
Abstract
Skin damage caused by radiation therapy (radiodermatitis) is a severe side effect of radiotherapy in cancer patients, and there is currently a lack of effective strategies to prevent or treat such skin damage. In this work, we show with several lines of evidence that plasminogen, a pro-inflammatory factor, is key for the development of radiodermatitis. After skin irradiation in wild-type (plg+/+) mice, the plasminogen level increased in the irradiated area, leading to severe skin damage such as ulcer formation. However, plasminogen-deficient (plg−/−) mice and mice lacking plasminogen activators were mostly resistant to radiodermatitis. Moreover, treatment with a plasminogen inhibitor, tranexamic acid, decreased radiodermatitis in plg+/+ mice and prevented radiodermatitis in plg+/− mice. Together with studies at the molecular level, we report that plasmin is required for the induction of inflammation after irradiation that leads to radiodermatitis, and we propose that inhibition of plasminogen activation can be a novel treatment strategy to reduce and prevent the occurrence of radiodermatitis in patients.
Similar content being viewed by others


Introduction
Approximately 50% of cancer patients receive some form of radiotherapy as a sole treatment or in combination with surgery or chemotherapy. Although radiotherapy techniques are continuously being developed, most patients still suffer from radiation-induced side effects which are mostly seen in tissues with rapidly proliferating cells such as the skin, gastrointestinal tract, and bone marrow1. In fact, the skin is affected to various degrees after any form of radiotherapy. The earliest visible skin reaction is erythema, which occurs in 90% of patients, and this can later evolve into desquamation or even into ulcers2. Radiation-induced dermatitis can be very painful and can severely affect the patient’s life quality3. The current strategies to treat radiation-induced dermatitis are suboptimal and include the application of dressings, antibiotics, and topical corticosteroids. Occasionally, severe wounds require skin grafting4.
The molecular mechanisms leading to radiation-induced dermatitis are not well understood, but DNA strand breaks and reactive oxygen species (ROS) induced by irradiation are considered to be the initial triggers for radiation-induced tissue damage1,5. These factors initiate cell death, especially in endothelial cells, fibroblasts, and keratinocytes. Radiation-induced damage to endothelial cells leads to the obstruction of the capillary lumen, ischemic damage, and vascular sclerosis;3,6 fibroblast dysfunction leads to defective collagen deposition and subsequent fibrosis; and damage to epithelial cells suppresses the formation of granulation tissue3,7. Signals sent by damaged cells and ROS induce the expression of transforming growth factor-β (TGF-β) and a burst of inflammation, both of which are known to be the main factors in the development of radiodermatitis6,8.
The pro-enzyme plasminogen, which is mainly produced in the liver and circulates in the blood at a relatively high concentration of 0.2 mg/ml9, is converted into the active protease plasmin by tissue-type plasminogen activator (tPA) or urokinase-type PA (uPA). Plasmin is a broad-spectrum protease that has an important role in fibrinolysis (the degradation of fibrin) and in the remodeling of the extracellular matrix10,11. Our previous studies have shown that plasminogen is a pro-inflammatory regulator that accumulates in wounds and accelerates wound healing12,13, but we have also shown that plasminogen plays a detrimental role in processes that involve excessive inflammation, such as sepsis14.
In the present study, we show that local irradiation of skin with ɣ-radiation in wild-type (plg+/+) mice induces plasminogen accumulation which is followed by the development of inflammation and subsequent skin damage. Notably, mice deficient in plasminogen (plg−/−) or mice lacking both plasminogen activators, PAs (tPA−/−;uPA−/−) are largely resistant to irradiation and do not develop radiodermatitis. Moreover, treatment with an inhibitor of plasminogen activation (tranexamic acid (TXA)) significantly delays the onset and decreases the severity of radiodermatitis in plg+/+ mice and completely prevents radiodermatitis in plg+/− mice. Our study also for the first time links plasminogen activation to TGF-β expression in vivo, suggesting that inhibition of plasminogen can be used to suppress TGF-β activation for the prevention of radiodermatitis. Taken together, our data show that the inhibition of plasminogen activation during and immediately after radiotherapy might be a potential treatment strategy to protect cancer patients from radiodermatitis and possible other tissue damage.
Materials and methods
Animals
Plg-heterozygous (plg+/−) mice15 in a C57BL/6 background were intercrossed to generate wild-type (plg+/+), heterozygous (plg+/−), and plg-deficient (plg−/−) mice. The mice were genotyped with a chromogenic assay16. Mice deficient in tPA or uPA17 were backcrossed for 10 generations with C57BL/6 mice, and tPA−/− and uPA−/− mice were then intercrossed to generate the tPA−/−;uPA−/− mice. The genotypes of these mice were determined by PCR analysis18. Approximately 10- to 14-week-old mice were used for the experiments. The animals were kept under standard laboratory conditions, and the Regional Ethics Committee of Umeå University approved all of the experimental protocols.
Radiation model
The dorsal skin of the mice was shaved 3 days prior to irradiation. For irradiation, the mice were anesthetized by intraperitoneal injection of 150 μl of a mixture containing 8% Ketaminol vet. (Intervet AB, Sollentuna, Sweden) and 5% Dormitor vet. (Orion Pharma AB, Espoo, Finland). The mice were placed inside a lead box with 2 cm thick walls to protect the whole body from radiation, and the dorsal skin was gently stretched out through a 4 cm gap at the bottom of the box and affixed with medical tape. The lead box with the mouse was then placed in a Gammacell 40 Exactor (Ashford, UK) that has two Cesium-137 sources. Radiation was given as a single dose of 1 Gy per min over 15 min (total dose of 15 Gy). The development of radiodermatitis was documented via digital photographs taken on different days after irradiation. The areas of the radiodermatitis lesions were quantified from the photographs using ImageJ (National Institute of Health, Bethesda, USA), and the severity of radiodermatitis was scored as 0 = no lesion, 1 = erythema, 2 = desquamation, and 3 = ulcer.
Morphological and immunohistochemical analyses
Skin from the irradiated area was removed, fixed in 4% paraformaldehyde, and embedded in paraffin. Sections with a thickness of 6 µm perpendicular to the skin surface were prepared. The sections were deparaffinized with xylene and rehydrated through washes in a graded ethanol series. Antigen retrieval was performed using citrate buffer at 95 °C.
For histological analyses, the sections were stained with Mayer’s hematoxylin (Histolab, Gothenburg, Sweden) using a standard protocol. 8-Oxo-2′-deoxyguanosine was stained with goat polyclonal antibody (Abcam, Cambridge, UK) followed by donkey anti-goat IgG HRP (Abcam, Cambridge, UK) and DAB substrate (Vector Laboratories, Burlingame, USA) and the sections were counterstained with Mayer’s hematoxylin. Images were taken using a Leica DC300F digital camera attached to a Leica DM LB microscope (Leica, Wetzlar, Germany). Epidermal thickness was then measured (three measurements per skin sample) from the microscopy images using Adobe Photoshop. Quantification of area stained for 8-oxo-2′-deoxyguanosine was performed using ImageJ software.
Macrophages were stained with rabbit anti-mouse CD68 polyclonal antibody (Abcam, Cambridge, UK) followed by Dylight 594-labeled anti-rabbit IgG antibody (Vector Laboratories, Burlingame, USA). Neutrophils were stained with the rat anti-mouse Ly-6B.2 monoclonal antibody clone 7/4 (AbD Serotec, Oxford, UK) and followed by staining with biotinylated goat anti-rat IgG antibody (Santa Cruz Biotechnology, USA) and streptavidin-Alexa Fluor 647-conjugated antibody (Thermo Fisher Scientific, Waltham, USA). Nuclear extracellular traps (NETs) were stained with rabbit anti-citrullinated histone H3 antibody (Abcam, Cambridge, UK) followed by Dylight 488-labeled goat anti-rabbit IgG antibody (Vector Laboratories, Burlingame, USA). Apoptotic cells were detected via TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) assay (In Situ Cell Death Detection Kit, Roche, USA). Proliferative cells were stained with rabbit antibody against mouse Ki67 protein (Thermo Fisher Scientific, Waltham, USA) followed by Dylight 594-conjugated anti-rabbit IgG antibody (Vector Laboratories, Burlingame, USA). Fibrinogen/fibrin was stained with goat anti-mouse fibrinogen antibody (Nordic Immunological Lab, Tilburg, The Netherlands) followed by rabbit anti-goat Alexa Fluor 555-conjugated antibodies (Thermo Fisher Scientific, Waltham, USA). Vessels were stained with rat anti-mouse CD31 antibody (cluster of differentiation 31 protein) (BD Bioscience, San Jose, USA) followed by goat anti-rat IgG-TR antibody (Santa Cruz Biotechnology, Texas, USA). For fluorescence staining, 4′,6-diamidino-2-phenylindole (DAPI; Thermo Fisher Scientific, Waltham, USA) was used for staining nuclei, and images were captured with a Zeiss Axio Imager Z1 (Zeiss, Oberkochen, Germany). Quantification of fluorescent area was performed using ImageJ software.
Analysis of skin extracts using ELISA
Dorsal skin samples from irradiated and non-irradiated mice were homogenized in lysis buffer (50 mM Tris-HCl buffer pH 8.0 with 120 mM NaCl, 1 mM EDTA, 6 mM EGTA, 1% NP-40, and 1 mM dithiothreitol) supplemented with PhosSTOP phosphatase inhibitors and cOmplete ULTRA mini protease inhibitor cocktail tablets (both from Roche, Basel, Switzerland). Skin samples were kept at −20 °C until use. The concentration of total protein in the extracts was quantified using a Pierce BCA protein assay kit according to the manufacturer’s instructions (Thermo Fisher Scientific, Waltham, USA). Mouse interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α) levels in the extracts were measured using specific enzyme-linked immunosorbent assay (ELISA) kits (eBioscience, San Diego, USA). Mouse plasminogen was quantified with a mouse plasminogen-specific ELISA kit (Omnio AB, Umeå, Sweden). IL-10 and IL-1β levels in skin extracts were measured using a mouse IL-10 ELISA kit (Abcam, Cambridge, UK) and a mouse IL-1β ELISA kit (Thermo Fisher Scientific, Waltham, USA) respectively.
Western blot analysis
For quantification of phospho-Smad2 (PSmad2) in skin extracts, the protein extracts were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis under reducing conditions using 4–12% gradient gels (NuPAGE, Thermo Fisher Scientific, Waltham, USA) and transferred to polyvinylidene difluoride membrane. PSmad2 (Ser465/467) was then detected with a rabbit monoclonal antibody (Cell Signaling Technology, Danvers, MA) followed by a goat anti-rabbit antibody coupled with peroxidase. Total Smad2 was detected with a rabbit monoclonal antibody (Cell Signaling Technology, Danvers, MA) and actin was detected with a monoclonal anti-actin antibody (Sigma-Aldrich, Saint Louis, USA). The protein bands were visualized with SuperBright ECL (Agrisera, Vännäs, Sweden) using ChemiDoc Touch Imaging System (Bio-Rad Laboratories, Hercules, USA). The protein band intensity was quantified using ImageJ software.
Quantitative RT-PCR
Skin samples were homogenized in TRIzol (Ambion, Carlsbad CA, USA) using Precellys CK28R tubes on a Precellys 24 homogenizer (both from Bertin Technologies, Lyon, France) according to the manufacturer’s instructions. Total RNA was extracted with a PureLink RNA Mini Kit (Ambion, Carlsbad CA, USA) according to the manufacturer’s instruction. A 2.5 µg aliquot of total RNA was reverse-transcribed using a SuperScript VILO cDNA Synthesis Kit (Invitrogen, Carlsbad CA, USA) and diluted threefold with diethylpyrocarbonate-treated water. Gene expression was analyzed using quantitative real-time PCR (RT-PCR) with the comparative CT method and with TATA-binding protein mRNA as the internal reference gene. The selection of the reference gene was based on data from RT² Profiler™ PCR Array Mouse Housekeeping Genes (Qiagen, Maryland, USA) and subsequent calculation of M-values using the geNorm software (Biogazelle NV, Gent, Belgium). The gene-specific primers and probes (TaqMan Gene Expression Assays) were obtained from Applied Biosystems (Foster City, USA), and 2× SsoAdvanced Universal Probes Supermix was from Bio-Rad (California, USA). Each sample was run in triplicate on the StepOnePlus Instrument (Applied Biosystems) using the real-time PCR conditions recommended for the 2× SsoAdvanced Universal Probes Supermix.
Treatment with TXA
Because TXA (Meda AB, Stockholm, Sweden) is orally active, it was dissolved at 20 mg/ml in drinking water. The total daily dose was estimated at 100 mg/day. The treatment was initiated 2 days before irradiation and continued until day 10 after irradiation.
Statistical analysis
In Figs. 1d, 6b, d, and Supplementary Figure 1, the differences between two groups were analyzed with the Mann–Whitney U-test, and the results are expressed as scatter dot plots with the median indicated as a line. For the rest of the figures, the differences between two groups were analyzed with two-tailed t-tests and the differences between multiple groups were analyzed using one-way ANOVA, and the results are expressed as the mean ± SEM. P < 0.05 was defined as the significance threshold.

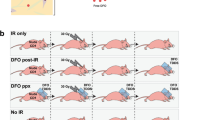
Representative photographs of the dorsal skin of three different plg+/+ (a), plg+/− (b), and plg−/− mice (c) at different time points after irradiation. Typical irradiated areas are marked on the photographs of the control plg+/− mice taken on day 0. Arrows show the wounds on the skin. d A comparison of the quality of dorsal skin in plg+/+ (n = 6), plg+/− (n = 6), and plg−/− (n = 6) mice at day 14 after irradiation. The skin damage scores are 0 = normal skin, 1 = erythema, 2 = desquamation, and 3 = ulceration. The mean values are indicated (black lines) and SEM is shown. *P < 0.05
Results
Plasminogen deficiency in mice protects against radiation-induced dermatitis
Radiodermatitis is caused by the induction of excessive inflammation8,19. Because we previously showed that plasminogen is a pro-inflammatory regulator12, we tested the hypothesis that plasminogen is involved in the development of radiodermatitis. Dorsal skin of plg+/+, plg+/−, and plg−/− mice was exposed to a single ɣ-radiation dose of 15 Gy, and Fig. 1 shows representative photographs of the dorsal skin of the mice on different days after irradiation. Approximately 9 to 10 days after irradiation, all plg+/+ mice developed erythema and then desquamation, and these wounds developed into ulcers between days 14 and 20 (Fig. 1a, d). The plg+/− mice, which have half the plasminogen level as that of plg+/+ mice, developed erythema and mild desquamation around days 9 to 10, which healed before day 20 and never progressed to a severe form of radiodermatitis (Fig. 1b, d, and Supplementary Fig. 1). In contrast, most of the plg−/− mice showed no signs of dermatitis at any time point after irradiation (Fig. 1c, d), and only a few plg−/− mice developed wounds (Fig. 1c, d, and Supplementary Fig. 1). Of the 39 plg−/− mice used in all of our studies, only 21% developed any degree of radiodermatitis. In summary, the data suggest that the development of radiodermatitis is strongly related to plasminogen levels.
Plasminogen is needed for the pathological changes associated with the development of radiodermatitis
As shown in Fig. 2a, b, and Supplementary Fig. 2, the thickness of the skin in plg+/+ mice increased gradually after irradiation, and at day 9 when erythema was observed the skin was approximately 4.6-fold thicker compared to non-irradiated skin (Fig. 2a, b). In sharp contrast, the thickness of the epidermis in plg−/− mice was not significantly different at any time point after irradiation (Fig. 2a, b). Moreover, thickening of the skin in plg+/+ mice suffering from radiodermatitis was accompanied by a significantly elevated number of blood vessels (shown by staining for CD31), which was not the case in plg−/− mice (Fig. 2c, d, and Supplementary Fig. 3).

a Representative images of hematoxylin and eosin (H&E)-stained skin sections from plg+/+ and plg−/− mice before and on day 9 after irradiation. Arrowheads and arrows point to the epidermis in plg+/+ and plg−/− mice, respectively. b The thickness of the epidermis in plg+/+ and plg−/− mice on different days after irradiation measured in H&E-stained skin sections. c Representative images of skin sections stained for CD31 (red) from plg+/+ and plg−/− mice on day 12 after irradiation. Arrowheads and arrows point to the blood vessels in plg+/+ and plg−/− mice, respectively. d Quantification of blood vessels in plg+/+ and plg−/− mice at different time points after irradiation. e Representative images of Ki67-stained (red) skin sections from plg+/+ and plg−/− mice on day 12 after irradiation. Arrowheads and arrows point to the proliferating cells in plg+/+ and plg−/− mice, respectively. f Quantification of proliferating cells in plg+/+ and plg−/− mice at different time points after irradiation. g Representative images of TUNEL (green) staining of skin sections from plg+/+ and plg−/− mice on day 1 and day 12 after irradiation. Arrowheads and arrows point to the apoptotic cells in plg+/+ and plg−/− mice, respectively. h Quantification of apoptotic cells in plg+/+ and plg−/− mice at different time points after irradiation. i Representative images of 8-oxo-2′-deoxyguanosine staining of skin sections from plg+/+ and plg−/− mice on day 1 and day 12 after irradiation. Arrowheads and arrows point to cells with oxidized DNA in plg+/+ and plg−/− mice, respectively. j Quantification of 8-oxo-2′-deoxyguanosine in plg+/+ and plg−/− mice at different time points after irradiation. For all quantifications, n ≥ 3 per genotype. In all immunostained sections, DAPI (blue) was used for staining the nuclei. Scale bar = 100 µm. *P < 0.05; **P < 0.01.
In agreement with the increased thickness of the epidermis, the number of proliferating cells in plg+/+ mice (as indicated by Ki67 staining) also gradually increased, with the highest rate of cell proliferation at day 12 after irradiation and coinciding with ulcer formation (Fig. 2e, f, and Supplementary Fig. 4). In contrast, the number of proliferating cells did not significantly increase in the irradiated plg−/− mice (Fig. 2e, f, and Supplementary Fig. 4).
Irradiation causes apoptosis due to the creation of DNA strand breaks20. Similar rates of apoptosis were observed in both plg+/+ and plg−/− mice at day 1 after irradiation, indicating that plasminogen has no effect on apoptosis at early time points after irradiation. However, at day 12 the number of apoptotic cells had increased by 8.5-fold in the plg+/+ mice, while the number of apoptotic cells had slightly decreased in the plg−/− mice compared to day 1 (Fig. 2g, h, and Supplementary Fig. 5). The high levels of apoptosis in plg+/+ mice on day 12 coincided with the development of radiodermatitis, and the lack of elevated apoptosis in plg−/− mice indicates that the development of radiodermatitis is suppressed in these mice.
Radiotherapy induces oxidative stress via induction of high levels of ROS. ROS are responsible for oxidation of cellular proteins and DNA, and are one of the major factors leading to cellular damage after irradiation21. To detect the level of oxidative stress in irradiated skin, we stained skin sections for 8-oxo-2′-deoxyguanosine that correlates with the damage of DNA caused by ROS22. As shown in Fig. 2i, j, and Supplementary Fig. 6, the same levels of DNA damage were detected in plg+/+ and plg−/− mice at day 1 after irradiation. At the later time points, the DNA damage increased significantly in plg+/+ mice, while it declined to normal levels in plg−/− mice, indicating that plasminogen deficiency preventes the formation of excessive oxidative stress in the later time points after irradiation.
Taken together, the above data show that in the absence of plasminogen, irradiation does not induce any of the visible pathological events that normally occur during the development of radiodermatitis.
Neutrophils and macrophages infiltrate the irradiated skin of plg + / + mice but not that of plg−/− mice
To determine if the lack of radiodermatitis in plg−/− mice is due to suppressed inflammatory cell infiltration after irradiation, we stained skin sections of both genotypes for neutrophils. We found no detectable neutrophils from day 0 to day 5 post irradiation in either genotype (Supplementary Fig. 7). In plg+/+ mice, neutrophils began to infiltrate the skin on day 9 post irradiation, and they reached very high numbers on day 12, which correlated with the development of ulcers (Fig. 3a, b). However, in plg−/− mice, almost no neutrophils were detected in the skin at any time point after irradiation (Fig. 3a, b). It is known that during inflammation neutrophils can release NETs (de-condensed chromatin linked with various cytotoxic proteins) that can cause tissue damage23. At day 12 post irradiation, no NETs were observed in the irradiated skin of plg−/− mice, which was in sharp contrast with the large numbers of NETs in the plg+/+ mice that appeared on day 12 when radiodermatitis developed (Fig. 3a, c, and Supplementary Fig. 7). These data suggest that plasminogen/plasmin is an indispensable trigger for the development of inflammation after irradiation. Without plasminogen, inflammation does not develop.

a Representative photographs of skin sections from plg+/+ and plg−/− mice on days 9 and 12 after irradiation stained for neutrophils (red), DAPI (blue), and NETs (citrullinated histone H3) (green). White arrowheads and arrows point to neutrophils in plg+/+ and plg−/− mice, respectively. Yellow arrowheads point to NETs in plg+/+ mice. b, c Quantification of neutrophils and NETs in the skin sections from plg+/+ and plg−/− mice on different days after irradiation (n ≥ 3 per genotype). d Representative immunostaining for macrophages (red) in sections from irradiated plg+/+ and plg−/− mouse skin on days 1 and 12 after irradiation. Arrowheads and arrows point to macrophages in plg+/+ and plg−/− mice, respectively. e Quantification of macrophages in the skin sections from plg+/+ and plg−/− mice on different days after irradiation (n ≥ 3 per genotype). Scale bar = 100 µm. f, g Expression of F4/80 and CD206 mRNA, respectively, in skin extracts from plg+/+ and plg−/− mice on different days after irradiation quantified by RT-PCR (n ≥ 4 per genotype). The red arrow indicates the day when dermatitis becomes visible in plg+/+ mice. *P < 0.05; **P < 0.01; ***P < 0.005; ****P < 0.001
It is well known that activated macrophages migrate to irradiated tissues24. As shown in Fig. 3d–f, in plg+/+ mice, the number of macrophages started to increase by days 1–5 post irradiation and continued to increase during following days which coincides with the development of erythema and ulcers. In contrast, the number of macrophages in plg−/− mice only increased slightly around days 5–9 and then returned to a low basal level after that (Fig. 3d–f and Supplementary Fig. 8). These data suggest that in plg+/+ mice the infiltration of macrophages occurred in two phases—the first phase was just after irradiation, and the second phase occurred at the time when erythema had developed and progressed into ulcers. In plg−/− mice, it is apparent that macrophage infiltration occurred only early after irradiation because these mice did not subsequently develop radiodermatitis. To test phagocytic ability of the macrophages, we measured the expression of CD206 (mannose receptor C type 1). CD206 expression was low in pro-inflammatory M1 macrophages, but high in anti-inflammatory M2 macrophages25. As shown in Fig. 3g, the expression of CD206 increased from days 3 to 5 in both plg+/+ and plg−/− mice, but from day 7 onward the expression of CD206 increased further and remained high throughout the development of radiodermatitis in plg+/+ mice, while it returned to low basal levels in plg−/− mice. This indicates a higher phagocytic activity in plg+/+ mice as compared to plg−/− mice that most likely is linked with an extended tissue damage in the plg+/+ mice. In addition, a fivefold increase of F4/80 expression at day 16 in plg+/+ mice was accompanied by only about a twofold increase of CD206 expression, suggesting that the number of pro-inflammatory M1 macrophages increased in radiation-induced wounds at day 16 (Fig. 3f, g).
Plasminogen accumulates in irradiated skin and induces the factors causing radiation-induced tissue damage
We have shown previously that plasminogen is transported to wounded skin by immune cells where its levels increase by approximately sevenfold12. Here, we have shown that the level of plasminogen in the irradiated plg+/+ mice increased gradually from day 1 after irradiation and reached the highest level (an approximately sevenfold increase) from days 9 to 12, after which it began to decrease (Fig. 4a).

a–j Skin samples were taken from plg+/+ and plg−/− mice on different days after irradiation (n ≥ 4 per time point), and specific mRNA and protein levels were measured using RT-PCR and protein-specific ELISA, respectively. a For statistical significance, levels of plasminogen in the skin of plg+/+ mice at different time points was compared to day 0. b, c IL-6 mRNA and protein levels. d, e IL-1-β mRNA and protein levels. f, g TNF-α mRNA and protein levels. h Protein levels of IL-10. i, j TGF-β and PAI-1 mRNA levels. k Fibrin(ogen) levels in skin sections of plg+/+ and plg−/− mice at different time points after irradiation (n ≥ 3 per genotype). *P < 0.05; **P < 0.01; ***P < 0.005; ****P < 0.001
The development of acute radiodermatitis is correlated with high levels of inflammatory cytokines24. As shown in Fig. 4b, c, the expression and protein level of IL-6 in plg+/+ mice began to increase from day 5 and reached the highest level from days 9 to 12 post irradiation when radiodermatitis developed. The expression and protein levels of IL1-β and TNF-α in these mice were low until day 7 post irradiation, but increased dramatically on day 9 (Fig. 4d–g). In contrast, in plg−/− mice these cytokines remained at basal levels at all time points after irradiation. The only exception was IL-10 for which protein levels were significantly higher in plg−/− mice during all time points after irradiation, compared to the plg+/+ mice. IL-10 is an anti-inflammatory cytokine which downregulates the production of pro-inflammatory cytokines after radiation exposure26. The relatively high levels of IL-10 in plg−/− mice may be, at least partially, responsible for the lack of inflammatory burst in irradiated skin in these mice. Since mRNA levels for IL-10 do not correlate with IL-10 protein levels27, the expression of mRNA for IL-10 is not shown here.
It is worth noting that TGF-β is one of the most important factors that contribute to tissue injury after irradiation19. As shown in Fig. 4i, the expression of TGF-β mRNA in the irradiated skin of plg+/+ mice occurred in two phases and followed the macrophage accumulation. The first peak of TGF-β expression was observed around day 5 (a 2.5-fold increase), and the second peak was seen from days 9 to day 16 (an approximately fourfold increase). In sharp contrast, no increase in TGF-β expression was detected in the irradiated skin of plg−/− mice, showing that in the absence of plasminogen TGF-β was not induced and thus could not initiate the inflammation that is required for radiodermatitis to develop. These data suggest that plasminogen may be involved in activation of the TGF-β signaling pathway. To support this conclusion, we have quantified phosho-Smad2 in extracts from irradiated skin by western blot. As shown in Supplementary Fig. 9, phosphorylation of Smad2 was increased in plg+/+ mice at days 9–16, whereas it was very low in plg−/− mice at these time points.
PAI-1 (Serpine1, plasminogen activator inhibitor type 1) is one of the molecules that are regulated by TGF-β28,29. In addition to being an inhibitor of plasminogen activators, PAI-1 can also regulate cell migration through a mechanism that is not dependent on its inhibitory activity30. TGF-β can induce the expression of PAI-1 via the Smad pathway, and high levels of PAI-1 have been implicated in intestinal radiation injury29,31. Here we found that the pattern of PAI-1 expression in the irradiated skin followed the expression of TGF-β. In plg+/+ mice, PAI-1 expression peaked first on day 5 (a 5-fold increase) and then from days 9 to 12 (an approximately 36-fold increase). In plg−/− mice, however, PAI-1 expression remained low at all time points after irradiation (Fig. 4j). This suggests that PAI-1 might also be involved in radiation-induced skin damage.
Irradiation and inflammation result in increased blood vessel permeability and leakage of plasma proteins into tissues32. Fibrin deposits appeared on day 1 after irradiation to the same extent in both plg+/+ and plg−/− mice. In plg−/− mice, this level remained stable over all subsequent days. However, in plg+/+ mice the fibrin levels decreased significantly on day 5 after irradiation, but increased again from days 9 to 12 concomitant with inflammation and ulcer formation (Fig. 4k, and Supplementary Fig. 10).
Plasminogen activation is necessary to induce inflammation and the expression of molecules that are involved in radiation-induced tissue damage
The above results indicate that plasminogen is required for the development of radiodermatitis. To confirm that plasminogen activation is important for this process, we used the tPA−/−;uPA−/− mice that have normal plasminogen levels but plasminogen cannot be activated to plasmin. Irradiated tPA−/−;uPA−/− mice did not develop any form of radiodermatitis at any time point after irradiation (Fig. 5a), and plasminogen levels in the irradiated skin of these mice were significantly lower than in irradiated tPA+/+;uPA+/+ mice (Fig. 5b), indicating that the total plasminogen accumulation partially depends on the activation of plasminogen into plasmin. However, the mRNA and protein levels of pro-inflammatory cytokines (IL1-β, IL-6, and TNF-α), protein levels of the anti-inflammatory cytokine IL-10, and the mRNA levels of TGF-β and PAI-1 in the irradiated skin from the tPA−/−;uPA−/− mice were all comparable to the levels in the plg−/− mice (Fig. 5c–g and Supplementary Fig. 11). Taken together, these data strongly suggest that plasmin, the active form of plasminogen, is responsible for inducing key factors that are involved in the development of radiodermatitis.

a Representative photographs of the dorsal skin of the tPA−/−;uPA−/− mice at different time points after irradiation. b Plasminogen levels in skin extracts from tPA+/+;uPA+/+ mice and tPA−/−;uPA−/− mice after irradiation measured by ELISA. c–g RT-PCR measurement of mRNA levels of IL1-β (c), IL-6 (d), TNFα (e), TGF-β (f), and PAI-1 (g) in irradiated skin of plg+/+, plg−/−, and tPA−/−;uPA−/− mice. *P < 0.05; **P < 0.01; ***P < 0.005
Inhibition of plasminogen activation by TXA delays the onset and decreases the severity of radiodermatitis in plg + / + mice and prevents radiodermatitis in plg+/− mice
Plasminogen binds to lysine residues on fibrin and on plasminogen-specific cell receptors, and it is then activated into plasmin by the PAs33. Lysine analogs, such as TXA, inhibit plasminogen activation and are used in the clinic to treat clotting disorders34. In this study, we added TXA to the drinking water of plg+/+ and plg+/− mice starting from 2 days before irradiation and ending on day 10 post irradiation. Plg+/+ mice treated with TXA showed delayed onset and reduced severity of radiodermatitis (Fig. 6a, c) compared to the irradiated plg+/+ mice without TXA. Moreover, in plg+/− mice treated with TXA the development of radiodermatitis was largely inhibited, and only low-grade erythema developed on day 14, which had healed by day 20 (Fig. 6b, d, and Supplementary Table 1).

plg+/+ and plg+/− mice were treated with TXA in their drinking water from 2 days before irradiation until day 10 post irradiation. Control mice were irradiated but did not receive TXA. a, b Representative photographs of the dorsal skin of irradiated plg+/+ mice treated with TXA, and a comparison of the quality of the dorsal skin in irradiated control and TXA-treated plg+/+ mice (n = 6 per time point). c, d Representative photographs of the dorsal skin of irradiated plg+/− mice treated with TXA, and a comparison of the quality of the dorsal skin in irradiated control and TXA-treated plg+/− mice (n = 6 per time point). Arrows show the wounded skin area. e Plasminogen levels in the skin extracts of control and TXA-treated plg+/+ mice on different days after irradiation. f–j RT-PCR measurement of mRNA levels of IL1-β (f), IL-6 (g), TNF-α (h), TGF-β (i), and PAI-1 (j) in the irradiated skin of control and TXA-treated plg+/+ and in plg−/− mice on different days after irradiation. *P < 0.05; **P < 0.01; ***P < 0.005; ****P < 0.001.
As expected, treatment with TXA led to a reduced accumulation of plasminogen in irradiated skin. On day 9, the plasminogen level in the irradiated skin of TXA-treated plg+/+ mice was approximately 4.5-fold lower than that of control irradiated plg+/+ mice (Fig. 6e). Because the TXA treatment ended on day 10, the plasminogen level in TXA-treated mice began to slowly increase on day 16 post irradiation, but this increase did not seem to be sufficient to induce radiodermatitis. The mRNA and protein levels for pro-inflammatory cytokines (IL1-β, IL-6, and TNF-α) were all low in TXA-treated plg+/+ mice and were comparable to those in irradiated plg−/− mice (Fig. 6f–h and Supplementary Fig. 12). Treatment of plg+/+ mice with TXA also resulted in decreased expression of TGF-β and PAI-1 (Fig. 6i, j). However, the low protein levels of the anti-inflammatory IL-10 observed in irradiated skin of plg+/+ mice increased significantly after TXA treatment (Supplementary Fig. 12).
Discussion
Because the molecular mechanisms behind the development of radiation-induced dermatitis are not fully understood, current treatments of radiation-induced wounds are still based on conventional wound treatments. Here, we provide compelling evidence indicating that plasminogen is a critical factor in the development of radiodermatitis. We found that the development of radiodermatitis in mice is initiated by the accumulation of plasminogen in the irradiated skin where it is activated to plasmin. We also showed that plasmin plays a critical role in recruiting neutrophils and macrophages and inducing the expression and activation of TGF-β and various inflammatory factors that are known to cause injury in healthy tissues. Finally, we found that genetic ablation of plasminogen or PAs prevents the formation of radiodermatitis in mice and that the inhibition of plasminogen activation by TXA significantly decreases the severity of radiodermatitis in plg+/+ mice and prevents radiodermatitis in plg+/− mice. Collectively, these results show that the expression of TGF-β and subsequent inflammatory reactions are not induced by irradiation in plg−/− mice and in mice in which plasminogen activation is prevented. This suggests that the inhibition of plasminogen/plasmin might be a novel treatment for preventing radiodermatitis in cancer therapy.
Based on our current findings and previously published data, we propose a new model for the role of plasmin in the development of skin damage after γ-irradiation (Fig. 7). Considering the timing of events, the interval between irradiation and the appearance of radiodermatitis can be divided into two phases. The first phase, which we call the “pre-inflammatory phase” (days 1 to 5 post irradiation), is characterized by a lack of inflammation. At the beginning of this phase, irradiation of the skin causes the production of ROS and induces vessel permeability32. Plasma diffuses from the vessels, transporting fibrinogen and plasminogen to the dermis. In the tissue environment, fibrinogen is rapidly converted to fibrin and binds to plasminogen33. Plasminogen then becomes activated to plasmin which degrades fibrin and produces various biologically active fibrin fragments35,36. Plasminogen also binds to cell-surface receptors where it is activated by cell-bound PAs37. Plasmin, together with ROS, activates latent TGF-β38,39 and the associated signaling pathways, and this leads to the enhanced expression of TGF-β. At this point, the “inflammatory phase” begins as plasmin, together with fibrin fragments and TGF-β, triggers the expression of pro-inflammatory proteins and drives the infiltration of inflammatory cells40. All of these events result in a burst of inflammation that is directly responsible for the development of radiodermatitis (Fig. 7). In plg−/− mice, the fibrin that is accumulated on day 1 after irradiation could not be degraded due to the lack of plasmin, and fibrin fragments therefore could not be formed. Together with the lack of TGF-β induction and activation in plg−/− mice, this might explain the lack of an inflammatory response and the absence of radiodermatitis in these mice after irradiation.

Inhibiting plasminogen activation by TXA (star) prevents the induction of the burst of inflammatory reaction
TGF-β is a primary factor involved in the tissue damage resulting from irradiation, and several strategies have been tested to target TGF-β signaling as a way to prevent tissue injury24, but such treatments have been unsuccessful due to numerous side effects. Our study has for the first time linked plasminogen activation to TGF-β expression, suggesting that inhibition of plasminogen might be an upstream event that can be used to suppress TGF-β activation for the prevention of radiodermatitis. Taking into account the necessity of plasmin for the development of radiodermatitis, we propose a novel preventive therapy using clinically approved lysine analogs (such as TXA or ɛ-aminocaproid acid) to inhibit plasminogen activation and thereby prevent or reduce the development of radiodermatitis in cancer patients undergoing radiotherapy. It is possible that the same strategy would also protect internal organs from radiation injury.
The molecular mechanisms for how plasminogen triggers inflammation remain an intriguing question that should be addressed in future studies. Answers to this question might lead to a better understanding of the downstream cascades of plasmin-mediated radiodermatitis, which might in turn lead to the development of novel methods for preventing radiodermatitis in cancer radiotherapy.
References
Ryan, J. L. Ionizing radiation: the good, the bad, and the ugly. J. Invest. Dermatol. 132, 985–993 (2012).
Singh, M., Alavi, A., Wong, R. & Akita, S. Radiodermatitis: a review of our current understanding. Am. J. Clin. Dermatol. 17, 277–292 (2016).
Dormand, E. L., Banwell, P. E. & Goodacre, T. E. Radiotherapy and wound healing. Int. Wound J. 2, 112–127 (2005).
Mendelsohn, F. A., Divino, C. M., Reis, E. D. & Kerstein, M. D. Wound care after radiation therapy. Adv. Skin. Wound Care. 15, 216–224 (2002).
Hubenak, J. R., Zhang, Q., Branch, C. D. & Kronowitz, S. J. Mechanisms of injury to normal tissue after radiotherapy: a review. Plast. Reconstr. Surg. 133, 49e–56e (2014).
Denham, J. W. & Hauer-Jensen, M. The radiotherapeutic injury--a complex ‘wound’. Radiother. Oncol. 63, 129–145 (2002).
Tibbs, M. K. Wound healing following radiation therapy: a review. Radiother. Oncol. 42, 99–106 (1997).
Lorimore, S. A., Coates, P. J., Scobie, G. E., Milne, G. & Wright, E. G. Inflammatory-type responses after exposure to ionizing radiation in vivo: a mechanism for radiation-induced bystander effects? Oncogene 20, 7085–7095 (2001).
Syrovets, T., Lunov, O. & Simmet, T. Plasmin as a proinflammatory cell activator. J. Leukoc. Biol. 92, 509–519 (2012).
Collen D. Ham-Wasserman lecture: role of the plasminogen system in fibrin-homeostasis and tissue remodeling. Hematology Am. Soc. Hematol. Educ. Program 2001,1-9 (2001).
Syrovets, T. & Simmet, T. Novel aspects and new roles for the serine protease plasmin. Cell. Mol. LIfe Sci. 61, 873–885 (2004).
Shen, Y. et al. Plasminogen is a key proinflammatory regulator that accelerates the healing of acute and diabetic wounds. Blood 119, 5879–5887 (2012).
Sulniute, R. et al. Plasminogen is a critical regulator of cutaneous wound healing. Thromb. Haemost. 115, 1001–1009 (2016).
Guo, Y., Li, J., Hagstrom, E. & Ny, T. Beneficial and detrimental effects of plasmin(ogen) during infection and sepsis in mice. PLoS One 6, e24774 (2011).
Ploplis, V. A. et al. Effects of disruption of the plasminogen gene on thrombosis, growth, and health in mice. Circulation 92, 2585–2593 (1995).
Ny, A. et al. Ovulation in plasminogen-deficient mice. Endocrinology 140, 5030–5035 (1999).
Carmeliet, P. et al. Physiological consequences of loss of plasminogen-activator gene-function in mice. Nature 368, 419–424 (1994).
Ny, A., Nordstrom, L., Carmeliet, P. & Ny, T. Studies of mice lacking plasminogen activator gene function suggest that plasmin production prior to ovulation exceeds the amount needed for optimal ovulation efficiency. Eur. J. Biochem. 244, 487–493 (1997).
Kim, J. H., Jenrow, K. A. & Brown, S. L. Mechanisms of radiation-induced normal tissue toxicity and implications for future clinical trials. Radiat. Oncol. J. 32, 103–115 (2014).
Borges, H. L., Linden, R. & Wang, J. Y. DNA damage-induced cell death: lessons from the central nervous system. Cell Res. 18, 17–26 (2008).
Robbins, M. E. & Zhao, W. Chronic oxidative stress and radiation-induced late normal tissue injury: a review. Int. J. Radiat. Biol. 80, 251–259 (2004).
Kasai, H. Analysis of a form of oxidative DNA damage, 8-hydroxy-2’-deoxyguanosine, as a marker of cellular oxidative stress during carcinogenesis. Mutat. Res. 387, 147–163 (1997).
Wong, S. L. et al. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat. Med. 21, 815–819 (2015).
Kim, J. H., Kolozsvary, A. J., Jenrow, K. A. & Brown, S. L. Mechanisms of radiation-induced skin injury and implications for future clinical trials. Int. J. Radiat. Biol. 89, 311–318 (2013).
Deng, W. et al. Mesenchymal stem cells promote CD206 expression and phagocytic activity of macrophages through IL-6 in systemic lupus erythematosus. Clin. Immunol. 161, 209–216 (2015).
Van Der Meeren, A., Squiban, C., Gourmelon, P., Lafont, H. & Gaugler, M. H. Differential regulation by IL-4 and IL-10 of radiation-induced IL-6 and IL-8 production and ICAM-1 expression by human endothelial cells. Cytokine 11, 831–838 (1999).
Powell, M. J., Thompson, S. A., Tone, Y., Waldmann, H. & Tone, M. Posttranscriptional regulation of IL-10 gene expression through sequences in the 3’-untranslated region. J. Immunol. 165, 292–296 (2000).
Doeuvre, L., Plawinski, L., Goux, D., Vivien, D. & Angles-Cano, E. Plasmin on adherent cells: from microvesiculation to apoptosis. Biochem. J. 432, 365–373 (2010).
Milliat, F. et al. Essential role of plasminogen activator inhibitor type-1 in radiation enteropathy. Am. J. Pathol. 172, 691–701 (2008).
Czekay, R. P. & Loskutoff, D. J. Plasminogen activator inhibitors regulate cell adhesion through a uPAR-dependent mechanism. J. Cell. Physiol. 220, 655–663 (2009).
Flaumenhaft, R., Abe, M., Mignatti, P. & Rifkin, D. B. Basic fibroblast growth factor-induced activation of latent transforming growth factor beta in endothelial cells: regulation of plasminogen activator activity. J. Cell. Biol. 118, 901–909 (1992).
Mittal, M., Siddiqui, M. R., Tran, K., Reddy, S. P. & Malik, A. B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 20, 1126–1167 (2014).
Cesarman-Maus, G. & Hajjar, K. A. Molecular mechanisms of fibrinolysis. Br. J. Haematol. 129, 307–321 (2005).
Mannucci, P. M. & Levi, M. Prevention and treatment of major blood loss. N. Eng. J. Med. 356, 2301–2311 (2007).
Senior, R. M., Skogen, W. F., Griffin, G. L. & Wilner, G. D. Effects of fibrinogen derivatives upon the inflammatory response - studies with human fibrinopeptide-B. J. Clin. Invest. 77, 1014–1019 (1986).
Richardson, D. L., Pepper, D. S. & Kay, A. B. Chemotaxis for human monocytes by fibrinogen-derived peptides. Br. J. Haematol. 32, 507–513 (1976).
Miles, L. A., Plow, E. F., Waisman, D. M. & Parmer, R. J. Plasminogen receptors. J. Biomed. Biotechnol. 2012, 130735 (2012).
Annes, J. P., Munger, J. S. & Rifkin, D. B. Making sense of latent TGFbeta activation. J. Cell. Sci. 116(Pt 2), 217–224 (2003).
Khalil, N., Corne, S., Whitman, C. & Yacyshyn, H. Plasmin regulates the activation of cell-associated latent TGF-beta 1 secreted by rat alveolar macrophages after in vivo bleomycin injury. Am. J. Respir. Cell Mol. Biol. 15, 252–259 (1996).
Barcellos-Hoff, M. H. How do tissues respond to damage at the cellular level? The role of cytokines in irradiated tissues. Radiat. Res. 150(5 Suppl.), S109–S120 (1998).
Acknowledgements
This study was supported by The Swedish Cancer Society, The Cancer Research Foundation in Northern Sweden, and the Medical Faculty of Umeå University. The authors thank Åsa Larefalk, Maria Ny, Aliyeh Moghadam, and Igor Iashchishyn for discussion and technical support.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
M.F., Y.S., M.B., M.J., M.W., and T.N. have filed a patent application based on some of the findings in this manuscript.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by I. Amelio
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fallah, M., Shen, Y., Brodén, J. et al. Plasminogen activation is required for the development of radiation-induced dermatitis. Cell Death Dis 9, 1051 (2018). https://doi.org/10.1038/s41419-018-1106-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-018-1106-8
This article is cited by
-
Green cocoon-derived sericin reduces cellular damage caused by radiation in human keratinocytes
Scientific Reports (2024)
-
Plasminogen is a master regulator and a potential drug candidate for the healing of radiation wounds
Cell Death & Disease (2020)
