
Abstract
Muscle LIM protein (MLP, CSRP3) is a key regulator of striated muscle function, and its mutations can lead to both hypertrophic cardiomyopathy (HCM) and dilated cardiomyopathy (DCM) in patients. However, due to lack of human models, mechanisms underlining the pathogenesis of MLP defects remain unclear. In this study, we generated a knockout MLP/CSRP3 human embryonic stem cell (hESC) H9 cell line using CRISPR/Cas9 mediated gene disruption. CSRP3 disruption had no impact on the cardiac differentiation of H9 cells and led to confirmed MLP deficiency in hESC-derived cardiomyocytes (ESC-CMs). MLP-deficient hESC-CMs were found to develop phenotypic features of HCM early after differentiation, such as enlarged cell size, multinucleation, and disorganized sarcomeric ultrastructure. Cellular phenotypes of MLP-deficient hESC-CMs subsequently progressed to mimic heart failure (HF) by 30 days post differentiation, including exhibiting mitochondrial damage, increased ROS generation, and impaired Ca2+ handling. Pharmaceutical treatment with beta agonist, such as isoproterenol, was found to accelerate the manifestation of HCM and HF, consistent with transgenic animal models of MLP deficiency. Furthermore, restoration of Ca2+ homeostasis by verapamil prevented the development of HCM and HF phenotypes, suggesting that elevated intracellular Ca2+ concentration is a central mechanism for pathogenesis of MLP deficiency. In summary, MLP-deficient hESC-CMs recapitulate the pathogenesis of HCM and its progression toward HF, providing an important human model for investigation of CSRP3/MLP-associated disease pathogenesis. More importantly, correction of the autonomous dysfunction of Ca2+ handling was found to be an effective method for treating the in vitro development of cardiomyopathy disease phenotype.
Similar content being viewed by others

Introduction
Muscle LIM protein (MLP), encoded by CSRP3, is primarily expressed in cardiomyocytes and skeletal muscle cells and consists of 194 amino acids with two LIM domains1. These LIM domains mediate interactions with a variety of proteins in different subcellular regions, including the cytoplasm and the nucleus. In the cytoplasm of cardiomyocytes, MLP has been found in the sarcomeres, intercalated disks, and costameres of striated myocytes. In addition, MLP can directly affect F-actin cytoskeleton dynamics by enhancing CFL2-dependent F-actin depolymerization2 and stabilizing actin filaments into bundles3. These studies suggest that MLP plays an integral role in sarcomeric structure. Previous studies have shown that mutations (missense, small insertions, and deletions) in CSRP3 are associated with hypertrophic cardiomyopathy (HCM)4,5 and dilated cardiomyopathy (DCM)6 in humans. In addition, MLP has been shown to be significantly downregulated in chronic human heart failure (HF)7. In mouse models, MLP-deficient mice develop dilated DCM with hypertrophy after birth, and are the first transgenic animal model of HF1. However, the underlying molecular mechanisms of CSRP3 mutation-induced cardiomyopathy are not well understood.
Despite the existence of genetically modified mouse models for MLP deficiency1,8, phenotypic results have shown significant discrepancies between identical CSRP3 mutations in animals and humans. For example, MLPW4R/+ transgenic mice are observed to develop features of HCM and HF8, whereas MLPW4R/+ is a known to be a benign polymorphism in humans carrying the mutation4,9. Efforts to obtain primary human cardiac tissue to resolve these discrepancies have not been successful due to difficulties in obtaining and culturing human cardiac tissue in vitro. Advances in disease modeling using human pluripotent stem cells (PSCs) in recent years have provided a novel pathophysiological approach to study the mechanics of disease and discover new targets for pharmacological intervention10.
We thus created a MLP-deficient cardiomyocyte model using from CSRP3−/− hESC-H9 cells, to elucidate the mechanism of cardiomyopathy and HF caused by MLP defects. We report here that MLP-deficient human embryonic stem cell-derived cardiomyocytes (hESC-CMs) recapitulate the phenotypes of HCM and HF. Mechanistically, impaired calcium handling is a major mechanism underlying the pathogenesis of MLP-associated HCM and HF, which can be prevented by pharmaceutical blockade of calcium entry into the cell.
Results
Generation of homozygous CSRP3 −/− hESCs
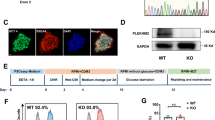
To generate the human PSC model of MLP deficiency, we designed a single-guide RNA targeting exon 3 of CSRP3 using the epiCRISPR/Cas9 gene editing system (Fig. 1a). H9 hESCs were then electroporated with a plasmid containing sgRNA and epiCRISPR/Cas911. After selection with puromycin, the resistant colonies were expanded for PCR screening verification. Among 12 clones, we found one homozygous CSRP3 knockout line with 1-nucleotide insertion resulting in a frame-shifted coding sequence with premature stop codon at 14th amino acid, resulting in homozygous null alleles (Fig. 1b). CSRP3−/− hESC colonies exhibited normal morphology (Fig. S1A). We further confirmed that CSRP3−/− lines expressed the human pluripotency markers TRA-1-81 and OCT4 (Fig. 1c) and had normal karyotype (Fig. S1B). Finally, CSRP3 knockout was not observed to affect the pluripotency of hESC evidenced by teratoma formation in vivo (Fig. S1C) and differentiation into all three embryonic germ layers (Fig. S1D). Moreover, we predicted the top ten off-target sites for sgRNA using an online analysis software (crispr.mit.edu) and found no off-target mutations in CSRP3−/− hESCs by DNA sequencing (data not shown)12.

a Structure of the CSRP3 locus showing localization of gRNA for epiCRISPR/Cas9 editing. b Sequence chromatograms demonstrate one base insertion in CSRP3 knockout (MLP KO) colonies. c Immunofluorescence staining of MLP KO colonies for the pluripotency markers TRA-1-81 and OCT4. Scale bar, 50 μm. d Immunoblot analysis of MLP in WT and MLP KO CMs at day 15. e Protocol for cardiac differentiation using small molecule-based methods. f, g Flow cytometry analysis for TNNT2 from representative WT and MLP KO differentiation protocols prior to purification at day 10. h Immunostaining for expression of MYL2 (green) and MYL7 (red) in WT and MLP KO CMS at day 30. i Quantification of ventricular marker MYL2 in WT (n = 382) and MLP KO CMS (n = 403). Scale bar, 50 μm. Results are presented as means ± S.E.M. of three independent experiments. ns, not significant, unpaired two-sided Student’s t test
MLP deficiency caused by CSRP3−/− does not affect cardiac differentiation of hESCs
Since MLP is primarily expressed in cardiomyocytes, we differentiated CSRP3−/− lines (MLP KO) and the parental H9 cells (WT) toward cardiomyocyte lineages (hESC-CMs) using small molecule-based protocols (Fig. 1e)13. At day 15 post differentiation, we validated the effect of CSRP3 knockout by western blot and observed absence of MLP protein in MLP KO hESC-CMs (Fig. 1d). Since MLP can serve as a positive regulator of myogenesis and promotes myogenic differentiation at the nucleus14, we next evaluated the impact of MLP deficiency on cardiac differentiation efficiency. Flow cytometry staining for cardiac Troponin T (cTnT) showed both the WT and MLP KO derived cells contained close to 85% positive cTnT cells by flow cytometry at day 10 post differentiation (Fig. 1f, g) and ~98% positive cTnT cells after purification at day 15 post differentiation (Fig. S1E, F). There was no statistical difference between the groups (P = 0.43). Double immunostaining for MYL2 and MYL7 showed both 83% of WT and MLP KO hESC-CMs were positive for the ventricular marker MYL2+ (Fig. 1h, i). These findings suggested that MLP deficiency has no significant effect on myocardial differentiation.
MLP-deficient hESC-CMs recapitulate HCM phenotypes in vitro
Since myofibrillar disarray is found in HCM patient specific hiPSC-CMs15 and have been shown to result in cardiomyocyte dysfunction16, we next examined the morphological changes of MLP-deficient CMs. Compared with WT hESC-CMs at day 30 post differentiation, immunostaining of cTnT and α-actinin of MLP-deficient hESC-CMs showed a significant higher frequency of sarcomeric disorganization, which was defined as disorganized sarcomeric α-actinin staining pattern observed in over 25% of the total cellular area with reference to previous studies17 (Figs. 2a, b and S2A). Multinucleation18 and increased cardiomyocyte size19 are also critical features of HCM in human cardiomyocytes20. We thus used a high-throughput (10,000 cells/sample) and nonsubjective flow cytometry method to measure hESC cardiomyocytes size via flow cytometry measurement of forward scatter using calibration spheres for cell size determination21. As previously reported22, only a small number of (<7%) WT hESC-CMs were observed to be binuclear or multinuclear, while MLP KO hESC-CMs exhibited significantly higher percentages of bi- or multi-nucleation (about 17%) at day 30 post differentiation (Fig. 2c, d). In addition, compared with WT hESC-CMs, MLP KO hESC-CMs gradually exhibited cell enlargement from day 22 post differentiation onward (Figs. 2e, f and S2B). These results suggest that MLP deficiency leads to cytoarchitectural features of HCM in hPSC-CMs.

a Immunostaining of sarcomeric α-actinin (green) and cTnT (red) demonstrates sarcomeric disarray in MLP KO CMs at day 30. Scale bar, 50 μm. b Compared with WT hESC-CMs (n = 189), a significant higher percentage of MLP KO hESC-CMS (n = 191) showed disorganized sarcomeric α-actinin staining pattern in greater than one fourth of the total cellular area. c Images of α-actinin/DAPI-immunostained hESC-CMs and d quantification of mono-, bi-, and multi-nucleation in WT (n = 267) and MLP KO (n = 259) hESC-CMs. Scale bar, 50 μm. e, f Calibration of forward scatter (FSC; 10,000 cells/sample, n = 3) showing an increased cellular size beginning 22 days post cardiac differentiation in MLP KO CMS. f Results are presented as means ± S.E.M. of three independent experiments. *P < 0.05; **P < 0.01; ns, not significant, unpaired two-sided Student’s t test
MLP deficiency leads to impaired calcium handling
Calcium handling plays a fundamental role in regulation of excitation–contraction in both skeletal and cardiac myocytes, dysfunction of which is commonly involved in the development of HCM and HF10,23. To examine the impact of MLP deficiency on calcium handling, we applied nickase Cas9 to introduce the green fluorescent calcium-modulated protein 6 fast type (GCaMP6f) calcium sensor into the AAVS1 locus of the WT and MLP KO H9 hESC lines as previously reported24 (Fig. 3a). Recording of calcium transients at days 15, 22, and 30 (Fig. 3b, c) show that compared with WT hESC-CMs, MLP KO hESC-CMs have slower beating rate (Figs. 3c and S3A). At day 15, the calcium release amplitude of MLP KO CMs was significantly higher than WT CMs, but gradually decreased (Fig. 3c, d). At day 30, compared with WT hESC-CMs, MLP KO hESC-CMs exhibited lower calcium release amplitude and longer time to peak (Fig. 3c–e). Beginning at day 22, MLP KO hESC-CMs started to demonstrate longer transient durations (Fig. 3c, f) and prolonged decay times (Fig. S3B) compared with WT hESC-CMs. Adjustment to heart rate variation did not impact prolonged transients of MLP KO hESC-CMs as compared with WT hESC-CMs (Fig. S3C). Interestingly, increased calcium release amplitude was observed to occur in MLP KO CMs before cellular hypertrophy (Figs. 2f and 3d), suggesting that abnormal Ca2+ handling may be a causal factor for the induction of HCM phenotype in MLP-deficient hESC-CMs.

a Schematic demonstrating the GCaMP-expression cassette that was integrated into AAVS1 of WT and MLP KO hESCs via nickase CRISPR/Cas9 editing. b Space-averaged calcium transients showing parameters measured for analysis of calcium handling. c Representative line-scan images in WT-GCaMP and MLP KO-GCaMP hESC-CMs at days 15, 22, and 30. d–f Quantification of peak, time to peak, and calcium transient duration in WT-GCaMP and MLP KO-GCaMP hESC-CMs (n = 15 cells per group). g Representative Ca2+ transients induced with 10 mM caffeine in Ca2+-free conditions between WT-GCaMP and MLP KO-GCaMP hESC-CMs at days 15, 22, and 30 (n = 5 cells per group). h–j Peak amplitude, transient duration, and decay time in caffeine-evoked Ca2+ transients WT-GCaMP and MLP KO-GCaMP hESC-CMs. Results are presented as means ± S.E.M. of three independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; ns, not significant, unpaired two-sided Student’s t test
To further profile calcium handling properties, we also measured caffeine-induced calcium release from the SR of WT and MLP KO hESC-CMs at days 15, 22, and 30 (Figs. 3g and S3D). Beginning at day 22, MLP KO hESC-CMs exhibited relatively smaller amplitudes, longer duration, and prolonged decay time compared with WT hESC-CMs (Figs. 3g, j and S3D), suggesting that MLP KO hESC-CMs have reduced SR calcium storage and impaired function of Ca2+ pumping and Ca2+ release channels in the SR and plasma membranes. Consistently, increased CACNA1C and RYR2 expression (Fig. S3E, F) and decreased SERCA 2a levels were observed in MLP KO hESC-CMs by qPCR (Fig. S3G) and western blot (Fig. S3H, I). These results indicate that MLP deficiency decreases the rate of calcium uptake and release in hESC-CMs as demonstrated in other models of HCM25,26 and HF27.
MLP-deficient cardiomyocytes exhibit elevated activity of hypertrophic signaling pathways
To understand how MLP deficiency leads to cardiomyopathy, we next accessed the expression of a panel of cardiomyopathy related genes at day 30 by qRT-PCR. Compared with WT hESC-CMs, MLP KO hESC-CMs displayed increased expression of genes involved in fetal development, calcium handling, hypertrophic signaling pathways, and autophagy (Fig. 4a–c). From this transcriptional data, we found significantly elevated activation of hypertrophic signaling in MLP KO hESC-CMs. To further examine signaling pathways involved28,29, we performed western blot for Ca2+-calcineurin–NFAT and Ca2+-Calmodulin-CaMKII. As compared with WT hESC-CMs, MLP KO hESC-CMs exhibited higher levels of P-CaMKII, which is a major mediator of hypertrophic signaling (Fig. 4d, e, g). Furthermore, MLP KO hESC-CMs exhibited higher level of MYH7, cTnT, and α-actinin, consistent with increased sarcomeric gene expression, which is a known feature of HCM (Fig. 4d, h–j)19.

a Heatmap showing changes in the expression of genes involved in HCM signaling pathways, calcium handling, fibrosis, and autophagy in WT and MLP KO hESC-CMs at day 30. b qRT-PCR analysis of HCM related genes in WT and MLP KO CMs at day 30. c Heatmap showing the expression of HCM associated genes at days 15, 22, and 30 of cardiac differentiation. d Immunoblot analysis of HCM signaling (calcineurin A, CaMKIIδ, and phosphorylated CaMKII) and sarcomere proteins (MYH7, α-actinin, and cTnT) in WT and MLP KO hESC-CMs at day 30. e–j Quantification of calcineurin A, CaMKIIδ, P-CaMKII, MYH7, α-actinin, and cTnT normalized by GAPDH in WT and MLP KO hESC-CMs. Results are presented as means ± S.E.M. of three independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; ns, not significant, unpaired two-sided Student’s t test
Because clinical symptoms of HCM usually take decades to manifest in affected individuals30, we also examined temporal gene expression in MLP KO hESC-CMs as compared with WT hESC-CMs by qRT-PCR. We found that from day 15 to day 30, the relative expression of hypertrophy related genes gradually increased in MLP KO hESC-CMs (Figs. 4c and S4A–G), and began to pick up in earnest around day 22 for genes such as MYH6, MYH7, MYL2, MYL7, and TNNT2, while NPPA and NPPB were significantly increased by day 30 (Figs. 4c and S4A–G).
MLP-deficient CMs develop mitochondrial dysfunction associated with HF
Previous studies have shown that cardiomyopathies and HF are commonly associated with pathological events such as mitochondrial dysfunction, energy depletion, increased generation of ROS, and cardiac dysfunction31,32. MLP-deficient mice have also been shown to exhibit regional absence of mitochondria and energy depletion33. In addition, increased intracellular basal [Ca2+] caused by MLP deficiency has been shown to damage mitochondria34. Thus, we next quantified mitochondrial content in MLP KO and WT hESC-CMs by comparing the amount of the mitochondrial-specific ND1 and ND2 genes to the housekeeping ACTB (encoding β-actin) at days 15, 22, and 3035. Beginning at day 30 post differentiation, MLP KO hESC-CMs exhibited a significant decrease in mtDNA/nDNA ratio compared with WT hESC-CMs (Fig. 5a). To further investigate the impacts of MLP deficiency on the metabolism of hESC-CMs, we next assessed mitochondrial content, total cellular ROS, and mitochondrial-specific ROS by flow cytometry using Mitotracker, Cell ROS green, and MitoSOX, respectively, at day 30. Compared with WT hESC-CMs, the mitochondrial content of MLP KO CMs was significantly reduced (Fig. 5b–d). Moreover, the staining pattern of Mitotracker in MLP KO hESC-CMs demonstrated that many mitochondria were punctate and fragmented (Fig. S5A) as compared with normal linear morphology in WT hESC-CMs, indicating that MLP KO hESC-CMs had hallmarks of mitochondrial damage. Flow cytometry assessment of cell ROS (Fig. 5e–g) and MitoSOX (Fig. 5h–j) staining demonstrated that both total cellular and mitochondrial-specific ROS are significantly elevated in MLP KO hESC-CMs, which is consistent with features of mitochondrial injury32.

a qPCR analysis of mitochondrial DNA (ND1 and ND2) to nuclear DNA (β-actin) ratio at days 15, 22, and 30 of cardiac differentiation (n = 9). b Mitotracker images and c, d quantification of Mitotracker green intensity obtained by flow cytometry demonstrates a significantly reduced fluorescence intensity in MLP KO hESC-CMs at day 30 as compared with WT hESC-CMs (n = 3). Scale bar, 50 μm. e–g Cell ROS green intensity and h–j MitoSox Red intensity suggests increase in ROS for MLP KO hESC-CMs as compared with WT hESC-CMs (n = 3). Scale bar, 50 μm. Results are presented as means ± S.E.M. of three independent experiments. **P < 0.01; ***P < 0.001; ****P < 0.0001; ns, not significant, unpaired two-sided Student’s t test
Since damaged mitochondria can lead to decreased ATP production and increased AMP/ATP ratio, we next determined the functional change of mitochondria by AMP-activated protein kinase (AMPK) activity, a “metabolic sensor”, that can be activated by ATP depletion and increased AMP/ATP ratio36. MLP KO hESC-CMs had a significantly higher P-AMPK levels compared with WT CMs at day 30 (Fig. S5B, C). These findings suggest that MLP deficiency may lead to HF phenotypes due to mitochondrial damage, increased ROS, and energy depletion.
MLP-deficient cardiomyocytes exhibit early failure after isoproterenol treatments
Chronic administration of β-adrenergic agonists, such as isoproterenol37, have been shown to aggravate HCM and induce HF in HCM models of disease38. Deletion of the β2-adrenergic receptor39 and overexpression of a β-adrenergic receptor kinase 1 inhibitor40 has been shown to prevent the development of cardiomyopathy in MLP-deficient mice. We thus examined whether treatment with ISO could accelerate the development of HCM disease phenotype in MLP KO hESC-CMs. One week of 10 μM ISO treatment from the day 15 of cardiac differentiation resulted in markedly increased calcium release amplitudes of WT hESC-CMs and significantly decreased calcium release amplitudes in MLP KO hESC-CMs (Fig. 6a–c). ISO treatment did not result in significant changes of time to peak in WT and MLP KO hESC-CMs (Fig. 6a, b, d). Although ISO treatment can reduce the duration of calcium transients both in WT and MLP KO hESC-CMs, the duration of calcium transients in MLP KO hESC-CMs was still maintained at higher levels than in WT hESC-CMs (Fig. 6a, b, e). Gene expression of cardiac hypertrophy related genes such as MYH6, TNNT2, NPPA, and NPPB was modestly induced in ISO-treated WT hESC-CMs, but was found to be markedly elevated in MLP KO hESC-CMs (Figs. 6f and S6A–G). These results suggest that β-adrenergic stimulation can exacerbate HCM disease phenotypes in MLP-deficient hESC-CMs.

a, b Representative line-scan images from WT and MLP KO hESC-CMs after 1 week of 10 μM ISO treatment beginning at day 15 of cardiac differentiation. c–e Quantification of peak, time to peak, and calcium transient duration in WT, MLP KO, WT/ISO, and MLP KO/ISO hESC-CMs (WT n = 13, WT/ISO n = 15, MLP KO n = 13, and MLP KO/ISO n = 12). e Heatmap representations of hypertrophy related gene expression in WT, MLP KO, WT/ISO, and MLP KO/ISO hESC-CMs (n = 3). Results are presented as means ± S.E.M. of three independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; ns, not significant, unpaired two-sided Student’s t test
Treatment of Ca2+ dysregulation prevents development of the HCM phenotype in MLP-deficient cardiomyocytes
As Ca2+ dysregulation was observed to occur before other hallmarks of HCM disease phenotypes, we next evaluated whether pharmaceutical inhibition of calcium entry with the L-type Ca2+ channel blocker verapamil could prevent the development of HCM phenotype in MLP KO hESC-CMs. Continuous addition of verapamil using therapeutic dosages (100 nM) for 10 days beginning at day 20 of cardiac differentiation was found to significantly improve impaired calcium handling function in MLP KO hESC-CMs, including increased rate of calcium uptake and release (Fig. 7a–d). Consistent with higher calcium amplitudes (Fig. 7b), increased SR Ca2+ storage capacity was also observed in verapamil-treated MLP KO hESC-CMs as compared with nontreated cells (Fig. S7A–C). Assessment of cell size and mitochondrial content by flow cytometry showed that administration of verapamil ameliorated cell enlargement (Fig. 7e, f) and mitochondrial damage (Fig. 7g, h) in MLP KO hESC-CMs. Treatment with verapamil was also found to downregulate the expression of genes related to cardiac hypertrophy (Figs. 7i and S7D) and reduce activation of the hypertrophic signaling pathways (Fig. 7j, k). These results suggest early intervention to treat calcium handling deficiencies can help prevent the development of HCM disease phenotypes in MLP-deficient hESC-CMs.

a Representative line-scan images in WT (blue), MLP KO (red), and MLP KO hESC-CMs treated with 100 nM verapamil (green) for 10 continuous days beginning at day 20 of cardiac differentiation (n = 13). b–d Quantification of peak, time to peak, and calcium transient duration in WT, MLP KO, and verapamil-treated MLP KO hESC-CMs (n = 12). e, f Forward scatter (FSC) and g, h Mitotracker green intensity obtained by flow cytometry in WT, MLP KO, and verapamil-treated MLP KO hESC-CMs. i Heatmap representations of hypertrophy related gene expression in WT, MLP KO, and verapamil-treated MLP KO hESC-CMs. j, k Immunoblot analysis of P-CaMKII and quantification of P-CAMKII normalized by GAPDH in WT, MLP KO, and verapamil-treated MLP KO hESC-CMs (n = 3). Results are presented as means ± S.E.M. of three independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; ns, not significant, unpaired two-sided Student’s t test
Discussion
In this study, we report the development of an in vitro human model for MLP-associated disease using CRISPR/Cas9 to genetically edit CSRP3−/− H9 hESCs and generate MLP KO cardiomyocytes. As prior preclinical models for MLP deficiency have been limited to transgenic animals, our system allows for the analysis of functional changes and molecular mechanisms underlying MLP deficiency in a human cardiac model of disease for the first time. Specifically, we show that MLP-deficient hESC-CMs develop disease phenotypes associated with HCM and that abnormal calcium handling is a central mechanism underlying disease pathogenesis. Treatment with the calcium channel blocker verapamil was found to prevent development of the disease phenotype in MLP-deficient CMs through maintenance of calcium homeostasis.
Given our findings in MLP KO hESC-CMs, we propose a model to illustrate the pathogenesis underlying MLP-associated HCM and HF (Fig. 8). Based on our findings, we suggest that disruption of intracellular Ca2+ homeostasis is an initial inciting event. Prior to the onset of HCM phenotypes, transcriptional expression of CACNA1C (encoding the alpha subunit of L-type calcium channel) was significantly elevated in MLP-deficient hESC-CMs, leading to higher levels of extracellular Ca2+ entering the cytoplasm during systole, and triggering the SR to release more Ca2+. Higher amplitude of calcium transients in MLP-deficient hESC-CMs was subsequently observed as early as day 15 of cardiac differentiation as compared with WT hESC-CMs. Similarly, SERCA expression was observed to be decreased at both the mRNA and protein levels in MLP-deficient hESC-CMs, resulting in reduced cytoplasmic Ca2+ transport back into the SR, and resulting in the elevation of diastolic [Ca2+]. As a result, SR Ca2+ load in MLP-deficient hESC-CMs was observed to be decreased because of increased RyR Ca2+ leak and diminished SR uptake. As a consequence of defective SR Ca2+ cycling, Ca2+ transients in MLP-deficient CMs show distinct alterations such as decreased amplitude, prolonged time to peak, reduced the rate of Ca2+ removal, and elevated resting [Ca2+], consistent with previously reported findings in failing CMs41. Prolonged calcium uptake and increased cytoplasmic calcium concentration in MLP-deficient hESC-CMs obstruct sarcomeric relaxation, leading to diastolic dysfunction.

Abnormal calcium handling and elevation of cytoplasmic Ca2+ concentrations are the central mechanism for pathogenesis of HCM and HF in MLP KO hESC-CMs. LTCC, L-type calcium channels; NCX, sodium-calcium exchange pump; CaM, Calmodulin; CnA, calcineurin
Several Ca2+-dependent pathways including calmodulin, CaMKII, and calcineurin have been shown to be key regulators of cardiomyopathy23. Cytoplasmic Ca2+ binds to calmodulin, which induces expression of ANF by calmodulin-binding transcriptional activator 2 (CAMTA2). In addition, CAMTA2 participates in hypertrophic response by binding to NKX2-542. Ca2+ binding to calmodulin also activates Ca2+-sensitive enzymes such as calcineurin and CaMKII, which have been shown to be overactivated in HCM and HF27,28. In line with these findings, MLP-deficient hESC-CMs were observed to have increased expression of calmodulin and phosphorylated CAMKII. In our cell models, overactivation of Ca2+-Calmodulin-CaMKII signaling in MLP-deficient CMs is most likely due to elevated cytoplasmic Ca2+. However, we found calcineurin was not increased at the protein level in MLP-deficient hESC-CMs. A possible explanation is that MLP, calcineurin, and calsarcin-1 can form a trimeric complex at the Z-disk and MLP is essential for calcineurin anchorage to the Z-disk43. Compartmentalization of calcineurin at the Z-disk is essential for coordinated dephosphorylation of NFAT transcription factors, which also are localized at the Z-disk44,45. Calcineurin was observed to be dislocated from the Z-disk in MLP-deficient hESC-CMs, which could inhibit the activation of Ca2+-calcineurin–NFAT signaling.
Resting Ca2+ can also be transported into the mitochondria by mitochondrial Ca2+ uniporters46. Elevated resting [Ca2+] in MLP-deficient CMs leads to even higher Ca2+ transport into the mitochondria, which has been shown to damage mitochondria34. MLP-deficient hESC-CMs demonstrate hallmarks of mitochondrial injury such as decreased mtDNA/nDNA ratio, Mitotracker green intensity, and reduced mitochondria density. Mitochondrial fragmentation was observed in MLP-deficient hESC-CMs, mimicking findings observed in patient HF47. Increased ROS were also observed in MLP-deficient hESC-CMs. Elevated mitochondrial ROS levels and ROS mediated damage has been demonstrated both in patients with HF and in preclinical models of the disease48,49. Increased ROS in MLP-deficient CMs can be explained by the several hypotheses. Frist, abnormal mitochondria observed in MLP-deficient CMs are a major source of ROS production. Second, increased energy demand due to inefficient sarcomeric ATP utilization in HCM31 require higher output of mitochondria, which could lead to further generation of ROS. It is known that Ca2+ uptake by SERCA, NCX, and Ca2+ ATPases in diastole are a process requiring significant energy consumption50. However, less ATP production due to reduction in mitochondrial density and injury in MLP-deficient hESC-CMs could hinder this process causing a further increase in cytoplasmic Ca2+, further forming a positive-feedback loop and thereby aggravating the progression of HCM and HF. Reduced ATP production and increased energy consumption in MLP-deficient CMs result in an increased AMP/ATP ratio, which can activate AMPK as evidenced by increased levels of phosphorylated AMPK.
Genetically modified PSC-derived CMs circumvent the limitations associated with transgenic animal models of human heart disease and have been used to model numerous genetic cardiovascular disorders in vitro, including HCM21 and long QT syndrome37. However, genetically modified PSC models are associated with several challenges in accurately modeling human heart disease. First, hESC-CMs are developmentally immature, and are characterized by transcriptional profiles more similar to fetal human CMs than adult CMs13. Second, hESC-CMs are not at the stage where they can model disease phenotypes at the tissue and organ levels such as interstitial fibrosis and endothelial cell dysfunction. Lastly, mutations in over 100 causal genes have been identified to engender cardiomyopathy in human patients, and our findings may only be specific to CSRP3 defects resulting in MLP deficiency. In spite of these limitations, our findings are the first to provide evidence of elevated cytoplasmic [Ca2+] as an initiating factor in the development of HCM and HF phenotypes caused by MLP deficiency in human CMs. These findings further demonstrate a possible mechanism to prevent development of this disease by early intervention via normalization of Ca2+ dysfunction.
Materials and methods
Cell culture and cardiac differentiation
H9 and derivative lines were maintained on feeder-free Matrigel (Corning, USA) and fed daily with E8 medium (Cellapy, China). Cells were routinely passaged every 3 days at 70–80% confluency using 0.5 mM EDTA in PBS without MgCl2 or CaCl2 (HyClone, USA). The cells were cultured at 37 °C with 5% CO2. hESCs were differentiated into ESC-CMs using a small molecule-based method as previously described13. hESC-CMs were purified with the lactate metabolic-selection method51.
Genome editing
Single-guide RNAs targeting CSRP3 were designed using online tools (http://crispr.mit.edu). Two sgRNAs (TGGGGCGGAGGCGCAAAATG and ATGGTAGACGGTCTTTTCAC) were tested for editing efficiency, and the first one was selected for genome editing in H9 cells. sgRNA was cloned into the epiCRISPR plasmid as previously reported11. Briefly, 2 × 106 H9 cells were disassociated using 0.5 mmol/L EDTA, then these cells were electroporated with 2.5 μg epiCRISPR plasmid in 82 μL stem cell P3 solution + 18 μL supplement (Lonza, Germany) using 4D nucleofector system with the CA137 program (Lonza). Transfected cells were seeded onto Matrigel-coated 10 cm petri dish and cultured in E8 medium with 10 µM Rho kinase inhibitor Y-27632 for the first 24 h. At day 3, cells were selected by puromycin (0.3 μg/mL). The surviving colonies were picked into a 24-well plate and expanded for PCR screening.
Generation of H9-GCaMP and CSRP3−/−-GCaMP: 2.5 μg AAVS1_sgRNA plasmid (Addgene, #100554, USA) and 2.5 μg pAAVS1-PC-GCaMP6f plasmid (Addgene, #73503) were electroporated together into H9 and CSRP3−/− ESCs. Methods for puromycin screening, selection, and identification of clones are as described above.
Immunostaining and imaging analyses
The ESC cells and ESC-CMs were fixed with 4% PFA, permeabilized in PBS containing 0.5% Triton X-100 (Sigma, USA) for 15 min, and blocked with 3% BSA (Sigma) for 60 min at room temperature. After that cells were incubated with primary antibodies overnight at 4 °C. Cells were then washed three times using PBS for 5 min each. Then cells were incubated with secondary antibodies diluted in 3% BSA for 1 h at room temperature. After three washes with PBS for 5 min each, cells were counterstained with DAPI (300 nM, Invitrogen, USA) for 5 min. Images were taken under Leica DMI 4000B. The full list of primary and secondary antibodies used and their appropriate dilution are provided in Table S2.
Flow cytometry
ESC-CMs were singularized with CardioEasy CM dissociation buffer (Cellapy). Cells were washed three times with PBS and fixed with chilled (4 °C) fixation buffer (BD Biosciences) for 10 min at room temperature. Next, the samples were incubated with the primary antibody (cTnT) for 30 min. After three washes with PBS, cells were incubated with the secondary antibody (Alexa Fluor 488) for 30 min. Then the samples were washed in PBS and assessed using FACS analysis (EPICS XL, Beckman).
Quantification of mitochondria and reactive oxygen species (ROS): Dissociated hESC-CMs were stained using 50 nM Mitotracker green (Beyotime, C1048, China), 10 μM Cell ROS green (Beyotime, S0033), and 2.5 μM MitoSOX Red (Invitrogen, #M36008). Live cells were incubated with these dyes (made in RPMI 1640, Corning) for 20 min at 37 °C and 5% CO2. After washed by PBS, cells were resuspended after centrifugation in PBS. Samples were stored on ice until flow cytometry was run on. The results were analyzed with FlowJo X program. Mean signal fluorescence intensity was used to compare mitochondrial content and ROS production in MLP KO CMs relative to WT CMs.
Ca2+ imaging
H9-GCaMP and CSRP3−/−-GCaMP derived cardiomyocytes were seeded onto confocal dishes. Intracellular calcium flux was imaged using a confocal microscope (Leica, TCS5 SP5, Germany) at 40×. Spontaneous Ca2+ transients were acquired at 37 °C and 5% CO2 using line-scan mode at a sampling rate of 1 ms/line. A total of 8192 lines were acquired for 8.192 s recoding. For detection of caffeine-evoked Ca2+ release, 10 mM caffeine in Ca2+ free solution was used to evoke sarcoplasmic reticulum (SR) Ca2+ transients in hESC-GCaMP-CMs. The results were analyzed with Image J and Igor.
RNA extraction and quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from about 2 × 105 cells with TRIzol (Life Technologies, USA) and treated with DNase I (Life Technologies) for about 30 min at 37 °C to eliminate DNA contamination. For qRT-PCR, 1 μg of RNA was reverse transcribed into cDNAs using the PrimeScriptTM reverse transcription system (Takara, Japan), following the manufacturer’s instructions. QRT-PCR was performed on iCycler iQ5 (Bio-Rad) using 2× SYBR Master Mix (Takara). The relative quantification of gene expression was carried out according to the ▵CT method. Primer sequences are listed in Table S1.
Western blots
Cell pellets were washed with cold PBS and resuspended in Mammalian Protein Extraction Reagent (Thermo, #78501, USA) with 5 mM EDTA (Thermo, #1861275), containing a protease inhibitor cocktail (Thermo, #1861278) and phosphatase inhibitor cocktail (Thermo, #1862495). Samples were incubated on ice for 30 min and vibrated every 10 min and then centrifuged at 12,000 rpm for 15 min. The protein concentration of the supernatant was measured by BCA method. Equal amount of denatured protein were resolved by electrophoresis 8–12% (depending on protein size) sodium dodecyl sulfate polyacrylamide gels. The gel was transferred onto a PVDF membrane at 300 mA for 90 min using gel transfer device (Bio-Rad). Then the membrane was blocked with 5% nonfat milk powder at room temperature for 1 h. Membranes were incubated with primary and secondary antibodies of appropriate concentrations listed in Table S2. All primary antibodies were incubated at 4 °C overnight, while secondary antibodies were incubated at room temperature for 1 h.
Data analysis and statistics
All data are presented as means ± standard errors of the means (S.E.M.). Statistical significance was evaluated using two-sided Student’s t test for two groups. Significant differences were considered when P < 0.05.
References
Arber, S. et al. MLP-deficient mice exhibit a disruption of cardiac cytoarchitectural organization, dilated cardiomyopathy, and heart failure. Cell 88, 393–403 (1997).
Papalouka, V. et al. Muscle LIM protein interacts with cofilin 2 and regulates F-actin dynamics in cardiac and skeletal muscle. Mol. Cell. Biol. 29, 6046–6058 (2009).
Hoffmann, C. et al. Human muscle LIM protein dimerizes along the actin cytoskeleton and cross-links actin filaments. Mol. Cell. Biol. 34, 3053–3065 (2014).
Bos, J. M. et al. Genotype-phenotype relationships involving hypertrophic cardiomyopathy-associated mutations in titin, muscle LIM protein, and telethonin. Mol. Genet. Metab. 88, 78–85 (2006).
Geier, C. et al. Mutations in the human muscle LIM protein gene in families with hypertrophic cardiomyopathy. Circulation 107, 1390–1395 (2003).
Mohapatra, B. et al. Mutations in the muscle LIM protein and alpha-actinin-2 genes in dilated cardiomyopathy and endocardial fibroelastosis. Mol. Genet. Metab. 80, 207–215 (2003).
Zolk, O., Caroni, P. & Bohm, M. Decreased expression of the cardiac LIM domain protein MLP in chronic human heart failure. Circulation 101, 2674–2677 (2000).
Knoll, R. et al. A common MLP (muscle LIM protein) variant is associated with cardiomyopathy. Circ. Res. 106, 695–704 (2010).
Geier, C. et al. Beyond the sarcomere: CSRP3 mutations cause hypertrophic cardiomyopathy. Hum. Mol. Genet. 17, 2753–2765 (2008).
Lan, F. et al. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem Cell 12, 101–113 (2013).
Xie, Y. et al. An episomal vector-based CRISPR/Cas9 system for highly efficient gene knockout in human pluripotent stem cells. Sci. Rep. 7, 2320 (2017).
Pankowicz, F. P. et al. Reprogramming metabolic pathways in vivo with CRISPR/Cas9 genome editing to treat hereditary tyrosinaemia. Nat. Commun. 7, 12642 (2016).
Burridge, P. W. et al. Chemically defined generation of human cardiomyocytes. Nat. Methods 11, 855–860 (2014).
Arber, S., Halder, G. & Caroni, P. Muscle LIM protein, a novel essential regulator of myogenesis, promotes myogenic differentiation. Cell 79, 221–231 (1994).
Tanaka, A. et al. Endothelin-1 induces myofibrillar disarray and contractile vector variability in hypertrophic cardiomyopathy-induced pluripotent stem cell-derived cardiomyocytes. J. Am. Heart Assoc. 3, e001263 (2014).
Kraft, T. et al. Familial hypertrophic cardiomyopathy: functional effects of myosin mutation R723G in cardiomyocytes. J. Mol. Cell. Cardiol. 57, 13–22 (2013).
Sun, N. et al. Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci. Transl. Med. 4, 130ra147 (2012).
Schneider, R. & Pfitzer, P. [Number of nuclei in isolated human myocardial cells]. Virchows Arch. B Cell Pathol. 12, 238–258 (1973).
Harvey, P. A. & Leinwand, L. A. The cell biology of disease: cellular mechanisms of cardiomyopathy. J. Cell Biol. 194, 355–365 (2011).
Arad, M., Seidman, J. G. & Seidman, C. E. Phenotypic diversity in hypertrophic cardiomyopathy. Hum. Mol. Genet. 11, 2499–2506 (2002).
Mosqueira, D. et al. CRISPR/Cas9 editing in human pluripotent stem cell-cardiomyocytes highlights arrhythmias, hypocontractility, and energy depletion as potential therapeutic targets for hypertrophic cardiomyopathy. Eur. Heart J. https://doi.org/10.1093/eurheartj/ehy249 (2018).
Denning, C. et al. Cardiomyocytes from human pluripotent stem cells: From laboratory curiosity to industrial biomedical platform. Biochim. Biophys. Acta 1863, 1728–1748 (2016).
Dewenter, M., von der Lieth, A., Katus, H. A. & Backs, J. Calcium signaling and transcriptional regulation in cardiomyocytes. Circ. Res. 121, 1000–1020 (2017).
Mandegar, M. A. et al. CRISPR interference efficiently induces specific and reversible gene silencing in human iPSCs. Cell Stem Cell 18, 541–553 (2016).
Knollmann, B. C. et al. Familial hypertrophic cardiomyopathy-linked mutant troponin T causes stress-induced ventricular tachycardia and Ca2+-dependent action potential remodeling. Circ. Res. 92, 428–436 (2003).
Frey, N., Luedde, M. & Katus, H. A. Mechanisms of disease: hypertrophic cardiomyopathy. Nat. Rev. Cardiol. 9, 91–100 (2011).
Lim, H. W. & Molkentin, J. D. Calcineurin and human heart failure. Nat. Med. 5, 246–247 (1999).
Chang, L. et al. Rad GTPase deficiency leads to cardiac hypertrophy. Circulation 116, 2976–2983 (2007).
Heineke, J. & Molkentin, J. D. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat. Rev. Mol. Cell Biol. 7, 589–600 (2006).
Maron, B. J. Hypertrophic cardiomyopathy: a systematic review. JAMA 287, 1308–1320 (2002).
Ashrafian, H., Redwood, C., Blair, E. & Watkins, H. Hypertrophic cardiomyopathy: a paradigm for myocardial energy depletion. Trends Genet. 19, 263–268 (2003).
Bayeva, M., Gheorghiade, M. & Ardehali, H. Mitochondria as a therapeutic target in heart failure. J. Am. Coll. Cardiol. 61, 599–610 (2013).
van den Bosch, B. J. et al. Regional absence of mitochondria causing energy depletion in the myocardium of muscle LIM protein knockout mice. Cardiovasc. Res. 65, 411–418 (2005).
Dong, Z., Saikumar, P., Weinberg, J. M. & Venkatachalam, M. A. Calcium in cell injury and death. Annu. Rev. Pathol. 1, 405–434 (2006).
Burkart, A. M. et al. Insulin resistance in human iPS cells reduces mitochondrial size and function. Sci. Rep. 6, 22788 (2016).
Fatkin, D. & Graham, R. M. Molecular mechanisms of inherited cardiomyopathies. Physiol. Rev. 82, 945–980 (2002).
Wang, Y. et al. Genome editing of isogenic human induced pluripotent stem cells recapitulates long QT phenotype for drug testing. J. Am. Coll. Cardiol. 64, 451–459 (2014).
Frey, N. et al. Mice lacking calsarcin-1 are sensitized to calcineurin signaling and show accelerated cardiomyopathy in response to pathological biomechanical stress. Nat. Med. 10, 1336–1343 (2004).
Fajardo, G. et al. Deletion of the beta2-adrenergic receptor prevents the development of cardiomyopathy in mice. J. Mol. Cell. Cardiol. 63, 155–164 (2013).
Rockman, H. A. et al. Expression of a beta-adrenergic receptor kinase 1 inhibitor prevents the development of myocardial failure in gene-targeted mice. Proc. Natl Acad. Sci. USA 95, 7000–7005 (1998).
Lehnart, S. E., Maier, L. S. & Hasenfuss, G. Abnormalities of calcium metabolism and myocardial contractility depression in the failing heart. Heart Fail. Rev. 14, 213–224 (2009).
Finkler, A., Ashery-Padan, R. & Fromm, H. CAMTAs: calmodulin-binding transcription activators from plants to human. FEBS Lett. 581, 3893–3898 (2007).
Heineke, J. et al. Attenuation of cardiac remodeling after myocardial infarction by muscle LIM protein-calcineurin signaling at the sarcomeric Z-disc. Proc. Natl Acad. Sci. USA 102, 1655–1660 (2005).
Vega, R. B., Bassel-Duby, R. & Olson, E. N. Control of cardiac growth and function by calcineurin signaling. J. Biol. Chem. 278, 36981–36984 (2003).
Liu, Y., Cseresnyes, Z., Randall, W. R. & Schneider, M. F. Activity-dependent nuclear translocation and intranuclear distribution of NFATc in adult skeletal muscle fibers. J. Cell Biol. 155, 27–39 (2001).
Bers, D. M. Cardiac excitation-contraction coupling. Nature 415, 198–205 (2002).
Jhun, B. S., O.-Uchi J., Adaniya, S. M., Cypress, M. W. & Yoon, Y. Adrenergic regulation of Drp1-driven mitochondrial fission in cardiac physio-pathology. Antioxidants (Basel) 7. https://doi.org/10.3390/antiox7120195 (2018).
Goh, K. Y. et al. Impaired mitochondrial network excitability in failing guinea-pig cardiomyocytes. Cardiovasc. Res. 109, 79–89 (2016).
Ide, T. et al. Mitochondrial electron transport complex I is a potential source of oxygen free radicals in the failing myocardium. Circ. Res. 85, 357–363 (1999).
Eisner, D. A., Caldwell, J. L., Kistamas, K. & Trafford, A. W. Calcium and excitation-contraction coupling in the heart. Circ. Res. 121, 181–195 (2017).
Tohyama, S. et al. Distinct metabolic flow enables large-scale purification of mouse and human pluripotent stem cell-derived cardiomyocytes. Cell Stem Cell 12, 127–137 (2013).
Acknowledgements
We gratefully acknowledge funding support from the National Natural Science Foundation of China Nos. 81570215 and 8142003.
Author information
Authors and Affiliations
Contributions
F.L., Y.W., A.S.L., and X.L. designed experiments. X.L., Y.L., F.W., R.B., S.M., T.D., W.-J.L., and H.Z. performed the experiments. X.L., Y.L., F.W., W.-J.L., and H.Z. analyzed the data. X.L., F.L., and A.S.L. wrote the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by I. Amelio
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, X., Lu, WJ., Li, Y. et al. MLP-deficient human pluripotent stem cell derived cardiomyocytes develop hypertrophic cardiomyopathy and heart failure phenotypes due to abnormal calcium handling. Cell Death Dis 10, 610 (2019). https://doi.org/10.1038/s41419-019-1826-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-019-1826-4
This article is cited by
-
PLEKHM2 deficiency induces impaired mitochondrial clearance and elevated ROS levels in human iPSC-derived cardiomyocytes
Cell Death Discovery (2024)
-
Thiamine-modified metabolic reprogramming of human pluripotent stem cell-derived cardiomyocyte under space microgravity
Signal Transduction and Targeted Therapy (2024)
-
Crispr-Based Editing of Human Pluripotent Stem Cells for Disease Modeling
Stem Cell Reviews and Reports (2024)
-
Losartan protects human stem cell-derived cardiomyocytes from angiotensin II-induced alcoholic cardiotoxicity
Cell Death Discovery (2022)
-
Phospholamban antisense oligonucleotides improve cardiac function in murine cardiomyopathy
Nature Communications (2021)
