycloaddition with indol-2-one. J Am Chem Soc. 2012;134:16941–3.
Zuo Z, Xie W, Ma D. Total synthesis and absolute stereochemical assignment of (−)-communesin F. J Am Chem Soc. 2010;132:13226–8.
Zuo Z, Ma D. Enantioselective total syntheses of communesins A and B. Angew Chem Int Ed. 2011;50:12008–11.
Han S-J, et al. A diastereodivergent synthetic strategy for the syntheses of communesin F and perophoramidine. Org Lett. 2014;16:3316–9.
Lathrop SP, Pompeo M, Chang W-T, Movassaghi M. Convergent and biomimetic enantioselective total synthesis of (−)-communesin F. J Am Chem Soc. 2016;138:7763–9.
Liang X, et al. Ir-catalyzed asymmetric total synthesis of (−)-communesin F. J Am Chem Soc. 2017;139:3364–7.
Park J, Jean A, Chen DY-K. Asymmetric total syntheses of communesin F and a putative member of the communesin family. Angew Chem Int Ed. 2017;56:14237–40.
Ishida T, Tsukano C, Takemoto Y. Synthesis of 2-iminoindolines via samarium diiodide mediated reductive cyclization of carbodiimides. Chem Lett. 2012;41:44–46.
Ishida T, Ikota H, Kurahashi K, Tsukano C, Takemoto Y. Dearomatizing conjugate addition to quinolinyl amidines for the synthesis of dehaloperophoramidine through tandem arylation and allylation. Angew Chem Int Ed. 2013;52:10204–7.
Ishida T, Takemoto Y. Synthetic study of perophoramidine: construction of pentacyclic core structure via SmI2-mediated reductive cyclization. Tetrahedron. 2013;69:4517–23.
Nanjo T, Tsukano C, Takemoto Y. Synthesis of 3,3-disubstituted 2-aminoindolenines by palladium-catalyzed allylic amidination with isocyanide. Synlett. 2014;25:1473–7.
Suetsugu S, Nishiguchi H, Tsukano C, Takemoto Y. Asymmetric synthesis of (−)-aurantioclavine via palladium-catalyzed intramolecular allylic amination. Org Lett. 2014;16:996–9.
Suetsugu S, Tsukano C, Takemoto Y. Synthetic Studies towards communesins: diastereoselective oxidative rearrangement of aurantioclavine derivatives. Eur J Org Chem. 2016;2016:108–15.
Tsukano C, Nakajima M, Hande SM, Takemoto Y. Palladium-catalyzed intramolecular carboborylation of 1,3-diene and synthesis of ABCD ring of communesins. Org. Biomol. Chem. 2018 (in press), https://doi.org/10.1039/C8OB02224K.
Ikota H, Tsukano C, Takemoto Y. Concise synthesis of (±)-aurantioclavine. Heterocycles. 2018;97:621–31.
Hande SM, Nakajima M, Kamisaki H, Tsukano C, Takemoto Y. Flexible strategy for syntheses of spirooxindoles using palladium-catalyzed carbosilylation and sakurai-type cyclization. Org Lett. 2011;13:1828–31.
Fukuyama T, Cheung M, Kan T. N-carboalkoxy-2-nitrobenzenesulfonamides: a practical preparation of N-Boc-, N-Alloc-, and N-Cbz-protected primary amines. Synlett. 1999;1999:1301–3.
Ishiyama T, Itoh Y, Kitano T, Miyaura N. Synthesis of arylboronates via the palladium(0)-catalyzed cross-coupling reaction of tetra(alkoxo)diborons with aryl triflates. Tetrahedron Lett. 1997;38:3447–50.
Papagni A, et al. Cross-coupling of 5,11-dibromotetracene catalyzed by a triethylammonium ion tagged diphenylphosphine palladium complex in ionic liquids. Organometallics. 2011;30:4325–9.
Hayashi R, Cook GR. Bi(OTf)3-catalyzed 5-exo-trig cyclization via halide activation. Tetrahedron Lett. 2008;49:3888–90.
Giera DS, Schneider C. InCl3-catalyzed allylic Friedel−Crafts reactions toward the stereocontrolled synthesis of 1,2,3,4-tetrahydroquinolines. Org Lett. 2010;12:4884–7.
Kargbo RB, Hashemi ZS, Roy S, Jin X, Herr RJ. Synthesis of 3-benzazepines and azepino[4,5-b]heterocyclic ring systems via intramolecular Friedel–Crafts cyclization. Tetrahedron Lett. 2013;54:2018–21.
Okada M, Sato I, Cho SJ, Dubnau D, Sakagami Y. Chemical synthesis of ComX pheromone and related peptides containing isoprenoidal tryptophan residues. Tetrahedron. 2006;62:8907–18.
Kawai N, Abe R, Uenishi J. Lewis acid-catalyzed intramolecular amination via 1,3-chirality transfer. Tetrahedron Lett. 2009;50:6580–3.
Kawai N, Abe R, Matsuda M, Uenishi J. Synthesis of chiral 1-substituted tetrahydroisoquinolines by the intramolecular 1,3-chirality transfer reaction catalyzed by Bi(OTf)3. J Org Chem. 2011;76:2102–14.
Acknowledgements
This work was supported by a Grant-in-Aid from JSPS KAKENHI (Grant Nos. JP17H05051 (CT), JP18H04407 (CT) and JP16H06384 (YT)) and a Grant-in-Aid from the Uehara Memorial Foundation, Japan (CT).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Dedication: Dedicated to Professor SJ Danishefsky and his great contribution to total synthesis of highly complex and biologically important natural products.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Nakajima, M., Tsukano, C., Yasui, M. et al. Synthesis of the ABCDG ring skeleton of communesin F based on carboborylation of 1,3-diene and Bi(OTf)3-catalyzed cyclizations. J Antibiot 72, 407–419 (2019). https://doi.org/10.1038/s41429-019-0142-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41429-019-0142-7
