
Abstract
Programmed cell death (PCD) is a fundamental biological process. Deficiency in MOSAIC DEATH 1 (MOD1), a plastid-localized enoyl-ACP reductase, leads to the accumulation of reactive oxygen species (ROS) and PCD, which can be suppressed by mitochondrial complex I mutations, indicating a signal from chloroplasts to mitochondria. However, this signal remains to be elucidated. In this study, through cloning and analyzing a series of mod1 suppressors, we reveal a comprehensive organelle communication pathway that regulates the generation of mitochondrial ROS and triggers PCD. We show that mutations in PLASTIDIAL NAD-DEPENDENT MALATE DEHYDROGENASE (plNAD-MDH), chloroplastic DICARBOXYLATE TRANSPORTER 1 (DiT1) and MITOCHONDRIAL MALATE DEHYDROGENASE 1 (mMDH1) can each rescue the ROS accumulation and PCD phenotypes in mod1, demonstrating a direct communication from chloroplasts to mitochondria via the malate shuttle. Further studies demonstrate that these elements play critical roles in the redox homeostasis and plant growth under different photoperiod conditions. Moreover, we reveal that the ROS level and PCD are significantly increased in malate-treated HeLa cells, which can be dramatically attenuated by knockdown of the human gene MDH2, an ortholog of Arabidopsis mMDH1. These results uncover a conserved malate-induced PCD pathway in plant and animal systems, revolutionizing our understanding of the communication between organelles.
Similar content being viewed by others

Introduction
Programmed cell death (PCD), a genetically controlled process of cell suicide, is a fundamental event of life, and originated before the advent of eukaryotes.1 Although similar attributes and common repressors have been found between plant and animal PCD,2,3 the uniqueness of plant PCD signaling pathways has emerged recently.4,5 PCD is now realized as essential for plant organogenesis and biotic/abiotic stress responses through its diverse functions in developmental patterning, cell differentiation, cell number regulation, and the hypersensitive response (HR).6,7,8,9,10 However, the understanding of different PCD signaling pathways in plants is still very limited.
Reactive oxygen species (ROS) provide a common thread and play multiple roles in many plant PCD systems.4,11 ROS can be generated from various subcellular sources, including mitochondria and chloroplasts. Chloroplasts are considered as the main source of ROS in the presence of light,12 and many types of plant PCD require functional chloroplasts and light.4 However, newly identified PCD components in mitochondria reveal the function of mitochondria in triggering and executing PCD in plants.4 ROS can act as PCD-inducing signaling molecules that trigger mitochondrial membrane permeabilization or the formation of the mitochondrial permeability transition pore (MPTP).4,13 ROS could also be elevated to a lethal level by cytochrome c release and the disruption of mitochondrial electron transport chain (mETC) in PCD execution.4,14,15 These findings demonstrate that both chloroplasts and mitochondria play essential roles in triggering and executing PCD. Communications between nucleus and organelles have been well-studied and these include anterograde (nucleus to organelle) and retrograde (organelle to nucleus) signaling pathways.16,17,18,19,20,21,22,23,24 However, fundamental challenges remain to be addressed, such as whether plant PCD requires communication between the two organelles and what is the nature of the signaling molecule(s) transduced between them.4
Our previous studies have shown that mosaic death 1 (mod1) in Arabidopsis thaliana is a cell death mutant,25 which presents an elevated ROS level.26 Unlike the lesion mimic mutants, such as accelerated cell death 2 (acd2)27 and lesion simulating disease 1 (lsd1),28 which exhibit runaway cell death lesions, mod1 shows a mosaic pattern of cell death in the leaf midvein and shoot meristem.25 The MOD1 gene encodes an enoyl-acyl carrier protein (ACP) reductase (ENR), a subunit of fatty acid synthase that catalyzes the de novo biosynthesis of fatty acids in plastids; its deficiency leads to PCD.25 Genetic studies on the suppressors of mod1 (soms) revealed that ROS accumulation and abnormal morphological phenotypes of mod1 plants could be rescued by mutations affecting the activity of mETC complex I (NADH dehydrogenase/NADH oxidase), and that the ROS generated from mitochondrial ETC, rather than plasma membrane NADPH oxidases, plays a crucial role in the mod1-triggered PCD.26 The discovery that the abnormalities in mod1 caused by the deficiency in fatty acid biosynthesis in the chloroplast can be restored by the decrease of mETC complex I activity suggests a signal produced in the chloroplast that ultimately induces ROS production by ETC in the mitochondrion. However, the identity of this signal and whether the signaling involves a direct communication with the mitochondrion are both unknown.29
To identify the signal transported from chloroplasts to induce ROS production in mitochondria, we now describe a screen for new mod1 suppressors derived from ethyl methanesulfonate (EMS) mutagenized Arabidopsis M2 seeds. This has identified three key components in a direct Chloroplast-To-Mitochondrion (CTM) communication. In-depth studies on these genes reveal that the accumulation of MOD1 substrate NADH and its indirect transport from the chloroplast to the cytosol and then to the mitochondrion through the malate shuttle are responsible for ROS induction in mETC. This pathway is essential for redox homeostasis and plant growth regulation under different photoperiod conditions. We further demonstrate the existence of a conserved pathway from the cytosol to the mitochondrion that regulates ROS and cell death in HeLa cells. These findings identify a long suspected communication pathway from chloroplasts to mitochondria in plants and its conserved cytosol-mitochondrion ROS- and cell death-inducing mechanism in animals.
Results
Screening for mod1 suppressors altered in the Chloroplast-To-Mitochondrion communication
In the previous study with a T-DNA insertion mutant library, we had screened and identified many soms, most of which were mETC complex I-deficient mutants.26 To identify the components of the CTM communication, we generated an EMS mutagenized mutant library with more than 100,000 M1 plants and screened for new soms that are not the result of a deficiency in mETC complex I. Among 1073 soms identified in screening the EMS mutagenized mutant library, only 95 soms (8.9%) did not affect mETC complex I activity, of which we analyzed 10 soms in-depth in this study (Fig. 1).

Screening for mod1 suppressors that do not affect mitochondrial complex I activities. a Comparison of phenotypes of som mutants with the wild type and mod1 plants at 30 days after germination (DAG). Scale bars, 1 cm. b In-gel assay of NADH oxidase activity with bir6-2 as a control. The dihydrolipoamide dehydrogenase activity was used as a loading control
SOM410 encodes a plastid-localized NAD-dependent malate dehydrogenase
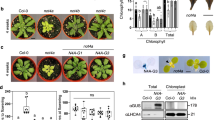
To understand the nature and functions of the genes mutated in these soms, we systematically mapped and cloned these genes. Among the 10 soms, the homozygous recessive suppressor som410 could fully suppress the mod1 morphological and biochemical phenotypes including plant height, cell death, and levels of H2O2 and O2−, showing a similar phenotype to that of the wild type (Fig. 2a–d). To clone SOM410, we took a map-based cloning approach and located the SOM410 gene in a 125-kb region on chromosome 3 (Fig. 2e). Through whole-genome resequencing of mod1 som410, we identified a C269T point mutation in At3g47520 gene in the mapped region (Fig. 2e). This mutation leads to an A90V amino acid substitution in the protein sequence. To verify that the suppression of the mod1 phenotypes was truly caused by this point mutation in At3g47520, the mod1 som410 mutant was complemented by a 3.1-kb genomic DNA fragment containing the At3g47520 gene. All the transformants (PSOM410:SOM410/mod1 som410, designated as mod1 som410 comp) resumed the mod1 phenotypes (Fig. 2a–d), demonstrating that the suppression of the mod1 phenotypes is caused by the mutation in At3g47520. In addition, we identified another two independent suppressors, som430+/- and som2167, which were mapped to the similar region as som410 and could partially or fully suppress the cell death in mod1. Whole-genome resequencing results showed that both of them have mutations in At3g47520. The som430+/- mutant has a C400T point mutation leading to a premature translation termination (Fig. 2e and Supplementary information, Figure S1a-e), whereas som2167 carries a C914T point mutation resulting in an A305V amino acid change (Fig. 2e and Supplementary information, Figure S2a and S2b). Therefore, these results further confirm that the mutations in At3g47520 are responsible for the suppression of the mod1 phenotypes in mod1 som410, mod1 som430+/-, and mod1 som2167.

SOM410 encodes and functions as a plastidial NAD-dependent malate dehydrogenase. a Phenotypes of Col-0, mod1, som410, mod1 som410, mod1 som410 comp (mod1 som410 complemented line), At3g47520+/CR, and mod1 At3g47520+/CR at 35 DAG. At3g47520+/CR and mod1 At3g47520+/CR were generated by CRISPR/Cas9 mutagenesis. Scale bars, 1 cm. b DAB-stained seedlings, showing the H2O2 levels. Scale bars, 1 cm. (c) NBT-stained seedlings, showing O2− levels. Scale bars, 1 cm. d Trypan blue-stained leaves. Scale bars, 1 cm. e Map-based cloning of SOM410 (top). The mutation sites in som410, som430, and som2167 DNA (middle) and protein (bottom) sequences are indicated with red, blue and green triangles, respectively. f Subcellular localization of SOM410-GFP in Arabidopsis mesophyll protoplasts of 35S:SOM410-GFP transgenic plants. Scale bars, 5 μm. g Comparison of total cellular MDH activities in the indicated plants. Values are means ± SD (n = 6), and different letters at top of each column indicate a significant difference at P < 0.05 determined by Tukey’s HSD test. h Transcript levels of SOM410 in the indicated plants, revealed by qRT-PCR using Actin as reference. Values are means ± SD (n = 3), and different letters above each column indicate a significant difference at P < 0.05 determined by Tukey’s HSD test. i Protein levels of SOM410 in the indicated plants, detected by immunoblotting with anti-SOM410 polyclonal antibodies
The At3g47520 (SOM410, SOM430, or SOM2167) locus encodes a plastidial NAD-dependent malate dehydrogenase (plNAD-MDH).30,31,32 Phylogenic analysis showed that the MDH domain is highly conserved across different plant species but the chloroplast transit peptide domain is less conserved (Supplementary information, Figure S3). To confirm the function of SOM410, we first examined its subcellular localization. The SOM410 protein was clearly localized in the chloroplast of the 35S:SOM410-GFP transgenic plants, which is in accord with its annotation and previous reports30,31,32 (Fig. 2f). We then measured and compared the total MDH activities among the relevant genotypes. The MDH activities were similar between the wild type and mod1, but they were significantly impaired in both som410 and mod1 som410 compared to the wild type and mod1 (Fig. 2g). As expected, the MDH activity was restored in all the complemented lines (mod1 som410 comp) compared to mod1 som410. We observed similar results among som430+/-, mod1 som430+/-, and complemented lines (mod1 som430+/- comp1 and 2) (Supplementary information, Figure S1i). These results confirm that SOM410 encodes a plastidial malate dehydrogenase.
To understand why the point mutation in som410 affects the MDH activity, we examined the expression levels of SOM410 by quantitative real-time PCR (qRT-PCR). We found no significant difference between wild type, mod1, som410, and mod1 som410, but in the complemented line the SOM410 transcript level was significantly increased (Fig. 2h). Moreover, we found that the SOM410 protein levels were obviously decreased in som410 and mod1 som410 compared with the wild type but strikingly increased in the complemented line (Fig. 2i), which is in line with the MDH activities in the corresponding lines. Taken together, these results suggest that the point mutation of som410 could reduce the stability of the SOM410 protein, which in turn leads to the deficiency of the MDH activity in chloroplasts.
To further understand the function of SOM410, we also attempted to generate At3g47520 loss-of-function mutants using the CRISPR/Cas9 system. We did not obtain any homozygous loss-of-function mutants of At3g47520, suggesting that the null mutation of SOM410 is lethal, which accords with previous reports that the T-DNA insertion mutant of plNAD-MDH was embryonic lethal at the globular-to-heart transition stage.30,32 However, we obtained a heterozygous mutant (At3g47520+/CR) with a T insertion at 178 bp downstream of ATG start codon, and this mutant partially rescued mod1 phenotypes (Fig. 2a–d and Supplementary information, Figure S2c). Similarly, the homozygous mutation of SOM430, which resulted in a premature translation termination of the protein, is also embryonic lethal. The segregation of mod1 som430+/- resulted in 303 suppressed to 160 non-suppressed plants in the self-pollinated progeny, fitting the theoretical segregation ratio of 2:1 (P-value = 0.58 by chi-square test). Moreover, overexpression of At3g47520 in mod1 som430 +/- resumed the mod1 phenotypes (Supplementary information, Figure S1a-d and S1f-h). Therefore, chloroplastic NAD-MDH is indispensable in plants, and partial loss of function of chloroplastic NAD-MDH in som410, som430+/-, and som2167 is sufficient for the suppression of mod1 phenotypes.
SOM787 encodes a chloroplastic dicarboxylate transporter
The suppressor som787 is another type of recessive mutant that fully rescued the mod1 phenotypes (Fig. 3a–d). We mapped the som787 mutation to a 477-kb region on chromosome 5. Through whole-genome resequencing, we identified a C952T point mutation in At5g12860 gene in the mapped region (Fig. 3e). This mutation results in a P318S amino acid substitution in protein sequence. The mod1 som787 mutant was complemented by a 4.2−kb genomic DNA fragment containing the At5g12860 gene, and all the transgenic plants (PSOM787:SOM787/mod1 som787, designated as mod1 som787 comp) were restored to the mod1 phenotypes (Fig. 3a–d). We also identified another two independent som mutants, som812 and som2073. Both som812 and som2073 were mapped to the same region as som787. som812 contained a C704T point mutation in At5g12860 leading to a P235L amino acid substitution (Fig. 3e and Supplementary information, Figure S4a and b), whereas som2073 carried a C1665T point mutation in At5g12860 leading to an S449F amino acid substitution (Fig. 3e and Supplementary information, Figure S4a and c). Therefore, the mutations in At5g12860 are responsible for the suppression of mod1 phenotypes. We additionally obtained a T-DNA insertion mutant of At5g12860, som787-2 (N877710), which contains a T-DNA insertion at 531 bp downstream of ATG start codon. The transcript level of At5g12860 is significantly decreased in som787-2 (Supplementary information, Figure S5). In contrast to the observation that som787 had no obvious abnormal phenotypes compared with Col-0, som787-2 plants were smaller in size, indicating that som787, som812 and som2073 are leaky mutants. The mod1 som787-2 double mutant showed similar morphology to som787-2, and was rescued for the mod1 phenotypes of cell death and accumulation of H2O2 and O2− (Fig. 3a–d).

Cloning and characterization of SOM787. a Phenotypes of Col-0, mod1, som787, mod1 som787, mod1 som787 comp (mod1 som787 complemented line), som787-2 (N877710), and mod1 som787-2 at 32 DAG. Scale bars, 1 cm. b DAB-stained seedlings. Scale bars, 1 cm. c NBT-stained seedlings. Scale bars, 1 cm. d Trypan blue-stained leaves. Scale bars, 1 cm. e Map-based cloning of SOM787 (top). The mutation sites in som787, som812, and som2073 DNA (middle) and protein (bottom) sequences are the indicated with red, blue and green triangles, respectively. f Subcellular localization of SOM787-GFP in Arabidopsis mesophyll protoplasts of 35S:SOM787-GFP transgenic plants. Scale bars, 5 μm. g Transcript levels of SOM787 revealed by qRT-PCR using Actin as reference. Values are means ± SD (n = 3), and different letters at top of each column indicate a significant difference at P < 0.05 determined by Tukey’s HSD test.
SOM787 is allelic to a previously reported gene, DICARBOXYLATE TRANSPORTER 1 (DiT1), which encodes a chloroplastic dicarboxylate transporter that functions as the malate/oxaloacetate (OAA) transporter or as the 2-oxoglutarate (2-OG)/malate transporter to export malate out of the chloroplast.33,34 Consistent with its transporting functions, we found that SOM787 is specifically localized in the chloroplast envelope membrane (Fig. 3f). The expression level of SOM787 is not affected in som787, indicating that the proline at 318 position (P318) is crucial for its normal function (Fig. 3g). Phylogenic analysis revealed that P318 and the dicarboxylate transporter domain of SOM787 are highly conserved across different plant species while the chloroplast transit peptide domain is less conserved (Supplementary information, Figure S6). Two paralogues of SOM787, DiT2.1 and DiT2.2, are found in Arabidopsis thaliana (Supplementary information, Figure S7a), of which only DiT2.1 has been reported as a glutamate/malate translocator able to transport malate into chloroplasts.34 We therefore crossed mod1 with two T-DNA insertion alleles of these two genes (dit2.1-1, dit2.1-2, dit2.2-1, and dit2.2-2). Morphological analysis showed that both mod1 dit2.1-1 and mod1 dit2.1-2 had severe growth phenotypes (Supplementary information, Figure S7b), similar to dit2.1-1 and dit2.1-2 as previously reported.34,35 Furthermore, neither dit2.2-1 nor dit2.2-2 could rescue the mod1 phenotypes (Supplementary information, Figure S7c). These results suggest that suppressing mod1 phenotypes by DiT1 mutations is closely related to the function of DiT1 as a transporter to export malate from chloroplasts.
SOM328 encodes a mitochondrial malate dehydrogenase
The suppressor som328 represents the third type of recessive mutants that rescued the mod1 phenotypes (Fig. 4a–d). We mapped som328 to a 211-kb region on chromosome 1. By whole-genome resequencing, we identified a G481A point mutation in At1g53240 gene in this region. This mutation leads to an R115H amino acid change in protein sequence (Fig. 4e). The mod1 som328 mutant was complemented by a 4.2-kb genomic DNA fragment containing At1g53240 (PSOM328:SOM328/mod1 som328, designated as mod1 som328 comp) and we found that the mod1 phenotypes were fully restored (Fig. 4a–d). Consistent with this result, we also found three additional suppressors, som369, som2169, and som2211, which all contained mutations in the At1g53240 gene. The som369 mutant has a G244A point mutation at the first nucleotide of intron 1 leading to abnormal splicing with dramatically decreased expression level and showed significantly impaired total MDH activity (Fig. 4e and Supplementary information, Figure S8); som2169 has a G1374A point mutation leading to the G250D amino acid change (Fig. 4e and Supplementary information, Figure S9a and b); and som2211 has a G1528A point mutation leading to the G270R amino acid change (Fig. 4e and Supplementary information, Figure S9a and c). These results demonstrate that the mutations in At1g53240 are responsible for the suppression of the mod1 phenotypes. In addition, we found that a T-DNA insertion mutant of At1g53240, som328-2 (N36263936), fully rescued the mod1 phenotypes (Fig. 4a–d).

Identification of SOM328 as a mitochondrial malate dehydrogenase, which is required for the generation of ROS and cell death in mod1. a Phenotypes of Col-0, mod1, som328, mod1 som328, mod1 som328 comp (mod1 som328 complemented line), som328-2 (N362639), and mod1 som328-2 at 32 DAG. Scale bars, 1 cm. b DAB-stained seedlings. Scale bars, 1 cm. c NBT-stained seedlings. Scale bars, 1 cm. d Trypan blue-stained leaves. Scale bars, 1 cm. e Map-based cloning of SOM328 (top). The mutation sites in som328, som369, som2169, and som2211 DNA (middle) and protein (bottom) sequences are indicated with red, blue, green and purple triangles, respectively. f Subcellular localization of SOM328-GFP in Arabidopsis mesophyll protoplasts of 35S:SOM328-GFP transgenic plants. Scale bars, 5 μm. g Transcript levels of SOM328 revealed by qRT-PCR with Actin as reference. Values are means ± SD (n = 3), and different letters at top of each column indicate a significant difference at P < 0.05 determined by Tukey’s HSD test. h Protein levels of SOM328 in the indicated plants, detected by immunoblotting with anti-SOM328 polyclonal antibodies. i Comparison of total cellular MDH activities in the indicated plants. Values are means ± SD (n = 6), and different letters at top of each column indicate a significant difference at P < 0.05 determined by Tukey’s HSD test
SOM328 is allelic to the previously reported gene, MITOCHONDRIAL MALATE DEHYDROGENASE 1 (mMDH1).36 Phylogenic analysis revealed that the malate dehydrogenase domain of SOM328 is highly conserved not only in different plant species but also in Caenorhabditis elegans, Drosophila melanogaster, and mammalian species, while the mitochondrial transit peptide domain is diverse (Supplementary information, Figure S10). To confirm the localization of the SOM328 protein in mitochondria, we examined its subcellular localization by stably expressing 35S:SOM328-GFP in the wild type (Col-0) and found that the GFP florescent signal co-localized with the mitochondria-specific marker, MitoTracker Red (Fig. 4f). Furthermore, we found that the point mutation in som328 did not affect its RNA level or the protein level, suggesting that the arginine at the 115 position may affect the mMDH enzyme activity (Fig. 4g, h). We therefore compared the total MDH activities in all relevant lines. No differences have been observed among the wild type, mod1 and complemented line (mod1 som328 comp). However, som328 or mod1 som328 showed significant decreased total MDH activities (Fig. 4i). Taken together, these results demonstrate that the mitochondrial malate dehydrogenase plays an essential role in causing mod1 phenotypes.
Malate shuttle is responsible for the organelle communication in PCD of mod1
The genetic and functional confirmation of the SOMs identified above allows us to draw a coherent picture of the CTM communication in mod1-triggered PCD (Fig. 5a). MOD1, an enoyl-ACP reductase, utilizes NADH to reduce the double bond in the fatty acyl group, and deficiency in MOD1 will favorably lead to the accumulation of the reducing equivalent NADH in the chloroplast and trigger plNAD-MDH to reduce OAA to malate. The over-produced malate in the chloroplast is then exported into the cytosol by DiT1, which is localized in the chloroplast inner envelope membrane. The cytosolic malate is in turn transported into the mitochondrion and oxidized to OAA by mMDH1, coupled to the generation of NADH. NADH then serves as the electron donor for the mETC to generate ROS. Therefore, it is likely that direct transport of malate from chloroplast to mitochondrion through the malate shuttle provides the chemical communication pathway by which the excess reducing equivalent in the chloroplast is conveyed to the mitochondrion, leading to the generation of ROS and PCD in mod1 plants.

Malate shuttle carries the Chloroplast-To-Mitochondrion signal in mod1. a Schematics of the locations and functions of SOMs. plNAD-MDH (plastidial NAD-dependent malate dehydrogenase) is encoded by SOM410, SOM430 or SOM2167; DiT1 (chloroplastic dicarboxylate transporter 1) by SOM787, SOM812 or SOM2073; and mMDH1 (mitochondrial malate dehydrogenase 1) by SOM328, SOM369, SOM2169, or SOM2211. b Changes of malate concentration in chloroplasts isolated from Col-0, mod1, mod1 som410, and som410. b–d Values are means ± SD (n = 3), and different letters at top of each column indicate a significant difference at P < 0.05 determined by Tukey’s HSD test. c Changes of malate concentration in chloroplasts isolated from Col-0, mod1, mod1 som787, and som787. d Changes of malate concentration in mitochondria isolated from Col-0, mod1, mod1 som328 and som328
If the malate shuttle is truly carrying the CTM communication signal of organelle communication, then the malate concentrations in the mod1 mutant and various suppressors should show corresponding changes in chloroplasts and mitochondria. We therefore measured and compared the malate concentrations in mod1 and mod1 som410, mod1 som787, or mod1 som328 (Fig. 5). Compared with mod1, the malate concentration in the chloroplasts of mod1 som410 was significantly decreased and restored to the wild-type level, reflecting the deficiency in conversion from OAA to malate as a result of the SOM410 mutation (Fig. 5b). In contrast, the malate concentration in the chloroplasts of mod1 som787 was significantly increased and much higher than that in the chloroplasts of mod1, because export of malate from the chloroplast was impaired (Fig. 5c). In mitochondria, the malate concentration in mod1 som328 is significantly elevated compared with mod1, because of the deficient conversion from malate to OAA as a result of the SOM328 mutation (Fig. 5d). Together, these data further demonstrate that the malate shuttle is responsible for the CTM communication from the chloroplast, the energy-generating organelle, to the mitochondrion, the energy-transforming organelle, to regulate ROS formation.
To investigate whether this CTM communication is involved in the cell death pathway of the lesion mimic mutants, we generated the double mutants by crossing different soms including som410, som787, som328, and som3 (an mETC complex I deficient mutant)26 with acd227 or lsd1 (N542687),37 two typical lesion mimic mutants. We found that the cell death phenotypes in acd2 and lsd1 are not suppressed by soms (Supplementary information, Figure S11). These results indicate that neither the CTM communication nor mETC-generated ROS are involved in the cell death regulation in acd2 and lsd1, and PCD triggered by mod1 is controlled by mechanisms different from that in acd2 and lsd1.
ROS by CTM communication affects plant growth under different photoperiod conditions
Photoperiod is a major environmental factor controlling plant growth and development. Photoperiod could affect a plant’s redox state and is considered as a crucial factor in the regulation of H2O2-induced cell death.38 However, the regulatory mechanism remains to be determined. The mod1 mutant was originally identified under continuous light condition, leading us to compare the mod1 phenotype under long-day condition (16-hr light/8-hr dark) and continuous light conditions (24-hr light). This revealed that both the PCD phenotype and ROS accumulation in mod1 become more severe under continuous light conditions (Fig. 6a, b). Moreover, the phenotypes of mod1 were still very severe under continuous light condition when the light intensity was decreased from 85 μmol m−2 s−1 to 55 μmol m−2 s−1 (Fig. 6b), indicating that the photoperiod condition affects the mod1 phenotypes and the ROS generation. Taken together with the findings that the interruption of the malate shuttle could suppress PCD and ROS accumulation in mod1, it follows that the CTM communication is involved in ROS accumulation under the continuous light conditions. We therefore grew the wild type and three suppressor mutants, som410, som787 and som328, under three different conditions (long-day condition with 85 μmol m−2 s−1 and continuous light conditions with either 85 or 55 μmol m−2 s−1). We found that ROS levels in Col-0 were higher under the continuous light conditions than long-day condition (Fig. 6c), indicating that the continuous light could induce ROS accumulation. More importantly, the ROS levels of three suppressor mutants were similar to that of Col-0 under the long-day condition, but lower than that of Col-0 under the continuous light conditions (Fig. 6c). This indicates that CTM communication plays an important role in regulating ROS accumulation under the continuous light conditions.

ROS generated through the malate shuttle affects plant growth under different photoperiod conditions. a Schematics of different growth conditions. L-85, long-day condition (16-hr light/8-hr dark) with a light intensity of 85 µmol m−2 s−1; C-85 or C-55, continuous illumination conditions (24-hr light) with a light intensity of 85 µmol m−2 s−1 or 55 µmol m−2 s−1. b Phenotypes and DAB staining of mod1 grown under different illumination conditions in a at 22 DAG. Scale bars, 1 cm. c Phenotypes and DAB staining of Col-0, som410, som787, and som328 grown under different illumination conditions in a at 22 DAG. Scale bars, 1 cm. d Aboveground fresh weight of Col-0, som410, som787, and som328 under different illumination conditions in a at 22 DAG. Values are means ± SD (n = 10). The asterisks represent significant difference between mutant and Col-0 grown under the same condition determined by Student’s t test. ns, no significant difference; *P < 0.05; **P < 0.01
Considering that the CTM communication plays an important role in regulating plant growth, we therefore compared plant size and aboveground fresh weight of som410, som787 and som328 with the wild type. We found that under both continuous light conditions plant size and fresh weight of all three suppressor mutants were remarkably larger (Fig. 6d), which accords with the decreased ROS levels in suppressor mutants (Fig. 6c). These results suggest that CTM communication plays an important role in regulating redox homeostasis and thereby affecting plant growth in response to different photoperiod conditions and that the impairment in the CTM communication could reduce the oxidative damages caused by continuous light.
Malate induces ROS generation and cell death in HeLa cells
It has been reported that animals and plants show some similar morphological and biochemical features of PCD.3 However, whether they share the same mechanism in regulating ROS generation and triggering PCD is still an open fundamental biological question. Since malate and NADH are ubiquitous primary metabolic molecules in all cells and the malate dehydrogenase domain and the function of mMDH is highly conserved across plants and animals, we asked whether the PCD pathway regulated by malate is conserved between plants and animals. To illustrate this, we used HeLa cells as a working system. Based on the report that the abundance of malate in different tissues of mice varies from ~60 to 386 ng/mg39 and considering the uptake efficiency, we treated the cells with 50 mM malate for 24 h and then measured the levels of ROS and cell death. The malate-treated cells showed a significantly increased ROS level detected by dihydroethidium (DHE) staining (Fig. 7a, b). Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) showed that the cell death levels were also significantly elevated in malate-treated cells (Fig. 7c, d). This is consistent with a previous study in human keratinocyte cell lines, in which treatment with exogenous malate could significantly increase the production of mitochondrial superoxide and induce PCD through mitochondria-dependent pathways.40 These results indicate that the ROS accumulation and PCD induced by malate are conserved processes in both plants and animals. More importantly, we examined whether mMDH also plays a conserved role in this process in animals. We identified human MDH2 gene, which is an ortholog of Arabidopsis mMDH1 and encodes a mitochondrial MDH. We then generated the MDH2 knockdown HeLa cells (siMDH2) by siRNA (Supplementary information, Figure S12). Remarkably, after treatment with malate for 24 h, both ROS and cell death levels were significantly decreased in siMDH2 cells compared with the control cells (siControl) (Fig. 7e–h), suggesting that MDH2 plays a vital role in regulating the malate-induced PCD pathway in human cells. Taken together, these results demonstrate that generation of ROS and PCD regulated by the malate shuttle are conserved processes in both plant and animal cells.

ROS induction and cell death triggered by malate in HeLa cells. a ROS levels revealed by dihydroethidium (DHE) staining in HeLa cells with or without malate treatment. Scale bars, 25 μm. b Quantitation of data in a. Values are means ± SD (n = 3) based on 3 independent experiments. The asterisks represent significant difference determined by Student’s t test. **P < 0.01. c Cell death levels revealed by TUNEL staining in HeLa cells with or without malate treatment. Scale bars, 50 μm. d Quantitation of data in c. Values are means ± SD (n = 3) based on three independent experiments. The asterisks represent significant difference determined by Student’s t test. **P < 0.01. e ROS levels revealed by DHE staining in control (siControl) and MDH2 knockdown HeLa cells (siMDH2) with or without malate treatment. Scale bars, 25 μm. f Quantitation of data in e. Values are means ± SD (n = 3) based on three independent experiments, and different letters at top of each column indicate a significant difference at P < 0.05 determined by Tukey’s HSD test. g Cell death levels revealed by TUNEL staining in control (siControl) and MDH2 knockdown HeLa cells (siMDH2) with or without malate treatment. Scale bars, 100 μm. h Quantitation of data in g. Values are means ± SD (n = 3) based on three independent experiments, and different letters at top of each column indicate a significant difference at P < 0.05 determined by Tukey’s HSD test
Discussion
Communication between the nucleus and organelles in eukaryotic cells plays vital roles in responding to different environmental stresses and in coordinating compartmentally localized materials and signals to regulate cell growth and differentiation. The signaling pathways from the nucleus to organelles (anterograde signaling) and from organelles to the nucleus (retrograde signaling) have been well studied over the past several decades.16,17,18,19,20,21,22,23,24 In recent years, studies of the communication between organelles have revealed its indispensable roles in executing cellular activities for almost all the life processes, for example, the communication or interaction of the endoplasmic reticulum (ER) with the Golgi, mitochondrion, chloroplast, or lysosome.41,42,43,44,45 Compared with animal cells, a higher plant cell not only contains a rigid cell wall and a large vacuole, but also possesses a unique phototrophic energy-generating system in the chloroplast that produces bioenergy for nearly all living organisms. Therefore, it is naturally believed that communication must exist between the chloroplast, an organelle generating chemical energy, and the mitochondrion, an organelle transforming chemical energy, and that such communication plays an essential role in regulating plant growth and development. Recent reports suggest that there is a CTM communication, direct or indirect, in mod1-triggered PCD in Arabidopsis thaliana,26,29 but its nature and mechanism are still elusive. In this work, we demonstrate that in plants, the malate shuttle conveys a direct communication signal from the chloroplast to mitochondrion presumably via the cytosol (Fig. 8a). Moreover, we further demonstrate that the pathway functions to deliver reducing equivalents for mitochondrial ROS generation, providing a communication system which reports the cellular redox status and mediates cell death in animal cells (Fig. 8b).

A proposed model of programmed cell death in plant and animal cells. a In plants, the deficiency of MOD1 leads to an increased level of NADH in the chloroplast, which drives OAA to be converted to malate by plNAD-MDH. Malate carrying the reducing equivalents is transported out of the chloroplast into the cytosol by DiT1, and then be transported into the mitochondrion by an unidentified transporter or transporters. In the mitochondrion, malate is converted to OAA by mMDH1, and simultaneously NADH is generated to provide electrons for mETC to induce the ROS formation and initiate the PCD process in the mod1 cells. b In HeLa cells, exogenous malate could be taken into the cytosol, and then transported into the mitochondrion by the malate/α-ketoglutarate (α-KG) transporter. In the mitochondrion, malate is converted to OAA by MDH2, and simultaneously NADH is generated to provide electrons for mETC to induce the ROS formation and initiate the cell death process, which is highly conserved with the PCD in mod1 plants
It has been well established that the malate shuttle is a fundamental mechanism to regulate the redox homeostasis among different compartmentalised organelles of a eukaryotic cell by indirectly transporting reducing equivalents across membranes.30,46,47,48,49,50 mod1 and soms provides us an ideal model system to investigate the mechanism underlying the communication between the chloroplast and mitochondrion. Under a long illumination condition, the reducing equivalent NADH appears to accumulate due to its reduced consumption resulting from a deficiency in the biosynthesis of fatty acids in mod1 chloroplasts. The accumulated reducing equivalent drives the formation of malate from OAA catalyzed by plNAD-MDH, the plastidial NAD-dependent malate dehydrogenase (Fig. 8a). The malate formed is then exported into the cytosol by a plastid-specific transporter DiT1 and subsequently transported into mitochondria by an unknown transporter. In the mitochondrial matrix, the malate is oxidized to OAA by mMDH, together with generation of the reducing equivalent NADH, which is used as the electron donor to mETC for ATP production by oxidative phosphorylation but will lead to ROS production in cells with redox imbalance (Fig. 8a). Malate functions as an important compound in cell metabolism and a carrier of the reducing equivalents, and it is the malate shuttle that plays an essential role in maintaining the redox homeostasis required for organelles. The malate shuttle could trigger the mitochondrial ROS generation and activate the downstream PCD pathway in response to different conditions that affect redox homeostasis. In higher plants, this mechanism ensures the reducing equivalents generated in the chloroplast are exported and utilized for the growth and development, and this may explain why the null mutation in plNAD-MDH is lethal.
The photoperiod is considered as a crucial factor in the regulation of H2O2-induced cell death.38 The Arabidopsis thaliana catalase 2 (cat2) mutant shows different levels of cell death under different photoperiod conditions due to the accumulation of H2O2.38 Hundreds of downstream genes show day length-dependent responses to intracellular H2O2 at transcript levels,51 and the H2O2-activated glutathione synthesis is influenced by the photoperiod in Arabidopsis.52 However, the origin of H2O2 in response to photoperiod changes is unclear. In this work, we discovered that CTM communication and mETC play essential roles in the H2O2 generation in response to photoperiod changes (Fig. 8a), which may result in the disturbance of the chloroplast NADH homeostasis and the triggering of cascade reactions. Our results also demonstrate that the malate shuttle is required for the photoperiod response of plants.
Fatty acids play critical roles in plant development, cell signaling, abiotic stress responses, and pathogen defense.53,54,55,56,57,58,59,60,61 Although it is reasonable to suggest that certain fatty acids or their derivatives in mod1 might provide the signal inducing ROS and PCD. The results presented here demonstrate that it is the accumulation of reducing equivalent and its transport system that trigger the generation of ROS to a harmful level and induce PCD. These results suggest that the biosynthesis of fatty acids is one of the major reducing equivalent consumers in chloroplasts, and in the cell as a whole.
Although different mutants of the CTM communication system can each reduce the oxidative damage caused by continuous light, they show varying degrees of growth deficiency. The som430 mutant, which results from a premature stop codon, is embryonic lethal, indicating that the function of plNAD-MDH is essential for plant growth. The T-DNA insertion mutant of DiT1, som787-2, shows remarkably reduced plant size and the point mutation in mMDH1 leads to a moderate but significant reduced plant size in som328. It is notable that the forward genetic screen for mod1 suppressors did not identify the mitochondrial malate/OAA transporter, which may be due to gene redundancy or lethality. Different levels of growth deficiencies in these mutants indicate that a complicated metabolic regulatory network is likely involved for the CTM communication. This needs to be further investigated to fully understand the crucial function of reducing equivalent homeostasis in plant growth and response to photoperiod.
Plants do not encode caspases and Bcl-2 family members that are required for animal apoptosis, however plant and animal PCD do share several similar morphological and biochemical features, such as cell shrinkage, chromatin condensation, nuclear fragmentation, endonuclease release, and common inhibitors,2,3,29,62,63 suggesting that both plant and animal PCD pathways may share a conserved mechanism that triggers and regulates PCD.63 This hypothesis is supported by evidence that treatment with malate can induce ROS generation and PCD in HeLa cells. This demonstrates that the malate-induced ROS generation and the PCD pathways are conserved and that mMDH is functionally conserved and essential for PCD pathways in both plants and animals (Fig. 8), even though the malate in mod1-triggered PCD originates in chloroplasts that do not exist in animal cells and translocation of malate from the cytosol to mitochondria is carried out by the malate-aspartate shuttle in animal cells and the malate-OAA shuttle in plant cells.
Materials and methods
Plant materials and cell culture
Seeds of Arabidopsis thaliana ecotype Columbia (Col-0), Landsberg erecta (Ler-0) and mod1 (in Columbia background) were from our own stocks. The T-DNA insertion mutants were from the Nottingham Arabidopsis Stock Centre (NASC) and their genotypes were confirmed by PCR analysis. Primers are listed in Supplementary information, Table S1. HeLa cells were cultured in the Dulbecoo’s modified Eagle Medium (DMEM) with 10% (v/v) Fetal Bovine Serum (FBS).
Plant growth conditions
Arabidopsis seeds were surface sterilized and sown on 0.5 × Murashige and Skoog (MS) medium plates containing 1.0% (w/v) sucrose and 0.65% (w/v) agar. After being vernalized at 4 °C for 3 days, seeds were germinated under long-day conditions (16-hr light/8-hr dark) and seedlings (7 days after germination) were transferred to soil and grown under long-day conditions (16-hr light/8-hr dark) with a light intensity of 80 to 120 μmol m−2 s−1 at 20–22 °C. For response to different photoperiod conditions, seeds of Col-0, mod1 and soms were sown and germinated in soil directly under long-day conditions (16-hr light/8-hr dark), or continuous illumination conditions (24-hr light) with a light intensity of 85 or 55 μmol m−2 s−1 at 20–22 °C. In screening for suppressors of mod1, M1 and M2 seeds were sown and germinated directly in soil under continuous illumination conditions (24-hr light) with a light intensity of 80 to 120 μmol m−2 s−1 at 20–22 °C for the phenotype observation.
Screening for suppressors
To identify the suppressor of mod1 (som), ~1.5 g mod1 seeds were mutagenized with ethyl methanesulfonate (EMS) as previously reported.64 M1 and M2 plants with the restored phenotypes were selected under continuous illumination. The mitochondrial complex I activity of soms was assayed as described previously.26,65 The soms without changes of complex I activities were selected for further studies.
Map-based cloning and whole-genome resequencing
For the map-based cloning of SOMs, mod1 in the Col-0 background was backcrossed 10 times to Ler to generate mod1 plants in the isogenic Ler background (mod1-Ler). To map the causal mutations in the soms, F2 populations were generated by crossing the soms with mod1-Ler. Individual plants showing restored phenotypes were selected from F2 populations for primary mapping. DNA from mod1 and soms was prepared for sequencing using an Illumina HiSeq X Ten with a 150 bp paired-end (PE) strategy. The reads were mapped to the Arabidopsis reference genome66 using Burrows-Wheeler Aligner (BWA) version 0.7.10-r789 with default parameters.67 The SNP calling was based on alignment results using the Genome Analysis Toolkit (GATK) version 3.1.1 and Picard package version 1.119.68 The SNPs were then compared between mod1 and soms, and all the nonsynonymous mutations in the mapping region of each som are listed in Supplementary information, Table S2.
Plasmid construction and plant transformation
For the genetic complementation test, fragments containing the full-length genomic DNA of SOM410 and SOM328 with their own native promoters were amplified, digested and ligated into the binary vector pCambia1300. The fragment containing the full-length genomic DNA of SOM787 with its native promoter was cloned into BamHI- and SalI- digested pCambia1300 using the Seamless Assembly Cloning Kit (Clone Smarter Technologies). At least two independent complemented lines were obtained and they showed similar phenotypes. For the subcellular localization, the fragment containing the full-length CDS of SOM328 was digested and ligated into a modified pWM101 vector.69 Fragments containing the full-length CDS of SOM410 and SOM787 were cloned into BamHI- and SalI- digested modified pWM101 using the Seamless Assembly Cloning Kit (Clone Smarter Technologies). For CRISPR/Cas9 mutagenesis, single-guide RNAs (sgRNA) binding in the coding sequence of the target gene were designed using the CRISPR-P tool (http://cbi.hzau.edu.cn/cgi-bin/CRISPR),70 and cloned into a binary vectors pHEE2A-TRI71 digested with BsaI. Two lines with different frameshift mutation were obtained, and showed similar phenotypes. The final binary vectors were introduced into Agrobacterium tumefaciens strain EHA105 and transformed into wild-type and mutant plants through the Agrobacterium-mediated floral dip method.72 Transformants were selected on 0.5 × MS plates containing 20 mg L−1 hygromycin. All primers used are listed in Supplementary information, Table S1.
RNA expression analysis
For semiquantitative and quantitative RT-PCR, leaves of 2- to 3-week-old plants were collected and immediately flash-frozen in liquid nitrogen. Total RNA was extracted using TRIzol (Invitrogen), and treated with the TURBO DNA-freeTM kit (Invitrogen) to eliminate contaminated genomic DNA from total RNA. A total of 2.5 μg RNA was used for cDNA synthesis using the SuperScript III First-Strand Synthesis System (Invitrogen). The synthesized cDNA was diluted 10-fold and a 2.0 μL aliquot was used for qRT-PCR in the reaction system of SsoFastTM EvaGreen supermix (Bio-Rad). All primer used are provided in Supplementary information, Table S1.
Detection of ROS
O2− and H2O2 were detected as previously described.26,73 Briefly, 3-week-old plants were immersed and infiltrated under vacuum for 10 min with nitroblue tetrazolium (NBT) staining solution (1 mg mL−1 NBT, 10 mM potassium phosphate buffer, 10 mM NaN3, pH 7.4) and freshly prepared 3,3′-diaminobenzidine (DAB) staining solution (1 mg mL−1 DAB, pH 3.8), respectively. After infiltration, stained plants were bleached in acetic acid:glycerol:ethanol (1:1:3, v/v/v) solution at 100 °C for 10 min, and then stored in 95% (v/v) ethanol until scanning.
Trypan blue staining
Trypan blue staining was performed as previously described.26,74 In brief, leaves were immersed with trypan blue staining solution (30 mL ethanol, 10 g phenol, 10 mL H2O, 10 mL glycerol, 10 mL lactic acid and 10 mg trypan blue), boiled for 2–3 min, and then cooled down at room temperature for 1 h. The samples were destained by boiling in chloral hydrate solution (2.5 g mL−1) for 20 min with 2–3 changes of the chloral hydrate solution at room temperature. Samples were finally stored in 50% (v/v) glycerol until photographs were taken.
Preparation of polyclonal antibodies
Fragments containing the full-length CDS of SOM410 and SOM328 were amplified and subcloned into pET-61-DEST. Recombinant SOM410 and SOM328 proteins were expressed in Escherichia coli BL21 cells and used to raise polyclonal antibodies in mouse. 1:3000 dilution of the anti-SOM410 serum and 1:6000 dilution of the anti-SOM328 serum were used for immunoblotting. All primers used are listed in Supplementary information, Table S1.
Preparation of leaf proteins
For immunoblotting analysis, extraction of total leaf proteins from 3- to 4-week-old plants was performed as previously described.26 For the total MDH activity assay, total leaf proteins from 2- to 3-week-old plants were extracted according to the previously reported method.75 Leaves were ground in extraction buffer (50 mM HEPES-KOH, pH 7.5, 1 mM EDTA, 5 mM DTT, 10% (v/v) glycerol and 0.5 mM phenylmethylsulfonyl fluoride). The homogenates were centrifuged at 20,000 g at 4 °C for 10 min, the supernatant was transferred into a new tube, quantified by the Bio-Rad protein assay, and used for the determination of the total MDH activity.
Assay of MDH activity
The total MDH activity was assayed as previously described.36 Briefly, freshly extracted total leaf proteins were added into a MDH reaction mixture (90 mM KH2PO4-KOH, pH 7.4, 0.05% (v/v) Triton X-100, 5 mM MgCl2, 750 mM OAA and 2 mM NADH), and the absorbance changes at 340 nm caused by the NADH oxidation to NAD+ were used to measure total MDH activity.
Isolation of mitochondria
Mitochondria were isolated as previously described76 with slight changes. Approximately 40 g leaves from 4-week-old plants were homogenized with a Warring blender in 160 mL pre-cooled grinding medium (0.3 M sucrose, 25 mM tetrasodium pyrophosphate, 1% (w/v) polyvinylpyrrolidone-40, 2 mM EDTA, 10 mM KH2PO4, 1% (w/v) bovine serum albumin (BSA), 20 mM sodium L-ascorbate, 1 mM DTT, 5 mM cysteine, pH 7.5) with three 15 s bursts at 30 s intervals. The homogenate was filtered through four layers of Miracloth and centrifuged at 1500×g for 5 min and the resulting supernatant was then centrifuged at 20,000×g for 15 min. The organelle pellet was washed by repeating the 1500×g and 20,000×g centrifugation steps in the washing medium (0.3 M sucrose, 10 mM TES, pH 7.5). The resulting pellet of crude organelles was carefully resuspended in 1–2 mL washing medium and gently layered over a Percoll discontinuous gradient consisting of 1.4-mL 18% (v/v) Percoll, 7-mL 27% (v/v) Percoll and 1.4-mL 50% (v/v) Percoll. The gradient was then centrifuged at 40,000×g for 45 min. The mitochondrial band that was seen as a yellow-brown band between 27% (v/v) and 50% (v/v) Percoll was carefully collected by pipette. The mitochondria collected were diluted with washing medium and centrifuged at 20,000×g for 10 min. After repeating this washing step 3 times, the final pellet was retained in a small volume of washing medium for later experiments.
Isolation of chloroplasts
Chloroplasts were isolated using a Chloroplast Isolation Kit (Sigma) following the manufacturer’s instructions with slight modification. Briefly, ~30 g leaves from 4-week-old plants were homogenized in the 120-mL pre-cooled isolation medium containing 0.4 M sorbitol, 50 mM HEPES-KOH, pH 7.6, 2 mM EDTA, 0.1% (w/v) sodium L-ascorbate and 0.1% (w/v) BSA, and the homogenate was filtered through four layers of Miracloth and centrifuged at 200×g for 3 min at 4 °C to remove unwanted whole cells and cell wall debris. The supernatant was centrifuged at 1000×g for 7 min to pellet the chloroplasts, the pellet was carefully resuspended in 1 to 2 mL of the washing medium (0.4 M sorbitol, 50 mM HEPES-KOH, pH 7.6, and 2 mM EDTA), and gently layered on top of a Percoll gradient consisting of 7-mL 40% (v/v) Percoll and 3.5-mL 80% (v/v) Percoll. After centrifugation at 3000×g for 15 min, the intact chloroplasts form a band at the interface between the 40% (v/v) and 80% (v/v) Percoll layers. The band was carefully collected, diluted with the washing medium, and centrifuged at 2000×g for 3 min. The chloroplasts were further washed twice and finally resuspended in a 0.5-mL washing medium for later experiments.
Malate measurement
Leaves from 4-week-old plants were ground into fine powder under liquid nitrogen, and ~30 mg of powder was extracted with 600 μL of acetonitrile:chloroform (7:3, v-v) using 20 ng of D3-Malice acid as internal standards for quantitation. After ultrasonic-assisted extraction in an ice-water bath for 1 h and centrifugation at 7000×g for 5 min, the supernatant was collected. 300 μL H2O was then added to the supernatant for two steps of liquid-liquid partitioning. The upper aqueous fractions were pooled and dried under a nitrogen stream. The samples were redissolved in 300 μL H2O and filtered through a 0.22 μm membrane for detection. LC-MS/MS detection was performed on a UPLC (Waters) combined with a 5500 Qtrap MS equipped with an ESI source (AB SCIEX), and 5 μL of each sample was injected onto a HSS T3 C18 column.
Measurements of malate concentration in freshly isolated mitochondria and chloroplasts were carried out in the same way. However, when preparing samples all the solvent volumes used were reduced to 1/6.
Assays of ROS and cell death in HeLa cells
HeLa cells were cultured in Dulbecoo’s modified Eagle Medium (DMEM) with 10% (v/v) Fetal Bovine Serum (FBS), followed by treatment with 50 mM malate (adjust pH to 7.4 by NaOH) for 24 h. For a negative control, HCl was added to DMEM until the pH was the same as the pH of 50 mM malate and the pH adjusted to 7.4 using NaOH. DHE staining was performed as previously described with slight modification.77 In brief, cells were washed twice with the Krebs Ringer buffer (KRB, 135 mM NaCl, 5 mM KCl, 1 mM CaCl2, 0.4 mM KH2PO4, 1 mM MgSO4, 20 mM HEPES, 5.5 mM Glucose, pH 7.4), followed by incubation for 15 min at 37 °C in KRB containing 2 μM DHE (Invitrogen). Fluorescence images were obtained using a confocal microscope at excitation and emission wavelengths of 552 nm and 605 nm, respectively. TUNEL staining was performed using an in situ cell death detection kit following the manufacture’s instruction (Sigma Aldrich), and cells were counterstained by DAPI. Fluorescence images were obtained using a confocal microscope at excitation wavelengths of 488 nm and 405 nm for TUNEL and DAPI, respectively.
HeLa cell transfection
MDH2 RNAi knockdown was performed by transfection of MDH2-siRNAs using Lipofectamine 2000 (Invitrogen), and RNAi knockdown for the negative control was performed by transfection of Control-siRNAs using Lipofectamine 2000 (Invitrogen), following the manufacturer’s instructions. Primers used are listed in Supplementary information, Table S1.
References
Ameisen, J. C. On the origin, evolution, and nature of programmed cell death: a timeline of four billion years. Cell Death Differ. 9, 367–393 (2002).
Dickman, M., Williams, B., Li, Y., de Figueiredo, P. & Wolpert, T. Reassessing apoptosis in plants. Nat. Plants 3, 773–779 (2017).
Vianello, A. et al. Plant mitochondrial pathway leading to programmed cell death. Physiol. Plant. 129, 242–252 (2007).
Van Aken, O. & Van Breusegem, F. Licensed to kill: mitochondria, chloroplasts, and cell death. Trends Plant Sci. 20, 754–766 (2015).
van Doorn, W. G. Classes of programmed cell death in plants, compared to those in animals. J. Exp. Bot. 62, 4749–4761 (2011).
Beers, E. P. Programmed cell death during plant growth and development. Cell Death Differ. 4, 649–661 (1997).
Coll, N. S., Epple, P. & Dangl, J. L. Programmed cell death in the plant immune system. Cell Death Differ. 18, 1247–1256 (2011).
Daneva, A., Gao, Z., Van Durme, M. & Nowack, M. K. Functions and regulation of programmed cell death in plant development. Annu. Rev. Cell Dev. Biol. 32, 441–468 (2016).
Lam, E. Controlled cell death, plant survival and development. Nat. Rev. Mol. Cell Biol. 5, 305–315 (2004).
Van Hautegem, T., Waters, A. J., Goodrich, J. & Nowack, M. K. Only in dying, life: programmed cell death during plant development. Trends Plant Sci. 20, 102–113 (2015).
Van Breusegem, F. & Dat, J. F. Reactive oxygen species in plant cell death. Plant Physiol. 141, 384–390 (2006).
Noctor, G., Veljovic-Jovanovic, S., Driscoll, S., Novitskaya, L. & Foyer, C. H. Drought and oxidative load in the leaves of C3 plants: a predominant role for photorespiration? Ann. Bot. 89, 841–850 (2002).
Gorlach, A., Bertram, K., Hudecova, S. & Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 6, 260–271 (2015).
Jabs, T. Reactive oxygen intermediates as mediators of programmed cell death in plants and animals. Biochem. Pharmacol. 57, 231–245 (1999).
Reape, T. J. & McCabe, P. F. Apoptotic-like regulation of programmed cell death in plants. Apoptosis 15, 249–256 (2010).
Ahlert, D., Ruf, S. & Bock, R. Plastid protein synthesis is required for plant development in tobacco. Proc. Natl. Acad. Sci. USA 100, 15730–15735 (2003).
Bailleul, B. et al. Energetic coupling between plastids and mitochondria drives CO2 assimilation in diatoms. Nature 524, 366–369 (2015).
Beck, C. F. Signaling pathways from the chloroplast to the nucleus. Planta 222, 743–756 (2005).
Chan, K. X., Phua, S. Y., Crisp, P., McQuinn, R. & Pogson, B. J. Learning the languages of the chloroplast: retrograde signaling and beyond. Annu. Rev. Plant Biol. 67, 25–53 (2016).
Denby, K. J., Jason, L. J., Murray, S. L. & Last, R. L. ups1, an Arabidopsis thaliana camalexin accumulation mutant defective in multiple defence signalling pathways. Plant J. 41, 673–684 (2005).
Ng, S. et al. Anterograde and retrograde regulation of nuclear genes encoding mitochondrial proteins during growth, development, and stress. Mol. Plant 7, 1075–1093 (2014).
Shao, N., Duan, G. Y. & Bock, R. A mediator of singlet oxygen responses in Chlamydomonas reinhardtii and Arabidopsis identified by a luciferase-based genetic screen in algal cells. Plant Cell 25, 4209–4226 (2013).
Wang, J. Z. et al. Initiation of ER body formation and indole glucosinolate metabolism by the plastidial retrograde signaling metabolite, MEcPP. Mol. Plant 10, 1400–1416 (2017).
Yang, L. et al. ABA-mediated ROS in mitochondria regulate root meristem activity by controlling PLETHORA expression in. Arab. PLoS Genet. 10, e1004791 (2014).
Mou, Z., He, Y., Dai, Y., Liu, X. & Li, J. Deficiency in fatty acid synthase leads to premature cell death and dramatic alterations in plant morphology. Plant Cell 12, 405–418 (2000).
Wu, J. et al. Deficient plastidic fatty acid synthesis triggers cell death by modulating mitochondrial reactive oxygen species. Cell Res. 25, 621–633 (2015).
Greenberg, J. T., Guo, A., Klessig, D. F. & Ausubel, F. M. Programmed cell death in plants: a pathogen-triggered response activated coordinately with multiple defense functions. Cell 77, 551–563 (1994).
Dietrich, R. A. et al. Arabidopsis mutants simulating disease resistance response. Cell 77, 565–577 (1994).
Yu, H. & Li, J. Plant and animal organelles in cell death. Oncotarget 6, 13852–13853 (2015).
Beeler, S. et al. Plastidial NAD-dependent malate dehydrogenase is critical for embryo development and heterotrophic metabolism in Arabidopsis. Plant Physiol. 164, 1175–1190 (2014).
Berkemeyer, M., Scheibe, R. & Ocheretina, O. A novel, non-redox-regulated NAD-dependent malate dehydrogenase from chloroplasts of Arabidopsis thaliana L. J. Biol. Chem. 273, 27927–27933 (1998).
Selinski, J. et al. The plastid-localized NAD-dependent malate dehydrogenase is crucial for energy homeostasis in developing Arabidopsis thalianaseeds. Mol. Plant 7, 170–186 (2014).
Kinoshita, H. et al. The chloroplastic 2-oxoglutarate/malate transporter has dual function as the malate valve and in carbon/nitrogen metabolism. Plant J. 65, 15–26 (2011).
Taniguchi, M. et al. Identifying and characterizing plastidic 2-oxoglutarate/malate and dicarboxylate transporters in Arabidopsis thaliana. Plant Cell Physiol. 43, 706–717 (2002).
Renné, P. et al. The Arabidopsis mutant dct is deficient in the plastidic glutamate/malate translocator DiT2. Plant J. 35, 316–331 (2003).
Tomaz, T. et al. Mitochondrial malate dehydrogenase lowers leaf respiration and alters photorespiration and plant growth in Arabidopsis. Plant Physiol. 154, 1143–1157 (2010).
Xu, W. et al. Endosperm and nucellus develop antagonistically in Arabidopsis Seeds. Plant Cell 28, 1343–1360 (2016).
Queval, G. et al. Conditional oxidative stress responses in the Arabidopsis photorespiratory mutantcat2 demonstrate that redox state is a key modulator of daylength-dependent gene expression, and define photoperiod as a crucial factor in the regulation of H2O2-induced cell death. Plant J. 52, 640–657 (2007).
Chouchani, E. T. et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515, 431–435 (2014).
Hsiao, Y. P. et al. Triggering apoptotic death of human epidermal keratinocytes by malic acid: involvement of endoplasmic reticulum stress- and mitochondria-dependent signaling pathways. Toxins 7, 81–96 (2015).
Bobik, K. & Burch-Smith, T. M. Chloroplast signaling within, between and beyond cells. Front. Plant Sci. 6, 781 (2015).
Bravo-Sagua, R. et al. Organelle communication: signaling crossroads between homeostasis and disease. Int. J. Biochem. Cell Biol. 50, 55–59 (2014).
Garrity, A. G. et al. The endoplasmic reticulum, not the pH gradient, drives calcium refilling of lysosomes. eLife 5, e15887 (2016).
Rowland, A. A. & Voeltz, G. K. Endoplasmic reticulum-mitochondria contacts: function of the junction. Nat. Rev. Mol. Cell Biol. 13, 607–625 (2012).
Schrader, M., Grille, S., Fahimi, H. D. & Islinger, M. Peroxisome interactions and cross-talk with other subcellular compartments in animal cells. Subcell. Biochem. 69, 1–22 (2013).
Heber, U. Metabolite exchange between chloroplasts and cytoplasm. Annu. Rev. Plant Biol. 25, 393–421 (1974).
Krömer, S. Respiration during photosynthesis. Annu. Rev. Plant Physiol. Plant Mol. Biol. 46, 47–70 (1995).
Scheibe, R. Malate valves to balance cellular energy supply. Physiol. Plant. 120, 21–26 (2004).
Sweetlove, L. J. & Fernie, A. R. The spatial organization of metabolism within the plant cell. Annu. Rev. Plant Biol. 64, 723–746 (2013).
Taniguchi, M. & Miyake, H. Redox-shuttling between chloroplast and cytosol: integration of intra-chloroplast and extra-chloroplast metabolism. Curr. Opin. Plant Biol. 15, 252–260 (2012).
Queval, G., Neukermans, J., Vanderauwera, S., Van Breusegem, F. & Noctor, G. Day length is a key regulator of transcriptomic responses to both CO2 and H2O2 in Arabidopsis. Plant Cell Environ. 35, 374–387 (2012).
Queval, G. et al. H2O2-activated up-regulation of Ggutathione in Arabidopsis involves induction of genes encoding enzymes involved in cysteine synthesis in the chloroplast. Mol. Plant 2, 344–356 (2009).
Bonaventure, G., Salas, J. J., Pollard, M. R. & Ohlrogge, J. B. Disruption of the FATB gene in Arabidopsis demonstrates an essential role of saturated fatty acids in plant growth. Plant Cell 15, 1020–1033 (2003).
Ding, W. et al. OsKASI, a β-ketoacyl-[acyl carrier protein] synthase I, is involved in root development in rice (Oryza sativa L.). Planta 242, 203–213 (2015).
Gao, J., Wallis, J. G. & Browse, J. Mutations in the prokaryotic pathway rescue the fatty acidbiosynthesis1 mutant in the cold. Plant Physiol. 169, 442–452 (2015).
Kachroo, A. & Kachroo, P. Fatty acid-derived signals in plant defense. Annu. Rev. Phytopathol. 47, 153–176 (2009).
Kachroo, A. et al. Plastidial fatty acid signaling modulates salicylic acid- and jasmonic acid-mediated defense pathways in the Arabidopsis ssi2 mutant. Plant Cell 15, 2952–2965 (2003).
Lim, G. H., Singhal, R., Kachroo, A. & Kachroo, P. Fatty acid- and lipid-mediated signaling in plant defense. Annu. Rev. Phytopathol. 55, 505–536 (2017).
Roudier, F. et al. Very-long-chain fatty acids are involved in polar auxin transport and developmental patterning in Arabidopsis. Plant Cell 22, 364–375 (2010).
Vijayan, P. & Browse, J. Photoinhibition in mutants of Arabidopsis deficient in thylakoid unsaturation. Plant Physiol. 129, 876–885 (2002).
Wu, G. Z. & Xue, H. W. Arabidopsis β-ketoacyl-[acyl carrier protein] synthase I is crucial for fatty acid synthesis and plays a role in chloroplast division and embryo development. Plant Cell 22, 3726–3744 (2010).
Bonneau, L., Ge, Y., Drury, G. E. & Gallois, P. What happened to plant caspases? J. Exp. Bot. 59, 491–499 (2008).
Dickman, M. B. & Fluhr, R. Centrality of host cell death in plant-microbe interactions. Annu. Rev. Phytopathol. 51, 543–570 (2013).
Kim, Y., Schumaker, K. S. & Zhu, J. K. EMS mutagenesis of Arabidopsis. Methods Mol. Biol. 323, 101–103 (2006).
Pineau, B., Layoune, O., Danon, A. & De Paepe, R. L-galactono-1,4-lactone dehydrogenase is required for the accumulation of plant respiratory complex I. J. Biol. Chem. 283, 32500–32505 (2008).
Lamesch, P. et al. The Arabidopsis Information Resource (TAIR): improved gene annotation and new tools. Nucleic Acids Res. 40, D1202–D1210 (2012).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Wang, L. et al. Strigolactone signaling in Arabidopsis regulates shoot development by targeting D53-Like SMXL repressor proteins for ubiquitination and degradation. Plant Cell 27, 3128–3142 (2015).
Lei, Y. et al. CRISPR-P: a web tool for synthetic single-guide RNA design of CRISPR-system in plants. Mol. Plant 7, 1494–1496 (2014).
Wang, Z. P. et al. Egg cell-specific promoter-controlled CRISPR/Cas9 efficiently generates homozygous mutants for multiple target genes in Arabidopsis in a single generation. Genome Biol. 16, 144 (2015).
Clough, S. J. & Bent, A. F. Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 16, 735–743 (1998).
Ramel, F., Sulmon, C., Gouesbet, G. & Couée, I. Natural variation reveals relationships between pre-stress carbohydrate nutritional status and subsequent responses to xenobiotic and oxidative stress in Arabidopsis thaliana. Ann. Bot. 104, 1323–1337 (2009).
Stone, J. M., Heard, J. E., Asai, T. & Ausubel, F. M. Simulation of fungal-mediated cell death by fumonisin B1 and selection of fumonisin B1-resistant (fbr) Arabidopsis mutants. Plant Cell 12, 1811–1822 (2000).
Ritte, G., Steup, M., Kossmann, J. & Lloyd, J. R. Determination of the starch-phosphorylating enzyme activity in plant extracts. Planta 216, 798–801 (2003).
Hrubec, T. C., Robinson, J. M. & Donaldson, R. P. Isolation of mitochondria from soybean leaves on discontinuous Percoll gradients. Plant Physiol. 77, 1010–1012 (1985).
Kalivendi, S. V. et al. 1-Methyl-4-phenylpyridinium (MPP+)-induced apoptosis and mitochondrial oxidant generation: role of transferrin-receptor-dependent iron and hydrogen peroxide. Biochem. J. 371, 151–164 (2003).
Acknowledgments
We thank Jian-Min Zhou (Institute of Genetics and Developmental Biology, Chinese Academy of Sciences) for critical comments on the manuscript, Weicai Yang (Institute of Genetics and Developmental Biology, Chinese Academy of Sciences) for providing binary vector pWM101 and assistance in isolating mitochondria and chloroplasts, Qijun Chen (China Agricultural University) for providing CRISPR/Cas9 binary vector pHEE2A-TRI, and Nottingham Arabidopsis Stock Centre (NASC) for providing som787-2 (N877710), som328-2 (N362639), dit2.1-1 (N663245), dit2.1-2 (N669411), dit2.2-1 (N661169), dit2.2-2 (N655976), and lsd1 (N542687) seeds. This work was supported by the National Natural Science Foundation of China (31521001, 31661143025), and the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB13030300).
Author information
Authors and Affiliations
Contributions
Y.Z., L.L., J.X., P.X., H.G., J.W., L.B., G.W., J.C., J.Z., H.Y., and X.H. performed the experiments and data analyses. Y.Z., H.Y., X.H., and J.L. designed the experiments. J.L., X.H. and H.Y. conceived the project. J.L., Y.Z. and H.Y. wrote the paper with input from L.L. and X.H. All authors have read, edited, and approved the content of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, Y., Luo, L., Xu, J. et al. Malate transported from chloroplast to mitochondrion triggers production of ROS and PCD in Arabidopsis thaliana. Cell Res 28, 448–461 (2018). https://doi.org/10.1038/s41422-018-0024-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41422-018-0024-8
Keywords
This article is cited by
-
DNA methylation and lipid metabolism are involved in GA-induced maize aleurone layers PCD as revealed by transcriptome analysis
BMC Plant Biology (2023)
-
A mitochondrial pentatricopeptide repeat protein enhances cold tolerance by modulating mitochondrial superoxide in rice
Nature Communications (2023)
-
Chemical imaging reveals diverse functions of tricarboxylic acid metabolites in root growth and development
Nature Communications (2023)
-
SnoRNP is essential for thermospermine-mediated development in Arabidopsis thaliana
Science China Life Sciences (2023)
-
The phytohormones underlying the plant lateral root development in fluctuated soil environments
Plant and Soil (2023)
