
Abstract
N6-methyladenosine (m6A), the most abundant internal modification in eukaryotic messenger RNAs (mRNAs), has been shown to play critical roles in various normal bioprocesses such as tissue development, stem cell self-renewal and differentiation, heat shock or DNA damage response, and maternal-to-zygotic transition. The m6A modification is deposited by the m6A methyltransferase complex (MTC; i.e., writer) composed of METTL3, METTL14 and WTAP, and probably also VIRMA and RBM15, and can be removed by m6A demethylases (i.e., erasers) such as FTO and ALKBH5. The fates of m6A-modified mRNAs rely on the functions of distinct proteins that recognize them (i.e., readers), which may affect the stability, splicing, and/or translation of target mRNAs. Given the functional importance of the m6A modification machinery in normal bioprocesses, it is not surprising that evidence is emerging that dysregulation of m6A modification and the associated proteins also contributes to the initiation, progression, and drug response of cancers. In this review, we focus on recent advances in the study of biological functions and the underlying molecular mechanisms of dysregulated m6A modification and the associated machinery in the pathogenesis and drug response of various types of cancers. In addition, we also discuss possible therapeutic interventions against the dysregulated m6A machinery to treat cancers.
Similar content being viewed by others
Introduction
It is well known that gene expression and cell growth/division are under sophisticated controls through genetic and epigenetic regulations. Abnormal genetic changes (e.g., gene mutation, deletion, amplification, or chromosomal translocation) and/or epigenetic abnormalities (e.g., DNA or histone modification changes) may lead to the development of cancers. In recent years, another layer of gene regulation at the RNA level, i.e., RNA epitranscriptomics,1 has gained increased attention and interest in the research community. Since 1960s, over 100 types of chemical modifications have been identified in protein-coding and non-coding RNAs.2,3,4 Of them, N6-methyladenosine (m6A) is the most abundant internal modification on eukaryotic mRNAs.5,6 The identification of the fat mass and obesity-associated protein (FTO) as a genuine demethylase of m6A modification7 indicated that m6A is a reversible and dynamic RNA modification, analogous to the well-studied reversible DNA and histone modifications.8 Subsequent high-throughput m6A sequencing studies revealed that m6A modifications may affect thousands of mRNAs and non-coding RNAs in each given type of cell, with a special enrichment in the 3′ untranslated regions (UTRs) near the stop codons of mRNAs.9,10
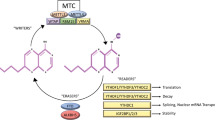
Methyltransferase-like 3 and 14 (METTL3 and METTL14) and their cofactors, Wilms tumor 1-associated protein (WTAP), VIRMA (KIAA1429), and RBM15, compose the m6A methyltransferase complex (MTC) that catalyzes m6A modification as the m6A writer.11,12,13,14,15,16 A set of m6A demethylases, such as FTO and ALKBH5, can remove m6A modification from RNA as m6A erasers and thus keep m6A modification in a dynamic balance.6,7,17 Members of the YT521-B homology (YTH) domain family of proteins, including YTHDF1, YTHDF2, YTHDF3, YTHDC1, and YTHDC2, have been identified as direct m6A readers.18,19,20,21,22,23 While YTHDF2, YTHDF3, and YTHDC2 may promote decay of target mRNAs, YTHDF1, YTHDF3, and YTHDC2 can promote translation of target mRNAs, and YTHDC1 likely impacts splicing and nuclear export of target mRNAs.18,19,20,21,22,23,24 Notably, in contrast to the decay-promoting functions of YTHDF2, YTHDF3, and YTHDC2, a recently identified new family of m6A readers, including IGF2BP1, IGF2BP2, and IGF2BP3, promote the stability (and also translation) of most of their target mRNAs (e.g., MYC).25 Eukaryotic initiation factor 3 (eIF3) could be considered as a reader of 5′ UTR m6A.26 It was reported that cytoplasmic METTL3 may also serve as a kind of m6A reader and promote translation of target mRNAs in certain cell types.27 Thus, dependent on the type of reader protein that recognizes the m6A modification of a given target mRNA, the stability of the target mRNA can be either decreased or enhanced, and translation, splicing, or nuclear transport of the target mRNA may also be affected. See Fig. 1 for a summary of the currently known m6A modification machinery.

Summary of m6A modification machinery. The m6A methyltransferase complex composed of METTL13, METTL14 and WTAP, probably also of VIRMA and RBM15, serves as m6A “writer”, demethylases (e.g., FTO and ALKBH5) serve as m6A “erasers”, and a set of m6A-binding proteins (e.g., YTHDF1/2/3, YTHDC1/2, IGF2BP1/2/3, METTL3 and eIF3) serve as m6A “readers” that determine the fate of target m6A-modified mRNA transcripts
During the past a few years, m6A modification in mRNAs or non-coding RNAs has been reported to play a critical role in virtually all major normal bioprocesses including self-renewal and differentiation of embryonic stem cells, tissue development (e.g., neurogenesis and hematopoiesis), response to heat shock or DNA damage, control of circadian clock, spermatogenesis, and maternal-to-zygotic transition, as well as primary microRNA processing, and RNA–protein interactions.9,10,13,17,18,19,23,26,28,29,30,31,32,33,34,35,36,37,38 More recently, extensive efforts have been exerted in investigating the biological impacts of dysregulated m6A modification and the associated machinery (i.e., m6A writer, eraser, and reader proteins) in various cancers.39 In this review, we focus on recent advances in the study of biological functions and underlying molecular mechanisms of dysregulated m6A modification and the associated regulatory proteins in the pathogenesis of various types of cancers, including leukemia, brain tumor, breast cancer, liver cancer, cervical cancer, and lung cancer. Moreover, we also discuss potential therapeutic strategies targeting dysregulated m6A machinery to treat the associated cancers.
FTO functions as an oncogenic m6A demethylase in leukemia and brain tumor
FTO became very famous a decade ago due to the strong association of single nucleotide polymorphisms (SNPs) located in its genomic locus with overweight and obesity in humans identified by large-scale, genome-wide association studies.40,41,42,43 Although there are some controversial discoveries regarding the link of FTO with overweight and obesity,44,45 mouse model studies did suggest a critical role of FTO in regulating fat mass, adipogenesis, and body weight,46,47,48 and there is also a link between the SNP risk genotype and increased FTO expression in human fibroblasts and blood cells.49,50 In addition, large-scale epidemiology studies demonstrate people with FTO SNPs or overweight/obesity have a higher risk of development of cancers such as breast, kidney, prostate, and pancreatic cancers, as well as leukemia, lymphoma and myeloma.51,52,53,54 However, the definitive role of FTO in cancer remained elusive.
To investigate the pathological role of FTO in cancer, we analyzed genome-wide gene expression profiling datasets of several large-cohorts of patients with primary acute myeloid leukemia (AML), and found that FTO is highly expressed in certain subtypes of AMLs including those carrying t(11q23)/MLL-rearrangements, t(15;17)/PML-RARA, FLT3-ITD, and/or NPM1 mutations.55 We next conducted in vitro and in vivo gain- and loss-of-function studies and demonstrated that forced expression of FTO enhanced human AML cell survival and proliferation, promoted leukemic oncogene (e.g., MLL-AF9) mediated transformation of normal hematopoietic stem/progenitor cells (HSPCs) and leukemogenesis, and inhibited all-trans retinoic acid (ATRA)-induced AML cell differentiation; the opposite was true when FTO expression was depleted.55 Thus, our data demonstrated that FTO plays an essential oncogenic role in cell transformation and leukemogenesis, as well as in drug response of AML cells. Importantly, we showed that FTO exerts its oncogenic role in AML in an m6A-dependent manner as an m6A demethylase.55 Briefly, FTO post-transcriptionally regulates the expression of its critical target RNAs, such as ASB2 and RARA, two genes that have been implicated in leukemia cell proliferation and drug response.56,57,58 We performed transcriptome-wide m6A-seq, luciferase reporter and mutagenesis assays, mRNA stability assays and gene-specific m6A-qPCR assays. Data were presented to show that FTO negatively regulates the expression of ASB2 and RARA through reducing the abundance of internal m6A modification, especially in the 3′ untranslated regions (3′-UTRs), which in turn leads to decreased stability of the target mRNA transcripts.55 Overall, our work provides compelling evidence showing the functional importance of m6A modification and FTO in tumorigenesis and drug response (see Fig. 2a).

FTO plays a critical oncogenic role in cancer as an m6A eraser and its function can be suppressed by R-2HG. a FTO functions as an oncogenic m6A demethylase in acute myeloid leukemia. b R-2HG targets the FTO/m6A/MYC/CEBPA axis and displays anti-tumor effects in leukemia and brain tumor
In brain tumor, Cui et al.60 reported that pharmaceutical inhibition of FTO by a chemical inhibitor (MA2, the ethyl ester form of meclofenamic acid (MA), a US Food and Drug Administration (FDA)-approved nonsteroidal anti-inflammatory drug that was shown to be a selective inhibitor of FTO59) suppresses tumor progression and substantially prolongs the lifespan of glioblastoma (GBM) stem cell (GSC)-grafted mice. Thus, FTO likely also plays a critical oncogenic role in self-renewal of GSCs and is required for the development of GBM.
R-2HG targets the FTO/m6A/MYC/CEBPA axis and displays anti-tumor effects in leukemia and brain tumor
R-2-hydroxyglutarate (R-2HG), produced at high levels by mutant isocitrate dehydrogenase 1/2 (IDH1/2) enzymes, which could be found in 10–20% of AML patients, ~80% of grade II-III gliomas and secondary GBM, and at a lower rate in other cancers, was reported as an oncometabolite.61,62,63,64,65,66,67,68 For instance, mutant IDH1 and its product R-2HG induce cytokine-independent growth and block erythropoietin (EPO)-mediated differentiation of TF-1 cells, a unique erythroleukemia cell line that is GM-CSF-dependent.68 Surprisingly, through analysis of the effects of R-2HG on cell viability, proliferation, apoptosis and cell cycle in 27 human leukemia cell lines, 15 primary AML samples, and 8 human GBM cell lines, we very recently found that R-2HG actually displays a broad and intrinsic anti-tumor activity in leukemia and glioma, causing decreased cancer cell viability/proliferation and increased cell-cycle arrest and apoptosis in a time- and dose-dependent manner in the vast majority of the tested samples.69 Exogenous R-2HG treatment showed no noticeable inhibitory effects on viability/proliferation of IDH-mutant AML cells, indicating these cells can tolerate the potential inhibitory effect of R-2HG. Moreover, we employed “human-in-mouse” xeno-transplantation leukemic models to evaluate the effect of R-2HG on leukemia progression in vivo. We found that both exogenous (in vivo injected) and endogenous (IDH1R132H-generated) R-2HG substantially inhibited leukemia progression in recipient mice xeno-transplanted with 2HG-sensitive AML cells (e.g., NOMO-1 or MA9.3ITD70), which was associated with reduced splenomegaly and inhibited engraftments in peripheral blood, bone marrow and spleen. However, no significant inhibitory effects were observed in mice xeno-transplanted with 2HG-resistant AML cells (e.g., MA9.3RAS70 or NB4 cells).69
Mechanistically, we identified FTO as a direct target of R-2HG and a main mediator of R-2HG-induced anti-tumor effects. R-2HG binds directly to FTO protein and inhibits its m6A demethylase activity, resulting in a significant increase of global m6A abundance in R-2HG-sensitive leukemia cells, and the effects of R-2HG is FTO-dependent. Notably, MYC is a direct and functionally essential target of FTO, and R-2HG treatment or FTO knockdown increases m6A level on MYC mRNA (especially at the 5′ UTR and middle exons), leading to mRNA decay and MYC down-regulation, and thereby suppression of MYC signaling.69 Interestingly, FTO transcription is controlled by CEBPA, and CEBPA mRNA is a direct target of FTO and is positively regulated by FTO in an m6A-dependent manner, so that there is a positive reciprocal regulation between FTO and CEBPA; as a result, R-2HG treatment can indirectly downregulate expression of both CEBPA and FTO through the FTO/m6A/CEBPA/FTO circuit.69 S-2HG, the enantiomer of R-2HG, exhibits similar effects to R-2HG by direct targeting FTO, causing increased global m6A modification and decreased leukemic cell proliferation/viability. Our data indicate that FTO/MYC homeostasis controls the sensitivity of leukemic cells to 2HG: a high abundance of FTO confers 2HG sensitivity in leukemic cells, whereas hyper-activation of MYC-associated signaling pathways renders leukemic cells resistant to 2HG; pharmaceutical or genetic inhibition of MYC signaling (e.g., by JQ1 or MYC shRNAs) can sensitize 2HG-resistant leukemic cells to (exogenous and endogenous) 2HG.69
Moreover, R-2HG also exhibits a synergistic effect with a series of first-line chemotherapy drugs such as ATRA, Azacitidine (AZA), Decitabine, and Daunorubicin. The inhibitory effect of R-2HG is also found in human brain tumor cells. Collectively, our results uncover a new mechanism involving an R-2HG⊣FTO⊣m6A⊣MYC/CEBPA axis that impacts cancer pathogenesis and drug response through post-transcriptional RNA methylation regulation (see Fig. 2b).69
Based on our work and those published by others, we presumed that endogenous R-2HG in IDH-mutant cancers most likely contributes to cancer initiation via inhibition of TET2 and probably also other epigenetic pathways. However, in IDH-wild-type AML cells, 2HG inhibits cancer proliferation in general. In low grade glioma and subsets of IDH-mutant AML cases in which the presence of 2HG leads to a more benign outcome, we suggest that 2HG contributes to cancer initiation via inhibiting TET2, but suppresses cancer progression/proliferation via inhibiting FTO/MYC signaling.69
FTO and R-2HG mainly target internal m6A rather than 5′ cap m6Am in leukemia
FTO has also been reported to demethylate 5′ cap N6,2′-O-dimethyladenosine (m6Am).71 However, we found that internal m6A abundance is approximately 20–30 times of the near 5′ cap m6Am abundance in human AML cells as detected by liquid chromatography-tandem mass spectrometry (LC-MS/MS) assays, and R-2HG treatment or FTO knockdown or overexpression in leukemia cells mainly affects internal m6A abundance69 (Su et al., 2018). In addition, we analyzed our m6A-seq data from human AML cells and found that over 95% of the m6A peaks affected by R-2HG treatment or FTO knockdown or overexpression are internal m6A, not 5′ cap m6Am69 (Su et al., unpublished). We also analyzed the fold changes of m6Am, Am, Cm, Gm, and Um-initiated mRNAs in leukemia cells, and found that m6Am-initiated mRNAs showed an even smaller fold change in expression than the other four groups of mRNAs upon R-2HG treatment or FTO overexpression. Even if increased m6Am through FTO inhibition by 2HG plays a role, it should lead to increased transcript stability,71 which is opposite to what was observed here for MYC and CEBPA as well as the observed cancer inhibition effect of 2HG.69 Moreover, our luciferase reporter and mutagenesis assays and gene-specific m6A-qPCR assays demonstrate that FTO demethylates the internal m6A, not potential cap m6Am, on target mRNA transcripts such as ASB2, RARA, MYC, and CEBPA.55,69 Taken together, the common internal m6A modifications, rather than the rare 5′ cap m6Am, are the main substrates of FTO that are responsible for FTO-mediated gene regulation and oncogenic role at least in leukemia. Of course, it is possible that m6Am might also be an important substrate of FTO in some other types of cells in which m6Am abundance is high.
ALKBH5 plays an oncogenic role as an m6A eraser in brain tumor and breast cancer
As the second m6A demethylase identified, ALKBH5 was reported by He and colleagues to affect mRNA export and RNA metabolism, and regulate spermatogenesis and apoptosis in mouse testes through targeting the p53 signaling pathway.17 Recently, it was reported that ALKBH5 functions as an oncoprotein in the pathogenesis of GBM and breast cancer, affecting the self-renewal and proliferation of relevant cancer stem cells.72,73 In brain tumors, ALKBH5 expression is aberrantly upregulated in GSCs and its increased expression is associated with poor outcome in patients with GBM.72 Elevated expression of ALKBH5 enhances self-renewal and proliferation of GSCs, while depletion of ALKBH5 expression significantly inhibits tumor development in nude mice intracranially implanted with GSCs.72 Mechanistically, ALKBH5 functions as an m6A demethylase, and enhances expression of its key target, FOXM1, by reducing m6A abundance on target mRNA transcripts (especially at the 3′ UTR); meanwhile, FOXM1-AS, a long non-coding RNA (lncRNA) that is located opposite to FOXM1 on human chromosome 12 with partial overlap, facilitates the interaction between ALKBH5 and nascent transcripts of FOXM1. As a functionally important target of ALKBH5, FOXM1 overexpression can reverse the effects of ALKBH5 or FOXM1-AS knockdown and reinstate the tumor growth of GSCs.72 Collectively, this study reveals that ALKBH5 enhances self-renewal and proliferation of GSCs and promotes tumorigenesis through regulating expression of FOXM1, with the aid of FOXM1-AS72 (see Fig. 3a).

ALKBH5 plays an oncogenic role in brain tumor and breast cancer. a ALKBH5 enhances self-renewal and proliferation of GSCs and promotes tumorigenesis through regulating expression of FOXM1 with the aid of FOXM1-AS. b HIF-induced ALKBH5 expression mediates the upregulation of pluripotency factor expression and the enrichment/specification of BCSCs in the hypoxic environment
It was also reported that hypoxia-stimulated HIF1α and HIF2α promote the expression of ALKBH5 in hypoxic breast cancer cells, and elevated expression of ALKBH5 promotes mRNA stability and expression of NANOG, a gene encoding a pluripotency factor, by catalyzing m6A demethylation.73 Ectopic expression of ALKBH5, under nonhypoxic conditions, significantly enhances NANOG expression and promotes enrichment of breast cancer stem cells (BCSCs), phenocopying the effects of hypoxia. Conversely, knockdown of ALKBH5 impairs hypoxia-induced NANOG expression and BCSC enrichment, and also impairs tumor formation in vivo.73 Thus, HIF-induced ALKBH5 expression mediates the upregulation of pluripotency factor expression and the enrichment/specification of BCSCs in the hypoxic tumor microenvironment through negative modulation of RNA methylation (see Fig. 3b). The same group showed further that both ALKBH5 and ZNF217 participate in the hypoxia-induced NANOG and KLF4 (another pluripotency factor gene) overexpression in breast cancer cells.74
METTL14 and METTL3 regulate normal and malignant hematopoiesis as m6A writers
As two major components of m6A MTC, the functions of METTL14 and METTL3 in normal and malignant hematopoiesis have been reported recently. We found that METTL14 is highly expressed in normal HSPCs and is downregulated during myeloid differentiation, and depletion of METTL14 expression further promotes terminal myeloid differentiation of normal HSPCs.75 METTL14 is also aberrantly overexpressed in human AMLs, especially those carrying t(11q23), t(15;17) and t(8;21), and forced expression of individual oncogenic fusion proteins resulting from such chromosomal translocations leads to upregulation of METTL14 expression. Moreover, we have demonstrated that METTL14 is required for both initiation and maintenance of AML and self-renewal of leukemia stem/initiation cells (LSCs/LICs).75 Mechanistically, METTL14 exerts its oncogenic role through m6A-dependent post-transcriptional regulation of its critical mRNA targets such as MYB and MYC, which encode master transcriptional regulators that are essential for self-renewal of normal HSPCs and LSCs/LICs;76,77,78,79,80 expression of METTL14 itself is negatively regulated by SPI1 (PU.1), a transcriptional master regulator of myelopoiesis.81 Notably, METTL14 promotes expression of MYB and MYC by increasing m6A abundance and enhancing stability of the target mRNA transcripts and likely also enhancing their translation.75 Collectively, our studies demonstrate that METTL14 plays an essential role in normal hematopoiesis and especially AML development and maintenance through blocking myeloid differentiation and promoting self-renewal of normal HSPCs and LSCs/LICs via an m6A-dependent mechanism involving the SPI1⊣METTL14-m6A-MYB/MYC signaling axis (see Fig. 4a).75 Our work also suggests that targeting METTL14, especially in combination with differentiation-inducing agents, may represent effective novel therapeutic strategies to treat AMLs with high levels of METTL14.75

METTL14 and METTL3 play oncogenic roles in leukemia. a METTL14 plays an essential oncogenic role in AML development and maintenance through regulating expression of critical targets (e.g., MYB and MYC) via an m6A-dependent mechanism. b METTL3 promotes AML cell proliferation and inhibits myeloid differentiation likely through promoting translation of its potential mRNA targets (e.g., MYC, and BCL2). c METTL3 is recruited to TSSs of target genes by CEBPZ, and its potential direct targets are SP1 and SP2, which regulate expression of MYC
As the main m6A methyltransferase, METTL3 has been shown recently to play a critical role in cell fate determination during the endothelial-to-hematopoietic transition (EHT) to specify the earliest HSPCs in vertebrate embryogenesis through an m6A-dependent mechanism.36 In zebrafish embryos, mettl3 is enriched in sorted endothelial cells and hemogenic endothelium, and loss-of-function of mettl3 by morpholino treatment14 or genetic knockout caused a significant decrease of m6A and a block of the emergence of HSPCs; a similar phenotype was observed in mice when Mettl3 was knocked down.36 Mechanistically, mettl3 deficiency causes continuous activation of Notch signaling, due to the suppression of YTHDF2-mediated mRNA decay of notch1a and rhoca in arterial endothelial cells, which in turn blocks EHT and thereby represses the generation of the earliest HSPCs.36
It was also reported recently that METTL3 plays an essential role in controlling myeloid differentiation of mammalian normal hematopoietic and leukemic cells.82 Forced expression of wild-type METTL3, but not a mutant METTL3 (with defect in catalytic activity), significantly promotes cell proliferation and inhibits cell differentiation of human cord blood-derived CD34 + HSPCs; the opposite is true when METTL3 is knocked down by shRNAs. METTL3 is highly expressed in AML compared to normal HSPCs or other types of cancers. Knockdown of METTL3 in human AML cell lines significantly induces cell differentiation and apoptosis and inhibits leukemia progression in mice xeno-transplanted with MOLM-13 AML cells. The biological function of METTL3 is likely attributed to the promotion of translation of its mRNA targets such as MYC, BCL-2, and PTEN in an m6A-dependent manner, although the exact molecular mechanism has not yet been defined (see Fig. 4b).82
A more recent study also demonstrated that METTL3 is critical for the maintenance of myeloid leukemia state.83 Interestingly, Barbieri et al.83 showed that METTL3 and METTL14 can both bind to chromatin, but mainly localize to the transcription start sites (TSSs) of distinct sets of coding genes that are featured with bimodal H3K4me3 peaks. The recruitment of METTL3 to TSSs of target genes is mediated by CEBPZ, a CCAAT-box binding factor. Promoter-bound METTL3 is required for m6A modification of associated transcripts, and METTL3 regulates translation, rather than transcription, of the relevant target transcripts.83 SP1 and SP2, which regulate expression of MYC,84 are two potential direct targets of METTL3 (see Fig. 4c).83
The functions of METTL3 and METTL14 in GBM and liver cancer are controversial
In GBM, Cui et al.60 reported that consistent with the increased m6A levels during the differentiation of GSCs, overexpression of wild-type METTL3, but not a catalytically inactive mutant of METTL3, significantly promoted differentiation of GSCs and inhibited the self-renewal and proliferation of GSCs. Conversely, depletion of METTL3 or METTL14 expression significantly enhanced GSC growth and self-renewal in vitro and promoted tumor progression in vivo.60 A number of GSC-associated genes (e.g., ADAM19) are putative targets of m6A modifications in GSCs that are probably responsible for the phenotypes caused by manipulating the expression of individual m6A writer or eraser genes.60 However, the opposite role of METTL3 in GBM was reported by a different group.85 They showed that METTL3 is highly expressed in GSCs and is downregulated during differentiation, associated with decreased levels of m6A during differentiation; silencing of METTL3 expression in GBM significantly inhibited tumor growth in mice and prolonged mouse survival, which is consistent with the observation that elevated expression of METTL3 was associated with poor survival in GBM patients; METTL3 knockdown also sensitized GSCs to γ-irradiation.85 SOX2 was identified as a functionally important target of METTL3, and METTL3-mediated m6A modification of SOX2 mRNA transcripts makes them more stable. Overall, this study suggests that METTL3 plays a critical oncogenic role in GSC maintenance and radioresistance.85
In liver cancer, Ma et al.86 reported that METTL14 plays a tumor-suppressor role in hepatocellular carcinoma (HCC), in which METTL14 and m6A levels were decreased compared to normal tissue or paratumor controls, with largely unchanged levels of METTL3 and WTAP. In analysis of 130 in-house HCC patient samples, they found that decreased expression of METTL14 was associated with poor prognosis in the patients; METTL14 knockdown enhanced HCC metastasis, and forced expression of METTL14 substantially suppressed HCC tumor invasion and metastasis, likely through m6A-dependent modulation of primary microRNA (e.g., mir-126) processing by interaction with DGCR8.86 In contrast, Chen et al.87 reported that METTL3 level was significantly higher and METTL14 level was slightly higher in HCC than in normal tissue, while WTAP level was unchanged; in analysis of TCGA HCC cohort dataset, they found that increased expression of METTL3 was associated with poor prognosis in the patients. They further showed that overexpression of METTL3 significantly promoted growth of HCC both in vitro and in vivo, while depletion of METTL3 expression substantially inhibited tumorigenesis and lung metastasis of HCC in vivo, likely through negative regulation of SOCS2 expression by an m6A- and YTHDF2-dependent mechanism.87 Similarly, they showed that METTL14 knockdown significantly suppressed HCC cell proliferation, migration and colony formation, and the opposite is true when METTL14 was overexpressed.87 Thus, they demonstrated that both METTL14 and METTL3 play oncogenic roles in HCC and are required for HCC growth and metastasis.87
METTL3 plays an oncogenic role in lung cancer as a potential m6A reader
METTL3 was also reported to be upregulated in lung adenocarcinoma and play an oncogenic role in promoting the growth, survival and invasion of human lung cancer cells.27 Interestingly, this study suggests that METTL3 may function as an m6A reader in cytoplasm and promote translation of its target mRNA transcripts (e.g., EGFR and TAZ) by interaction with the translation initiation machinery27 (see Fig. 5). Nevertheless, METTL3′s catalytic activity might be still required for its function in promoting translation of m6A-containing target transcripts, because its targets need to be modified with m6A in nucleus first before their translation is enhanced in cytoplasm.

METTL3 plays an oncogenic role in lung cancer. METTL3 enhances the growth, survival, and invasion of lung cancer cells through promoting translation of target mRNA transcripts (e.g., EGFR and TAZ)
IGF2BP proteins play oncogenic roles in cancers as m6A readers
Thus far, the most well documented m6A readers are the YTH domain-containing proteins including YTHDF1, YTHDF2, YTHDF3, YTDHDC1, and YTHDC2. Of them, YTHDF2, YTHDF3, and YTHDC2 promote decay of m6A-modified mRNAs.18,20,23 Interestingly, in contrast to what could be predicted by the mRNA decay mechanism mediated by YTHDF2, YTHDF3, or YTHDC2, our recent data showed that the vast majority portion of mRNA transcripts with a significant decrease in m6A abundance caused by overexpression of FTO tend to be downregulated in leukemia cells, likely due to decreased RNA stability along with reduced m6A abundance.55 In addition, through analysis of publically available datasets and our own experimental datasets, we found that a significant proportion of m6A-modified mRNA transcripts tend to be downregulated upon knockdown of m6A writers (METTL3 and/or MELLT14). Thus, we presumed that there could be alternative m6A reader(s) that promote mRNA stabilization.
Indeed, through both m6A-oligo-pulldown/mass spectrometry assays and in silico m6A-binding protein prediction analysis, we have recently identified the insulin-like growth factor-2 (IGF2) mRNA-binding proteins 1, 2, and 3 (IGF2BP1/2/3) as a new family of m6A readers, which selectively recognize m6A-modified mRNAs with a consensus of GG(m6A)C25. We show that IGF2BPs promote the stability and storage of their target mRNAs (e.g., MYC, FSCN1, TK1, and MARCKSL1) in an m6A-dependent manner in normal and stress conditions, likely by recruiting mRNA stabilizers such as HuR and MATRIN3. Different from the previously identified m6A reader proteins that contain a YTH domain,9,18,21,88 IGF2BP proteins contain six canonical RNA-binding domains, including two RNA recognition motif domains on the N-terminus and four KH domains on the C-terminal regions.89,90 Our data indicate that the KH3-4 di-domains of IGF2BP proteins are the most critical domains for their binding to m6A-modified target mRNAs and for their biological functions. Remarkably, over 3000 mRNA transcripts were identified as targets of each individual IGF2BP proteins, with over 5000 mRNAs being targeted by at least one protein and more than 2000 being co-targeted by all three IGF2BP proteins, highlighting the broad impact of the IGF2BP proteins as m6A readers that globally regulate gene expression at the post-transcriptional level. Notably, the binding sites of IGF2BP proteins are highly enriched in the 3′ end of target mRNAs. In addition, our data suggest that IGF2BP proteins are likely also involved in translation initiation/elongation of target mRNAs.25
We also showed that knockdown of individual IGF2BP genes significantly inhibited cell growth/proliferation, colony formation, and migration and invasion of human cervical cancer (Hela) and liver cancer (HepG2) cells. Such function of IGF2BP proteins relies on their role as m6A readers. MYC is a critical target of IGF2BPs in cancers, and its depletion mimics the phenotypes caused by IGF2BP depletion while its overexpression can rescue the effects of IGF2BP depletion.25 Collectively, IGF2BPs elicit oncogenic functions as m6A readers in promoting proliferation, migration, and invasion of cancer cells through post-transcriptionally regulating the stability and also translation of their key target mRNAs (e.g., MYC). Our work reveals a new facet of m6A reading and also suggests IGF2BPs as potential targets for anti-cancer therapy (see Fig. 6).

IGF2BP1/2/3 proteins play oncogenic roles in cancers. IGF2BP1/2/3 proteins promote proliferation, migration, and invasion of cancer cells through post-transcriptionally regulating the stability and translation of key target mRNAs (e.g., MYC)
Conclusions and perspectives
Despite still being in the infant stage, recent studies of m6A in cancers have revealed that m6A modification and the associated regulatory proteins play critical roles in a variety of cancers (see Table 1 for a summary). The m6A writers and erasers, relative to readers, have been better studied in cancers. Interestingly, a given m6A regulatory protein may play a similar role across different types of cancers. For example, FTO functions as an oncoprotein in both leukemia and GBM55,60 and ALKBH5 plays an oncogenic role in both breast cancer and GBM.72,73 Notably, while the oncogenic roles of METTL3 and METTL14 in AML have been confirmed by different groups,75,82,83 their reported functions in brain and liver cancers are controversial.60,85,86,87 The different roles of a given gene (e.g., METTL3 and METTL14) in the same cancer type (e.g., GBM and HCC) reported by different groups might be due to genetic/epigenetic heterogeneities of the cancer cell lines and primary tumor specimens used by different groups, and thus further systematical studies are warranted to clarify the discrepancies and better understand the factors that affect the functions of a given gene in different cellular contexts.
One may expect that m6A writer and eraser proteins function oppositely in a given type of cancer. However, this is not always the case. For instance, while FTO plays an essential oncogenic role in AML as an m6A eraser,55,69 three components of the m6A MTC including METTL3,82,83 METTL1475, and WTAP91 also function as oncoproteins in AML. Consistent with this, it is well known that TET2 (a DNA demethylase) and DNMT3A (a DNA methyltransferase) both function as tumor suppressors in myeloid malignancies in which they both are frequently associated with loss-of-function mutations92,93; furthermore, they can work cooperatively in repressing lineage differentiation of hematopoietic stem cells.94 Therefore, it is not unusual that a writer and an eraser of the same epigenetic modification (e.g., m6A RNA modification or DNA methylation) may play similar functional roles in the same cancer cell context, probably through regulating distinct sets of target genes. Alternatively, they may also target the same set of genes and cause similar biological consequences through different mechanisms. Indeed, we found that MYC is a critical target of and positively regulated by both FTO and METTL14.69,75 FTO mainly modulates m6A abundance on the 5′-terminal and middle exons of MYC mRNA;69 in contrast, METTL14 overexpression or depletion mainly affects m6A abundance in the 3′-region of MYC,75 likely due to the compensation effect of FTO on m6A modification of the other regions of MYC mRNA, because FTO expression is also positively regulated by METTL14 through an indirect mechanism (Su et al., unpublished data). There is a ~250- nucleotide cis-acting element termed as coding region instability determinant (CRD) in the 3′-region of MYC, which is required for regulating the stability of MYC mRNA.95 We showed that IGF2BP proteins preferentially recognize and bind to the m6A-modified CRD region of MYC mRNA, thereby stabilizing MYC mRNA and promoting translation;25 in contrast, YTHDF2 preferentially recognizes and binds to m6A-modified 5′-terminal and middle exons of MYC mRNA and thereby promotes mRNA decay69 (Su et al., unpublished data); this model is illustrated in Fig. 7. Moreover, while FTO preferentially recognizes and binds to m6A modifications on the 5′-terminal and middle exons of MYC mRNA, ALKBH5 preferentially recognizes and binds to m6A modifications on the 3′-region of MYC mRNA (Su et al., unpublished data). Interestingly, ALKBH5 was reported previously to be frequently associated with DNA copy number loss in AML, especially in AML carrying p53 mutations, implying that it may play a tumor-suppressor role in AML.96 Overall, different m6A erasers and readers may preferentially bind to distinct regions of the same mRNA transcripts and lead to different fates of the target transcripts. For instance, while FTO promotes the stability of MYC mRNA through inhibition of YTHDF2-mediated RNA decay due to decreased m6A abundance on the 5′-terminal and middle exons of MYC mRNA,69 METTL14 also promotes the stability and translation of MYC mRNA through IGF2BPs-mediated RNA stability/translation enhancement due to increased m6A abundance on the 3′-region of MYC mRNA.25,75 Similarly, METTL3 was also shown to be able to promote translation of MYC mRNA82 and also probably indirectly regulate MYC transcription.83

Model of YTHDF2- and IGF2BP1/2/3-mediated m6A-dependent post-transcriptional regulation of MYC expression. IGF2BP1/2/3 proteins preferentially bind to m6A sites in the 3′ end region of MYC and enhance RNA stability and promote RNA translation; in contrast, YTHDF2 protein preferentially binds to m6A sites in the 5′ end and middle regions of MYC and promotes RNA decay (based on Su et al., unpublished data)
A number of target genes of the aforementioned m6A regulators have been identified or implicated (see Table 1), and their expression is post-transcriptionally affected by the m6A regulators through m6A-dependent mechanisms, such as increased RNA decay or stability, and/or enhanced RNA translation. Many of such targets have been validated to be functionally important targets that upon appropriate manipulations can largely mimic or rescue the phenotype caused by the manipulation of a given m6A regulator. It is always very important to identify the most essential targets that are largely or even fully responsible for the effects of manipulation of a given m6A regulator. On the other hand, it would also be important to better understand the global effects of manipulation of individual m6A regulators, which may affect expression of hundreds or even thousands of downstream targets.
In addition, as the fates of m6A-modified RNA transcripts are ultimately determined by the types of m6A reader proteins that recognize and bind to the transcripts, it would be also important to identify the reader proteins that bind to and regulate expression of the functionally important targets. Actually, different readers may target distinct sets of transcripts, but in some cases different readers may preferentially bind to distinct regions of the same transcripts or even competitively bind to the same regions of the same transcripts. Therefore, in order to better understand m6A-mediated regulation of mRNA transcripts, it would be important to know which regions of the mRNA transcripts are m6A-modified and what type(s) of readers bind to the modified region(s).
The important roles of m6A regulatory proteins observed in various cancers suggest that they are potential therapeutic targets of cancer therapy. For example, given the essential role of FTO in leukemia and GBM,55,60,69 targeting FTO holds therapeutic potential to treat such cancers. Indeed, several FTO small-molecule inhibitors have been developed to inhibit the catalytic activity of FTO.59,97,98,99 MA59 has been shown to be able to inhibit GBM tumor progression in vivo.60 We showed that by inhibition of FTO catalytic activity and expression, 2HG can significantly suppress survival/proliferation of leukemic cells in vitro and substantially inhibit leukemia progression in vivo.69 Therefore, either FTO inhibitors or 2HG (or its analogs) can be applied to the clinic to treat IDH1/2 wild-type GBM and leukemia, especially those with FTO overexpression; in treating IDH-mutant cancers, combinational application of both IDH-mutant inhibitors and FTO inhibitors could lead to a more beneficial outcome than using IDH-mutant inhibitors alone, as suppression of R-2HG production by IDH-mutant inhibitors alone may cause rebounded expression/function of FTO and thus may lead to relapse.
With regard to METTL3, the situation is more complicated. METTL3 was reported to play an oncogenic role in both AML82 and lung cancer.27 Nonetheless, METTL3 may also have other functions independent of its catalytic activity in lung cancer, although such function was not reported in AML.27,82,83 Thus, development of inhibitors to target METTL3’s catalytic activity may not be sufficient to inhibit its overall functions.
In the future, development of more selective and potent inhibitors for FTO and other m6A regulatory proteins may lead to the development of effective novel therapeutic strategies to treat various cancers. In particular, the combinations of such inhibitor(s) with other therapeutic agents may represent more effective therapies to treat cancers that are resistant to currently available therapies. Indeed, we found that there is a synergistic effect between R-2HG and standard therapeutic agents such as ATRA, AZA, Decitabine, and Daunorubicin.69 Consistently, it was reported previously that leukemia patients with IDH mutations are more sensitive to treatment with AZA or Decitabine,100 ATRA,101 or standard chemotherapy (e.g., Daunorubicin),102,103 than those without. Similarly, our data69 and previous studies104 showed that glioma cells carrying IDH mutations are also more sensitive to Temozolomide, a common chemotherapy agent for brain tumor treatment. Therefore, it is important to test different combinations for different types of cancers to achieve the optimal therapeutic effects with minimal side effects in a manner of precision medicine.
References
He, C. Grand challenge commentary: RNA epigenetics? Nat. Chem. Biol. 6, 863–865 (2010).
Machnicka, M. A. et al. MODOMICS: a database of RNA modification pathways—2013 update. Nucleic Acids Res. 41, D262–D267 (2013).
Liu, N. & Pan, T. N-6-methyladenosine-encoded epitranscriptomics. Nat. Struct. Mol. Biol. 23, 98–102 (2016).
Fu, Y., Dominissini, D., Rechavi, G. & He, C. Gene expression regulation mediated through reversible m(6)A RNA methylation. Nat. Rev. Genet. 15, 293–306 (2014).
Roundtree, I. A., Evans, M. E., Pan, T. & He, C. Dynamic RNA modifications in gene expression regulation. Cell 169, 1187–1200 (2017).
Zhao, B. S., Roundtree, I. A. & He, C. Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 18, 31–42 (2017).
Jia, G. et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 7, 885–887 (2011).
Jia, G., Fu, Y. & He, C. Reversible RNA adenosine methylation in biological regulation. Trends Genet. 29, 108–115 (2013).
Dominissini, D. et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206 (2012).
Meyer, K. D. et al. Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell 149, 1635–1646 (2012).
Bokar, J. A., Shambaugh, M. E., Polayes, D., Matera, A. G. & Rottman, F. M. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA 3, 1233–1247 (1997).
Liu, J. et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 10, 93–95 (2014).
Wang, Y. et al. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat. Cell Biol. 16, 191–198 (2014).
Ping, X. L. et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 24, 177–189 (2014).
Schwartz, S. et al. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5’ sites. Cell Rep. 8, 284–296 (2014).
Patil, D. P. et al. m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature 537, 369–373 (2016).
Zheng, G. et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell. 49, 18–29 (2013).
Wang, X. et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505, 117–120 (2014).
Wang, X. et al. N(6)-methyladenosine modulates messenger RNA Translation Efficiency. Cell 161, 1388–1399 (2015).
Shi, H. et al. YTHDF3 facilitates translation and decay of N6-methyladenosine-modified RNA. Cell Res. 27, 315–328 (2017).
Xiao, W. et al. Nuclear m(6)A Reader YTHDC1 regulates mRNA Splicing. Mol. Cell 61, 507–519 (2016).
Li, A. et al. Cytoplasmic m6A reader YTHDF3 promotes mRNA translation. Cell Res. 27, 444–447 (2017).
Hsu, P. J. et al. Ythdc2 is an N6-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res. 27, 1115–1127 (2017).
Roundtree, I. A. et al. YTHDC1 mediates nuclear export of N6-methyladenosine methylated mRNAs. Elife 6, e31311 (2017).
Huang, H. et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol. 20, 285–295 (2018).
Meyer, K. D. et al. 5’ UTR m(6)A promotes Cap-independent translation. Cell 163, 999–1010 (2015).
Lin, S., Choe, J., Du, P., Triboulet, R. & Gregory, R. I. The m(6)A methyltransferase METTL3 promotes translation in human cancer cells. Mol. Cell. 62, 335–345 (2016).
Zhao, X. et al. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 24, 1403–1419 (2014).
Liu, N. et al. N(6)-methyladenosine-dependent RNA structural switches regulate RNA–protein interactions. Nature 518, 560–564 (2015).
Alarcon, C. R., Lee, H., Goodarzi, H., Halberg, N. & Tavazoie, S. F. N6-methyladenosine marks primary microRNAs for processing. Nature 519, 482–485 (2015).
Geula, S. et al. Stem cells. m6A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science 347, 1002–1006 (2015).
Chen, T. et al. m(6)A RNA methylation is regulated by microRNAs and promotes reprogramming to pluripotency. Cell Stem Cell 16, 289–301 (2015).
Zhou, J. et al. Dynamic m(6)A mRNA methylation directs translational control of heat shock response. Nature 526, 591–594 (2015).
Xiang, Y. et al. RNA m6A methylation regulates the ultraviolet-induced DNA damage response. Nature 543, 573–576 (2017).
Zhao, B. S. et al. m6A-dependent maternal mRNA clearance facilitates zebrafish maternal-to-zygotic transition. Nature 542, 475–478 (2017).
Zhang, C. et al. m6A modulates haematopoietic stem and progenitor cell specification. Nature 549, 273–276 (2017).
Xu, K. et al. Mettl3-mediated m(6)A regulates spermatogonial differentiation and meiosis initiation. Cell Res. 27, 1100–1114 (2017).
Lin, Z. et al. Mettl3-/Mettl14-mediated mRNA N(6)-methyladenosine modulates murine spermatogenesis. Cell Res. 27, 1216–1230 (2017).
Deng, X., Su, R., Feng, X., Wei, M. & Chen, J. Role of N(6)-methyladenosine modification in cancer. Curr. Opin. Genet. Dev. 48, 1–7 (2017).
Frayling, T. M. et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 316, 889–894 (2007).
Dina, C. et al. Variation in FTO contributes to childhood obesity and severe adult obesity. Nat. Genet. 39, 724–726 (2007).
Scuteri, A. et al. Genome-wide association scan shows genetic variants in the FTO gene are associated with obesity-related traits. PLoS Genet. 3, e115 (2007).
Yang, J. et al. FTO genotype is associated with phenotypic variability of body mass index. Nature 490, 267–272 (2012).
Smemo, S. et al. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature 507, 371–375 (2014).
Claussnitzer, M. et al. FTO obesity variant circuitry and adipocyte browning in humans. N. Engl. J. Med. 373, 895–907 (2015).
Fischer, J. et al. Inactivation of the Fto gene protects from obesity. Nature 458, 894–898 (2009).
Church, C. et al. Overexpression of Fto leads to increased food intake and results in obesity. Nat. Genet. 42, 1086–1092 (2010).
Merkestein, M. et al. FTO influences adipogenesis by regulating mitotic clonal expansion. Nat. Commun. 6, 6792 (2015).
Berulava, T. & Horsthemke, B. The obesity-associated SNPs in intron 1 of the FTO gene affect primary transcript levels. Eur. J. Hum. Genet. 18, 1054–1056 (2010).
Karra, E. et al. A link between FTO, ghrelin, and impaired brain food-cue responsivity. J. Clin. Invest. 123, 3539–3551 (2013).
Soderberg, K. C. et al. Overweight, obesity and risk of haematological malignancies: a cohort study of Swedish and Finnish twins. Eur. J. Cancer 45, 1232–1238 (2009).
Castillo, J. J., Mull, N., Reagan, J. L., Nemr, S. & Mitri, J. Increased incidence of non-Hodgkin lymphoma, leukemia, and myeloma in patients with diabetes mellitus type 2: a meta-analysis of observational studies. Blood 119, 4845–4850 (2012).
Li, G., Chen, Q., Wang, L., Ke, D. & Yuan, Z. Association between FTO gene polymorphism and cancer risk: evidence from 16,277 cases and 31,153 controls. Tumour Biol. 33, 1237–1243 (2012).
Hernandez-Caballero, M. E. & Sierra-Ramirez, J. A. Single nucleotide polymorphisms of the FTO gene and cancer risk: an overview. Mol. Biol. Rep. 42, 699–704 (2015).
Li, Z. et al. FTO plays an oncogenic role in acute myeloid leukemia as a N6-methyladenosine RNA demethylase. Cancer Cell 31, 127–141 (2017).
Kohroki, J. et al. ATRA-regulated Asb-2 gene induced in differentiation of HL-60 leukemia cells. FEBS Lett. 505, 223–228 (2001).
Guibal, F. C. et al. ASB-2 inhibits growth and promotes commitment in myeloid leukemia cells. J. Biol. Chem. 277, 218–224 (2002).
Glasow, A., Prodromou, N., Xu, K., von Lindern, M. & Zelent, A. Retinoids and myelomonocytic growth factors cooperatively activate RARA and induce human myeloid leukemia cell differentiation via MAP kinase pathways. Blood 105, 341–349 (2005).
Huang, Y. et al. Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res. 43, 373–384 (2015).
Cui, Q. et al. m6A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. 18, 2622–2634 (2017).
Rohle, D. et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 340, 626–630 (2013).
Wang, F. et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science 340, 622–626 (2013).
Xu, W. et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 19, 17–30 (2011).
Zhao, S. et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science 324, 261–265 (2009).
Figueroa, M. E. et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18, 553–567 (2010).
Lu, C. et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483, 474–478 (2012).
Okoye-Okafor, U. C. et al. New IDH1 mutant inhibitors for treatment of acute myeloid leukemia. Nat. Chem. Biol. 11, 878–886 (2015).
Losman, J. A. et al. R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 339, 1621–1625 (2013).
Su, R. et al. R-2HG exhibits anti-tumor activity by targeting FTO/m(6)A/MYC/CEBPA signaling. Cell 172, 90–105 (2018). e123.
Wunderlich, M. et al. AML cells are differentially sensitive to chemotherapy treatment in a human xenograft model. Blood 121, e90–e97 (2013).
Mauer, J. et al. Reversible methylation of m6Am in the 5’ cap controls mRNA stability. Nature 541, 371–375 (2017).
Zhang, S. et al. m6A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell 31, 591–606 (2017). e596.
Zhang, C. et al. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m6A-demethylation of NANOG mRNA. Proc. Natl Acad. Sci. USA 113, E2047–E2056 (2016).
Zhang, C. et al. Hypoxia-inducible factors regulate pluripotency factor expression by ZNF217- and ALKBH5-mediated modulation of RNA methylation in breast cancer cells. Oncotarget 7, 64527–64542 (2016).
Weng, H. et al. METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m(6)A modification. Cell Stem Cell 22, 191–205 (2018). e199.
Ramsay, R. G. & Gonda, T. J. MYB function in normal and cancer cells. Nat. Rev. Cancer 8, 523–534 (2008).
Zuber, J. et al. An integrated approach to dissecting oncogene addiction implicates a Myb-coordinated self-renewal program as essential for leukemia maintenance. Genes Dev. 25, 1628–1640 (2011).
Jin, S. et al. c-Myb binds MLL through menin in human leukemia cells and is an important driver of MLL-associated leukemogenesis. J. Clin. Invest. 120, 593–606 (2010).
Wall, M. et al. Translational control of c-MYC by rapamycin promotes terminal myeloid differentiation. Blood 112, 2305–2317 (2008).
Delgado, M. D. & Leon, J. Myc roles in hematopoiesis and leukemia. Genes Cancer 1, 605–616 (2010).
Dakic, A. et al. PU.1 regulates the commitment of adult hematopoietic progenitors and restricts granulopoiesis. J. Exp. Med. 201, 1487–1502 (2005).
Vu, L. P. et al. The N6-methyladenosine (m6A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat. Med. 23, 1369–1376 (2017).
Barbieri, I. et al. Promoter-bound METTL3 maintains myeloid leukaemia by m(6)A-dependent translation control. Nature 552, 126–131 (2017).
Geltinger, C., Hortnagel, K. & Polack, A. TATA box and Sp1 sites mediate the activation of c-myc promoter P1 by immunoglobulin kappa enhancers. Gene Expr. 6, 113–127 (1996).
Visvanathan, A. et al. Essential role of METTL3-mediated m(6)A modification in glioma stem-like cells maintenance and radioresistance. Oncogene 37, 522–533 (2018).
Ma, J. Z. et al. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N(6) -methyladenosine-dependent primary microRNA processing. Hepatology 65, 529–543 (2017).
Chen, M. et al. RNA N6-methyladenosine methyltransferase METTL3 promotes liver cancer progression through YTHDF2 dependent post-transcriptional silencing of SOCS2. Hepatology (2017). https://doi.org/10.1002/hep.29683. [Epub ahead of print]
Wang, X. et al. N-6-methyladenosine modulates messenger RNA translation efficiency. Cell 161, 1388–1399 (2015).
Bell, J. L. et al. Insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs): post-transcriptional drivers of cancer progression? Cell. Mol. Life Sci. 70, 2657–2675 (2013).
Nielsen, J. et al. A family of insulin-like growth factor II mRNA-binding proteins represses translation in late development. Mol. Cell Biol. 19, 1262–1270 (1999).
Bansal, H. et al. WTAP is a novel oncogenic protein in acute myeloid leukemia. Leukemia 28, 1171–1174 (2014).
Ley, T. J. et al. DNMT3A mutations in acute myeloid leukemia. N. Engl. J. Med. 363, 2424–2433 (2010).
Delhommeau, F. et al. Mutation in TET2 in myeloid cancers. N. Engl. J. Med. 360, 2289–2301 (2009).
Zhang, X. et al. DNMT3A and TET2 compete and cooperate to repress lineage-specific transcription factors in hematopoietic stem cells. Nat. Genet. 48, 1014–1023 (2016).
Doyle, G. A. et al. The c-myc coding region determinant-binding protein: a member of a family of KH domain RNA-binding proteins. Nucleic Acids Res. 26, 5036–5044 (1998).
Kwok, C. T., Marshall, A. D., Rasko, J. E. & Wong, J. J. Genetic alterations of m6A regulators predict poorer survival in acute myeloid leukemia. J. Hematol. Oncol. 10, 39 (2017).
Chen, B. et al. Development of cell-active N6-methyladenosine RNA demethylase FTO inhibitor. J. Am. Chem. Soc. 134, 17963–17971 (2012).
Zheng, G. et al. Synthesis of a FTO inhibitor with anticonvulsant activity. ACS Chem. Neurosci. 5, 658–665 (2014).
Singh, B. et al. Important role of FTO in the survival of rare panresistant triple-negative inflammatory breast cancer cells facing a severe metabolic challenge. PLoS ONE 11, e0159072 (2016).
Emadi, A. et al. Presence of isocitrate dehydrogenase mutations may predict clinical response to hypomethylating agents in patients with acute myeloid leukemia. Am. J. Hematol. 90, E77–E79 (2015).
Boutzen, H. et al. Isocitrate dehydrogenase 1 mutations prime the all-trans retinoic acid myeloid differentiation pathway in acute myeloid leukemia. J. Exp. Med. 213, 483–497 (2016).
Patel, J. P. et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N. Engl. J. Med. 366, 1079–1089 (2012).
Chou, W. C. et al. The prognostic impact and stability of isocitrate dehydrogenase 2 mutation in adult patients with acute myeloid leukemia. Leukemia 25, 246–253 (2011).
Wang, J. B., Dong, D. F., Wang, M. D. & Gao, K. IDH1 overexpression induced chemotherapy resistance and IDH1 mutation enhanced chemotherapy sensitivity in glioma cells in vitro and in vivo. Asian Pac. J. Cancer Prev. 15, 427–432 (2014).
Acknowledgements
We apologize to colleagues whose work could not be included due to space limitations. This work was supported in part by a grant (No. 81603149 to X.D.) from National Nature Science Foundation of China, as well as the National Institutes of Health (NIH) R01 Grants CA214965 (J.C.), CA211614 (J.C.), and CA178454 (J.C.). Z.L. is supported by an American Cancer Society (ACS) Research Scholar Award. J.C. is a Leukemia & Lymphoma Society (LLS) Scholar.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
We have a patent filed based on our R-2HG/FTO work (to J.C. and R.S.). The remaining authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Deng, X., Su, R., Weng, H. et al. RNA N6-methyladenosine modification in cancers: current status and perspectives. Cell Res 28, 507–517 (2018). https://doi.org/10.1038/s41422-018-0034-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41422-018-0034-6
This article is cited by
-
The epigenetic downregulation of LncGHRLOS mediated by RNA m6A methylase ZCCHC4 promotes colorectal cancer tumorigenesis
Journal of Experimental & Clinical Cancer Research (2024)
-
LINC00115 promotes chemoresistant breast cancer stem-like cell stemness and metastasis through SETDB1/PLK3/HIF1α signaling
Molecular Cancer (2024)
-
N6-methyladenosine demethyltransferase FTO mediated m6A modification of estrogen receptor alpha in non-small cell lung cancer tumorigenesis
Oncogene (2024)
-
Multigenerational paternal obesity enhances the susceptibility to male subfertility in offspring via Wt1 N6-methyladenosine modification
Nature Communications (2024)
-
Overcoming therapeutic resistance in oncolytic herpes virotherapy by targeting IGF2BP3-induced NETosis in malignant glioma
Nature Communications (2024)
