
Abstract
USP22, the deubiquitinating subunit of the SAGA transcriptional cofactor complex, is a member of an 11-gene “death-from-cancer” signature. USP22 has been considered an attractive therapeutic target since high levels of its expression were associated with distant metastasis, poor survival, and high recurrence rates in a wide variety of solid tumors, including colorectal cancer (CRC). We sought to investigate the role of Usp22 during tumorigenesis in vivo using a mouse model for intestinal carcinogenesis with a tissue-specific Usp22 ablation. In addition, we assessed the effects of USP22 depletion in human CRC cells on tumorigenic potential and identified underlying molecular mechanisms. For the first time, we report that USP22 has an unexpected tumor-suppressive function in vivo. Intriguingly, intestine-specific Usp22 deletion exacerbated the tumor phenotype caused by Apc mutation, resulting in significantly decreased survival and higher intestinal tumor incidence. Accordingly, human CRC cells showed increased tumorigenic properties upon USP22 reduction in vitro and in vivo and induced gene expression signatures associated with an unfavorable outcome in CRC patients. Notably, USP22 loss resulted in increased mTOR activity with the tumorigenic properties elicited by the loss of USP22 being reversible by mTOR inhibitor treatment in vitro and in vivo. Here, we demonstrate that USP22 can exert tumor-suppressive functions in CRC where its loss increases CRC burden by modulating mTOR activity. Importantly, our data uncover a tumor- and context-specific role of USP22, suggesting that USP22 expression could serve as a marker for therapeutic stratification of cancer patients.
Similar content being viewed by others


Introduction
Due to early diagnosis by colonoscopy, the mortality rate of colorectal cancer (CRC) has decreased over the last decades [1, 2]. However, disease recurrence and metastasis remain major challenges, making CRC a major medical and economic problem due to its high prevalence. Glinsky et al. identified an 11-gene so-called death-from-cancer gene expression signature displaying a “stem cell-like” profile, which defined a patient subgroup characterized by distant metastasis, poor survival, and high recurrence rates of human malignancies. One of the genes contained within this signature is the Ubiquitin-Specific Protease 22 (USP22) [3,4,5]. USP22 is a ubiquitin hydrolase which, together with Ataxin 7 (ATXN7), Ataxin 7-Like 3 (ATXN7L3), and Enhancer of Yellow 2 homolog (ENY2), constitute the deubiquitinating module of the SAGA (Spt-Ada-Gcn5 acetyltransferase) transcriptional cofactor complex [6]. Within this complex USP22 removes ubiquitin from the core histones H2B and H2A. While H2B monoubiquitination at lysine 120 (H2Bub1) is associated with active gene expression, the monoubiquitination of H2A is associated with gene silencing [6, 7]. Recently, it was demonstrated that several functions of USP22 can be taken over by its paralogs USP27X and USP51. For instance, USP22, USP27X, and USP51 compete for interaction with ATXN7L3 and ENY2 in order to regulate H2Bub1 levels [8]. In addition, USP22 deubiquitinates and thereby stabilizes several nonhistone substrates, including various key players in the development and progression of human malignancies such as Sirtuin 1 [9, 10], Mouse Double Minute 4 [11], MYC [7], and Nuclear Factor of Activated T cells 2 [12]. In addition, the removal of monoubiquitination from H2B is a further mechanism whereby USP22 may function during tumorigenesis. In fact, H2Bub1 was described as a tumor-suppressive mark in CRC because it was demonstrated that decreased H2Bub1 levels were associated with advanced tumor grade and poor patient survival [13]. Accordingly, several studies, based either on mRNA levels or immunohistochemical (IHC) analyses, reported that increased USP22 expression was correlated with poor prognosis in CRC patients [14,15,16,17]. For instance, USP22 protein levels were evaluated using IHC in CRC biopsies and patients with positive USP22 staining showed lower 5-year disease-free survival rates (25.8%) than individuals with negative staining (70.5%) [16]. Based on these findings, USP22 has been suggested to represent an attractive therapeutic target in CRC [18].
To date the role of USP22 in CRC was mainly addressed by IHC analysis of human tumor samples and in vitro using CRC cell lines. However, few studies have investigated the function of USP22 in vivo, since the complete ablation of the Usp22 gene results in early embryonic lethality at E10.5 of the post-implantation stage in mice [10] most likely due to defective placental development [19]. Previously, we overcame the embryonic lethal effect of a complete loss of this gene by generating a global Usp22 hypomorphic mouse line. Notably, while Usp22 loss did not influence the gross intestinal morphology, cell differentiation and lineage specification were affected in the small intestine. Here, we aimed to build upon these findings and investigate the effect of intestinal Usp22 deletion in a genetic mouse model of CRC. In addition, we sought to experimentally test the recently discussed concept that USP22 might exert context-dependent functions during tumorigenesis [20]. Intriguingly, we demonstrate that USP22 exerts a biological context-dependent tumor-suppressive function in CRC in vitro and in vivo.
Materials and methods
Generation of mice and genotyping
All animal work was performed in accordance with the institutional regulations for care and use of laboratory animals and approved by the Lower Saxony State Office for Consumer Protection and Food Safety (registration numbers: G11/540). The mouse line containing an Adenomatous Polyposis Coli (Apc) mutation (Apc1638N/+) was a kind gift from F. Bosman (Erasmus University Medical Center Rotterdam, The Netherlands) and genotyped as described [21]. The mouse line harboring a conditional Usp22 allele was generated based on a mouse line we previously described [21]. Briefly, animals with a global Usp22 reduction were generated using a “knockout first” embryonic stem cell line (Usp22tm1a(KOMP)Wtsi C57BL6) obtained from the University of California-Davis Knockout Mouse Project Repository. To generate mice with a tissue-specific, conditional loss of Usp22, the lacZ, and neomycin resistance cassettes were removed by FLP-mediated excision. Usp22loxP mice were crossed with Villin-CreERT2 and Apc1638N mouse lines (in a C57BL/6N background) to achieve an intestinal knockout and to investigate its role in tumorigenesis. Numbers of Apc wild type experimental mice analyzed throughout the study were n = 15 (Usp22wt/wt), n = 14 (Usp22wt/fl), n = 8 (Usp22fl/fl) and of Apc1638N/+ mice n = 9 (Usp22wt/wt), n = 8 (Usp22wt/fl), n = 7 (Usp22fl/fl). Genotyping was performed by preheating PCR samples to 95 °C for 3 min. The respective DNA fragments were amplified for 35–40 cycles and final elongation took place at 72 °C for 10 min. The respective programs utilized for Usp22 were: 95 °C for 30 s, 67 °C for 30 s, 72 °C for 1 min (35 cycles); Apc1638N: 95 °C for 30 s, 58 °C for 30 s, 72 °C for 1 min (40 cycles); and for Villin-CreERT2: 95 °C for 15 s, 56 °C for 15 s, 72 °C for 30 s (35 cycles). Primers for genotyping are listed in Table S1.
Tamoxifen treatment of mice
To induce the Villin-CreERT2-mediated intestinal knockout of Usp22, mice were intraperitoneally injected with a dose of 1 mg tamoxifen per day for 5 consecutive days at the age of 4 weeks. For this purpose, 5% tamoxifen (w/v) was dissolved in 100% ethanol. This solution was mixed 1:10 with autoclaved sunflower oil directly prior to injection.
Xenograft experiments
To monitor growth behavior of human CRC cells deficient for USP22 in vivo, HCT116 cells with a CRISPR/Cas9-mediated USP22 knockout were analyzed in a xenograft experiment (registration number: 15/2039). For each animal, 1 million tumor cells in a total volume of 25 μl of a 1:1 mixture of McCoy’s medium and BD Matrigel Matrix High Concentration, Growth Factor Reduced (BD Bioscience) were stored on ice until transplantation. To compare the growth pattern of HCT116 USP22 wild type and knockout cells, 3–4-month-old SCID mice were analyzed. To test the effect of everolimus treatments in HCT116 USP22 wild type and knockout tumors, 8-week-old NMRIFoxn1nu/Foxn1nu (Janvier) mice were analyzed. Animals were anesthetized by isoflurane inhalation (2–3%, forene) and cell suspensions were injected under sterile conditions subcutaneously with a 0.3 ml Micro-Fine syringe (BD Bioscience) into the abdominal flanks. After tumors were palpable, mouse weight and tumor size were measured daily. Everolimus treatment was performed as described earlier [22]. Briefly, 10 mM everolimus (Selleckchem) dissolved in DMSO was stored at −20 °C and added dropwise to 0.5% Tween-80 in PBS prior to injection. Animals received 10 mg/kg everolimus daily intraperitoneally for 14 days.
Histology
Hematoxylin and eosin (H&E) and IHC staining were performed as previously described [23]. Briefly, intestine sections with 5 µm thickness were de-paraffinized in xylol for 20 min and rehydrated in ethanol (100, 90, and 70%) for 5 min each. For H&E staining, slides were rinsed with water, nuclei were stained in Mayer’s hematoxylin solution for 1 min and slides were washed under running tap water for 5 min. Counterstaining was performed using eosin for 5–10 min. For IHC, rehydrated sections were boiled in 10 mM citric acid buffer. Sections were quenched for endogenous peroxidases with 5% H2O2 in PBS and blocked with 10% fetal bovine serum (FBS) in PBS. Primary antibodies (Table S2) were diluted in 5% FBS in PBS and incubated in a humidified chamber overnight at 4 °C. Sections were incubated with biotinylated secondary antibodies (1:200) and ExtrAvidin-Peroxidase (Sigma Aldrich; 1:1000) each for 1 h. Staining was developed using 3,3′-diaminobenzidin-tetrahydrochloride and counterstaining was performed using hematoxylin. Slides were washed, dehydrated in ethanol (70, 90, and 100%), incubated in xylol and mounted.
Determination of Histo-score (H-score)
Epithelial damage and the rate of lymphocyte infiltration were scored as previously described: normal intestinal epithelium (0), mild lymphocyte infiltration pushing crypts apart from each other (1), lymphocyte accumulations destroying crypt structure (2), no epithelial lining on top of the lymphocyte accumulation (3). The percentage of damaged tissue was multiplied by the respective score (0–3) and the sum was divided by the maximum H-score of 30 [24].
Cell culture and CRISPR/Cas9-mediated deletion of USP22
Human HCT116 CRC cells were grown in McCoy’s medium supplemented with 10% FBS, 100 units/ml penicillin and 100 μg/ml streptomycin at 37 °C and 5% CO2. siRNA (GE Dharmacon siGENOME; Table S3) transfections were performed using Lipofectamine® RNAiMAX (Invitrogen) according to the manufacturer’s instructions. To achieve a permanent CRISPR/Cas9-mediated deletion of USP22 in HCT116 cells, 400,000 cells were seeded per well of a six-well plate. Single guide RNAs (sgRNAs) targeting USP22 were designed using the E-CRISP algorithm from the German Cancer Research Center (http://www.e-crisp.org); sgRNA1: 5′-CACCGGTGTTTGGCAGCTCATGCCC-3′, sgRNA2: 5′-CACCGTTAGAGAGACCTGGCGGTGG-3′. The selected guide RNA sequences were then cloned into the pSpCas9(BB)-2A-GFP (PX458, Addgene) vector. HCT116 cells were transfected with the plasmid containing Cas9 and GFP sequences as well as a sgRNA using Lipofectamine® 2000 according to the manufacturer’s instructions. After 24 h, cells were washed with PBS and medium supplemented with antibiotics was added. After 48 h of transfection, the cells were sorted based on their fluorescence intensity via fluorescence-activated cell sorting at the Cell Sorting Core Facility, Department of Hematology and Medical Oncology, University Medical Center Göttingen. Single highly fluorescent cells were sorted into the wells of a 96-well plate. Single cell clones were cultivated and propagated. The loss of USP22 was confirmed by western blot and qRT-PCR. To test the correlation between USP22 and mammalian target of rapamycin (mTOR) activity, cells were treated with 100 nM everolimus (Selleckchem) for 24 h or 100 nM rapamycin (Calbiochem) for 6 h. As a negative control, DMSO was added to the cells.
Cell characterization assays
HCT116 cells were seeded for characterization assays 24 h post siRNA transfection. To assess proliferation rates, 2000 cells were seeded onto 96-well assay plates (Corning Life Sciences) and cell confluence was measured every 24 h using a Celigo® S Adherent Cell Cytometer (Nexcelom Bioscience LLC). Clonogenic properties were evaluated by seeding 500 cells onto a six-well plate and staining colonies with crystal violet after 3–4 days. Anchorage-independent growth was examined using a soft agar colony formation assay. For this purpose, a base layer of 0.8% agarose in complete cell culture medium was poured into six-well plates and 10,000 cells resuspended in 0.48% agarose in medium were added into each well. After covering with complete culture medium, cells were maintained at 37 °C with 5% CO2 and stained once the colonies were macroscopically visible. The migration potential of cells was assessed using a transwell migration assay as previously described [25]. Well areas covered by cells were quantitated using FIJI.
Western blotting and qRT-PCR
Protein isolation, western blot analysis, RNA extraction, reverse transcription, and qRT-PCR were performed as previously described [26]. The primary antibodies for western blot and primers for qRT-PCR are listed in Supplementary Tables S2 and S4, respectively.
Chromatin immunoprecipitation (ChIP)
To assess PRKAA2 occupancy by H3K9ac, HCT116 wild type and USP22 knockout cells were fixed in 1% formaldehyde in PBS for 20 min and quenched with 125 mM glycine for 5 min. Cells were washed twice with ice-cold PBS, scraped and washed with Nelson Buffer (150 mM NaCl, 20 mM EDTA (pH 8.0), 50 mM Tris/HCl (pH 7.5), 0.5% (v/v) NP-40, 1% Triton-X-100, 20 mM NaF and protease inhibitors). The nuclear pellet was lysed using Lysis Buffer (20 mM EDTA, 150 mM NaCl, 1% (v/v) NP-40, 0.1% (w/v) SDS, 0.5% (w/v) sodium deoxycholate, 20 mM NaF, 50 mM Tris/HCl (pH 8.0), and protease inhibitors) and sonicated for 15 cycles (30 s on/off). Samples were precleared with 50% Sepharose 4B (GE Healthcare) and incubated with the respective antibodies overnight (H3K9ac,C15410004, Diagenode; IgG, C15410206, Diagenode). Sepharose beads with Protein A (GE Healthcare) were added and incubated at 4 °C for 2 h and immune complexes were washed twice with Wash Buffer (20 mM EDTA, 500 mM LiCl, 1% (v/v) NP-40, 20 mM NaF, 1% (w/v) sodium deoxycholate, 100 mM Tris/HCl (pH 8.5)) and twice with TE buffer. Next, samples were incubated with RNAse A (Qiagen) in 10 mM Tris/HCl (pH 8.0) at 37 °C for 30 min. For de-crosslinking, samples were incubated in 20 mM EDTA, 2% SDS, 100 mM Tris/HCl (pH 8), and 20 μg Proteinase K at 65 °C overnight. Samples were extracted with Roti® phenol/chloroform/isoamyl alcohol (Roth) and DNA was precipitated with ethanol. The corresponding input samples were used as controls and primer sequences for subsequent qRT-PCR are listed in Table S5.
Library preparation and next-generation sequencing
To evaluate transcriptome-wide effects of USP22 loss by mRNA-seq, HCT116 cells were transfected in triplicate with nontargeting control or USP22 siRNAs. In parallel, HCT116 wild type and USP22 knockout cells were analyzed. RNA integrity was verified on an agarose gel and libraries were prepared using the TruSeq RNA Library Preparation Kit v2 (Illumina®). Library insert size and quality were estimated using an Agilent Bioanalyzer 2100 and mRNA-seq of pooled libraries was performed at the Transcriptome and Genome Analysis Laboratory, Göttingen, using HiSeq 4000 (Illumina®). Bcl output files (Basecaller software, Illumina®) were demultiplexed to fastq files using CASAVA v1.8.2. Quality control of fastq files was performed using FastQC and reads were mapped to the human reference genome (GRCh38.p10) using HISAT2 (version 2.0.3.3) on Galaxy. Read count files were generated using the featureCounts tool (version 1.4.6.p5) and analyzed for differential gene expression analysis using DESeq2 (version 2.11.39). All mRNA-seq data have been deposited at ArrayExpress (http://www.ebi.ac.uk/arrayexpress, accession number: E-MTAB-7393). Significantly differentially regulated genes were selected based on the Benjamini–Hochberg adjusted p-value ≤ 0.05. Gene set enrichment analysis (GSEA) [27, 28] was performed using default settings (1000 permutations for gene sets, Signal2Noise metric for ranking genes). Custom GSEA gene sets with genes significantly regulated after rapamycin treatment were obtained from a previous study in HCT116 cells [29] and genes associated with CRC patient prognosis were obtained from the Human Protein Atlas website [30].
Statistical analyses
All graphs have been prepared with GraphPad Prism version 5.04 (GraphPad Software, Inc.). Statistical analysis was performed using Student’s t-test or one-way analysis of variants and Tukey Post hoc test (α = 0.05).
Results
USP22 suppresses CRC tumorigenesis in vivo
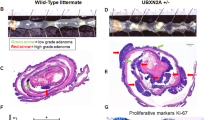
Based on the hypothesis that high Usp22 expression elevates tumor burden, we examined the effects of a tissue-specific, conditional Usp22 genetic deletion in a murine model for intestinal tumorigenesis. For this purpose, we generated mice with a conditional Villin-CreERT2-mediated intestinal Usp22 deletion in the presence or absence of a heterozygous Apc1638N truncation mutation. The intestinal loss of Usp22 alone only marginally affected the overall survival of mice (Fig. 1a). Surprisingly, in the background of an Apc mutation, Usp22 loss significantly reduced survival rates where Apc1638N/+, Usp22fl/fl mice died 14–33 weeks after tamoxifen injection, while wild type (aged 22–64 weeks) or Usp22 heterozygous (27–71 weeks) animals survived significantly longer. In addition, intestinal-specific Usp22 deletion was also associated with a higher weight loss after tamoxifen injection (Fig. 1b) and shorter colons (Fig. 1c), as frequently observed in inflammatory bowel disease patients [31]. Intriguingly, in Apc1638N/+, Usp22fl/fl mice the number (Fig. 1d) as well as the size (Fig. 1e) of colorectal tumors was significantly increased compared with litter mates. The reduction of USP22 was verified in normal, healthy intestinal epithelium and in tumor material at the protein level (Fig. 1f, g, respectively) as well as in intestinal epithelium at the mRNA level (Fig. 1h). In summary, loss of Usp22 in the intestine resulted in reduced survival and significantly increased tumor burden. These findings reveal previously unknown tumor-suppressive functions of USP22 in solid tumors in vivo.

USP22 exerts tumor-suppressive functions in colorectal cancer in vivo. The consequences of an intestinal-specific deletion of Usp22 were tested in Apc wild type (Usp22wt/wt, n = 15; Usp22wt/fl, n = 14; Usp22fl/fl, n = 8) and Apc1638N/+ mice type (Usp22wt/wt, n = 9; Usp22wt/fl, n = 8; Usp22fl/fl, n = 7). a In Villin-CreERT2 mice carrying an Apc1638N/+ mutation, the conditional Usp22 deletion led to reduced overall survival rates. b The intestinal deletion of Usp22 resulted in increased weight loss and c reduced colon length. Mean ± SEM, one-way ANOVA. Villin-CreERT2, Apc1638N/+, Usp22fl/fl mice displayed elevated tumor number (d) and size (e) in the colon. Mean ± SEM, one-way ANOVA; box plot shows 5–95 percentile. The reduction of USP22 at the protein level was confirmed in healthy intestinal epithelium (f) and tumor (g), and at the mRNA level (h) in healthy intestinal epithelium. Mean ± SEM, one-way ANOVA
Intestinal deletion of Usp22 increases aggressive tumor growth in vivo
When assessing H&E-stained colon sections, we identified several tumor areas spreading beyond the inner muscular lining of the colon exclusively in Usp22fl/fl animals (Fig. 2a, b), indicating that the deletion of Usp22 in Apc-mutated mice not only increased tumor number and size in the colon, but also unexpectedly enabled aggressive tumor growth. In addition, mice with an intestinal-specific Usp22 deletion displayed epithelial damage in the colon. This effect was quantitated by determining the H-Score, reflecting normal epithelium (0), as well as segments with mild (1), medium (2), or severe (3) damage (Fig. 2c). Intriguingly, in contrast to wild-type animals, both Usp22+/fl and Usp22fl/fl mice showed a lower degree of intact epithelium (Fig. 2d, e). To evaluate the underlying mechanisms of enhanced tumor growth following Usp22 deletion, we stained colon sections of Apc1638N/+ mice with Ki67 and CD31 as markers for proliferating and endothelial cells, respectively (Fig. 2f). Indeed, both cell proliferation and angiogenesis were elevated in Usp22fl/fl animals. These findings suggest that not only tumor number and size, but also tumor aggressiveness is elevated after Usp22 loss.

Reduced USP22 expression elevates aggressive colorectal tumor growth. a Colorectal tumors of Villin-CreERT2, Apc1638N/+, Usp22fl/fl displaying aggressive growth. Scale bar: 100 µm. b Approximately 5% of tumors found in Usp22fl/fl mice and 16% of cancer lesions in Apc1638N/+, Usp22fl/fl mice showed spreading beyond the inner muscular lining. Mean ± SEM. c Scoring system to assess no (0), mild (1), medium (2), or severe (3) epithelial damage. Scale bar: 100 µm. Animals with reduced intestinal Usp22 expression showed a higher degree of epithelial damage (d) and, therefore, a higher H-Score (e). Mean ± SEM, one-way ANOVA. f Immunohistochemistry for Ki67 and CD31 was performed on colon sections isolated from Apc1638N/+ mice. The loss of Usp22 was associated with higher tumor proliferation and vascularization. Scale bar: 100 µm. Mean ± SEM, t-test
USP22 reduction increases tumorigenic potential of human CRC cells in vitro
In order to further investigate USP22 function in CRC cells, we examined the effects of USP22 depletion on cell growth characteristics in HCT116 cells in vitro. In support of our in vivo findings, we observed that the siRNA-mediated knockdown of USP22 (siUSP22) accelerates the proliferation of HCT116 colorectal carcinoma cells compared with controls (siControl; Fig. 3a). In addition, the migratory properties, anchorage-independent growth and clonogenic potential were all elevated after the depletion of USP22 (Fig. 3b). Consistent with both our in vivo and in vitro results, analysis of human patient tumor gene expression data generated by the TCGA Research Network (http://cancergenome.nih.gov/) demonstrated that low USP22 expression tends to correlate with poor prognosis in rectal adenocarcinoma (Fig. 3c). Thus, we sought to determine how decreased USP22 levels affect global gene expression and signaling pathways. For this purpose, we performed mRNA-seq analyses in control siRNA transfected and USP22-depleted HCT116 cells. Remarkably, using custom gene sets associated with favorable and unfavorable outcome in CRC [27, 28], GSEA revealed that gene expression patterns associated with an unfavorable outcome in CRC patients were highly upregulated while genes associated with a favorable outcome were downregulated following USP22 depletion (Fig. 3d). These data clearly indicate that low USP22 expression promotes tumorigenic properties of CRC cells in vitro and affects gene expression patterns associated with patient prognosis.

USP22 loss elevates tumorigenic features of human CRC cells and its transcriptome-wide effects correlate with poor prognosis. The knockdown of USP22 in HCT116 cells (n = 3) enhanced proliferation (a), as well as cell migration, anchorage-independent growth and clonogenic potential (b) in three independent experiments. Scale bar: 1 mm. Mean ± SEM, t-test. c Publically available TCGA data were evaluated for USP22-associated survival of rectal cancer patients, indicating poor prognosis in association with low USP22 expression. d mRNA-seq was performed to evaluate the transcriptome-wide effect of USP22 depletion in HCT116 cells. Gene sets were generated based on genes associated with favorable (upper panel) and unfavorable (lower panel) outcome in CRC as published on the Human Protein Atlas website [27, 28, 30]. GSEA revealed that genes associated with an unfavorable prognosis are enriched in USP22-depleted cells
USP22 deletion enhances tumorigenic potential in vitro and in vivo
To assess the consequences of a permanent deletion of USP22 in CRC cells, we took advantage of the CRISPR/Cas9 gene editing technology. We simultaneously transfected HCT116 cells with two sgRNAs targeting intronic sequences flanking the region containing exons 3–5 of USP22, thereby resulting in the excision of this segment (Fig. 4a). This generated a frameshift and loss of the USP22 protein (Fig. 4b). While the knockout of USP22 in two individual clones (#1 and #2) did not affect cell morphology (Fig. 4c), it enhanced proliferation, similar to siRNA transfection (Fig. 4d). We next examined the effects of USP22 deletion on in vivo tumor growth by injecting these cells subcutaneously into immune-deficient mice. Consistent with the in vitro analyses and genetic deletion models, USP22 knockout accelerated HCT116 tumor growth in this xenograft model (Fig. 4e, f), though effects were less pronounced compared with the Apc-driven tumor mouse model. Consistent with the conditional genetic deletion studies, USP22-deficient HCT116 tumors showed increased vascularization (Fig. 4g, h). Together, these data support that loss of USP22 promotes a more aggressive growth of CRC cells.

A CRISPR/Cas9-mediated USP22 knockout enhances CRC cell growth in vitro and in vivo. a HCT116 cells were transfected with two sgRNAs targeting USP22 in a Cas9- and GFP-containing vector. To allow selection, the construct contained a GFP-reporter. These sgRNAs target intronic regions upstream of exon 3 and downstream of exon 5 of the USP22 gene and thereby generate a frameshift of any resulting spliced mRNA transcript and thereby a complete loss of function. b The loss of USP22 at the protein level was confirmed in CRISPR/Cas9 clones #1 and #2. c, d The permanent deletion of USP22 did not affect the morphology of HCT116 cells (c; scale bar: 100 µm) but did result in increased proliferation (d). Mean ± SEM. e, f Parental and USP22−/− HCT116 cells were injected subcutaneously into immunodeficient mice and the loss of USP22 resulted in increased tumor size. Scale bar: 0.5 mm. g, h Immunohistochemistry was performed for CD31 to compare vascularization in USP22+/+ and USP22−/− tumors (g). Scale bar: 100 µm. In the xenograft approach, the loss of USP22 in HCT116 cells increased the relative number of vessels per tumor (h). Mean ± SEM, t-test
USP22 deficiency promotes mTOR activation
A deeper analysis of our mRNA-seq data revealed that several mTOR signaling-associated genes were regulated following USP22 depletion in HCT116 cells. The mTOR pathway is an attractive therapeutic target since it affects cell growth, proliferation, and survival in numerous human malignancies including CRC [32]. To assess the effect of USP22 loss on mTOR-associated genes in more detail, we generated a list of genes downregulated by the mTOR inhibitor rapamycin in HCT116 cells [29] and performed GSEA [27]. Indeed, this analysis revealed that genes whose expression normally decrease after rapamycin treatment were enriched in USP22 knockdown cells (Fig. 5a), suggesting that reduced USP22 expression may promote the activity of the mTOR pathway. Genes controlled by mTOR signaling and upregulated after USP22 reduction included Interleukin 20 Receptor subunit beta (IL20RB), Phosphoglycerate Dehydrogenase (PHGDH), and Discs Large Homolog 4 (DLG4; Fig. 5b). Indeed, using qRT-PCR we verified that USP22 knockout increases the mRNA expression of these factors and that this effect can be rescued by treating cells with the clinically approved mTOR inhibitor (mTORi) everolimus (Fig. 5c). We next sought to determine the mechanism by which mTOR pathway activity was activated following depletion of USP22 and identified the PRKAA2 gene, encoding the AMP-activated Protein Kinase-alpha 2 (AMPKα2), as being USP22-dependent (Fig. 5d). The downregulation of Prkaa2 was confirmed in intestinal epithelial cells of Usp22 knockout mice (Fig. 5e). Notably, AMPKα2 was reported to suppress tumor growth via inhibition of mTOR pathway activity [29]. Thus, we hypothesized that loss of USP22 may directly lead to increased mTOR activity via this mechanism involving decreased AMPKα2 expression. Thus, we examined the phosphorylation status of the mTOR target ribosomal protein S6 using western blot and confirmed that S6 phosphorylation was, indeed, increased following USP22 depletion (Fig. 5f). Importantly, this effect could be reversed by treatment with the mTORi everolimus and rapamycin. In order to confirm that mTOR activity is also increased in our in vivo genetic mouse model of intestinal tumorigenesis, we also examined S6 phosphorylation in colorectal tumor lysates from Apc1638N/+, Usp22wt/wt and Usp22fl/fl mice. Indeed, pS6 levels were also increased in Usp22-deficient Apc1638N tumors (Fig. 5g). Next, we sought to investigate how the deficiency of the deubiquitinating protein USP22 reduces PRKAA2 mRNA levels and promotes mTOR activity. We hypothesized these effects may be related to the function of USP22 within the SAGA complex and performed ChIP for H3K9ac, which is mediated by the GCN5 acetyltransferase subunit of SAGA. Consistent with our hypothesis, H3K9ac occupancy on PRKAA2 was 20-fold decreased in cells lacking USP22 (Fig. 5h). Moreover, the knockdown of GCN5 also resulted in decreased PRKAA2 mRNA levels (Fig. 5i, Supplementary Fig. S1E). In summary, our findings indicate, that USP22 deficiency results in reduced GCN5-mediated H3K9ac levels, thereby reducing PRKAA2 mRNA expression and elevating mTOR activity.

USP22 loss increases mTOR activity in CRC cells. a Genes downregulated in rapamycin-treated HCT116 cells [29] were summarized in a gene set and compared with HCT116 USP22 control and knockdown cells using GSEA [27]. Genes downregulated upon rapamycin treatment were enriched in USP22-depleted cells. b A subset of genes downregulated in rapamycin-treated HCT116 cells is highly expressed after USP22 reduction (IL20RB, PHGDH and DLG4). mTOR-related effects were evaluated in vitro in three independent experiments. c As verified using qRT-PCR, IL20RB, PHGDH, and DLG4 expression was induced by USP22 deficiency, while this effect was rescued by everolimus treatment (n = 3). Mean ± SEM, one-way ANOVA. d mRNA levels of PRKAA2 were reduced in HCT116 USP22 knockdown cells (n = 3) as well as in e Villin-CreERT2, Usp22fl/fl mice. Mean ± SEM, t-test. f In HCT116 cells, while mTOR activity was decreased using the mTORi everolimus and rapamycin, reduced USP22 levels resulted in increased mTOR activity which could be reversed by mTORi treatment as reflected by pS6 levels using western blot. g Increased mTOR activity was confirmed at the protein level in colorectal tumor material isolated from Apc-mutated Usp22 knockout mice. h As detected by ChIP-qRT-PCR in two independent experiments, H3K9ac occupancy is significantly reduced on the PRKAA2 gene upon the deletion of USP22. As a control, a H3K9ac-negative site was tested (n = 4). The red dotted lines represent the average ChIP-qPCR signal for the negative control IgG. Mean ± SEM, t-test. i The knockdown of the SAGA complex acetyltransferase subunit GCN5 reduced PRKAA2 mRNA levels (n = 3). Mean ± SEM, t-test
USP22 loss sensitizes colorectal cancer cells towards mTOR inhibitors
To evaluate the targetability of USP22-deficient tumors, we aimed to inhibit the mTOR pathway. Intriguingly, treatment of HCT116 cells with everolimus significantly decreased the proliferation of USP22-depleted cells while only marginally affecting control transfected cells (Fig. 6a). Consistently, the increased clonogenic potential of USP22-depleted cells was also reversed by everolimus treatment (Fig. 6b, c). Moreover, silencing the mTORC1 key component RPTOR similarly reduced the proliferation and clonogenic capacity of USP22 knockout cells (Supplementary Fig. S1A–C) and reversed the upregulation of mTORi-responsive genes (i.e., IL20RB, PHGDH, and DLG4) (Supplementary Fig. S1D). To confirm the sensitivity of USP22-deficient tumors towards mTOR inhibition, HCT116 USP22 wild type and knockout cells were transplanted into immunodeficient mice in a xenograft approach. Once tumors were palpable, animals were treated with everolimus or vehicle solution and tumor size was measured daily. In agreement with our previous observations, USP22 loss promoted tumor growth. However, these effects could be rescued by treating animals with everolimus, whereby USP22-deleted tumors showed 71% reduction in tumor growth following everolimus treatment compared with only 27% reduction in USP22 wild type cells (Fig. 6d–f). These findings suggest that activation of the mTOR pathway after the loss of USP22 in vitro and in vivo introduces a therapeutic vulnerability making USP22-deficient CRC cells particularly sensitive to mTOR inhibition.

USP22 deficiency sensitizes CRC cells towards mTOR inhibition. a mTORi treatment only mildly affected proliferation of parental HCT116 cells, while the growth of USP22 knockdown cells was reduced by everolimus treatment (n = 8). b, c The clonogenic potential of HCT116 USP22-depleted cells was significantly decreased after mTORi treatment (n = 3) as shown by the relative percentage of well area covered by colonies. Scale bar: 1 mm. Mean ± SEM, one-way ANOVA. d Two different clones of USP22+/+ and USP22−/− HCT116 cells were injected subcutaneously into immunodeficient mice and the accelerated growth of USP22 knockout tumors was rescued by everolimus treatment (n = 14–16 tumors per genotype and treatment). Tumor weight (e) and size (f) of USP22+/+ and USP22−/− tumors. Mean ± SEM, one-way ANOVA. Scale bar: 1 cm
Discussion
To date, several studies have reported that USP22 overexpression promotes tumor development and progression, suggesting an oncogenic role of USP22. Therefore, based on the literature, USP22 would represent an attractive target in cancer therapy, including CRC, prompting researchers to seek to generate and optimize USP22 inhibitors [18]. In fact, in tumor biology, USP22 has almost exclusively been studied in the context of its putative tumor-promoting function. Apart from a single recently published article [33], no experimental study investigated the possibility that USP22 could exert tumor-suppressive functions in solid tumors under certain conditions. Importantly, our work supports a highly context-dependent function of USP22 during carcinogenesis and suggests that USP22 function in cancer is not as black and white as previously assumed. We demonstrate that simultaneous Apc mutation and intestinal Usp22 deletion in an in vivo genetic model for CRC resulted in decreased survival and increased colorectal tumor burden. Moreover, Villin-CreERT2, Apc1638N/+, Usp22fl/fl mice even showed aggressive carcinoma growth with tumor cells spreading beyond the inner muscular lining. Since no macroscopically visible metastases were detected, it is possible that these were only pseudocarcinomatous invasions. This unexpected tumor-suppressive function of USP22 was further confirmed in the widely investigated human CRC cell line HCT116 in vitro and in xenograft tumors. Thus, our data suggest that while inhibiting USP22 may be beneficial for some tumor types or subgroups, a better understanding of USP22 in tumor biology will be indispensable for proper patient stratification, as the same treatment could have devastating effects on CRC patients since it may potentially accelerate tumor growth and lead to the formation of invasive carcinomas. Consistent with our data, a very recent KrasG12D/+-based mouse model for myeloid leukemia also implied a tumor-suppressive role of USP22 [33]. Together, the context-dependent specificity of USP22 function in tumorigenesis detected in our studies supports the possibility that some tumors may be sensitive towards USP22 inhibitors while clearly displaying that such inhibitors may have devastating effects in patients with other tumor types.
Our findings demonstrating a tumor-suppressive role of USP22 in CRC may appear at first glance to be at odds with a plethora of publications exclusively describing USP22 as a tumor-promoting factor. Importantly, a large number of the studies demonstrating elevated USP22 levels in cancer patient samples were based on either microarray analyses or IHC staining. However, as recently discussed [20], appropriate controls were missing in several of these studies and some of the antibodies used for the IHC approaches are very likely to be cross-reactive. For example, USP27X was recently shown to function as an alternative deubiquitinating enzyme in the SAGA complex [8]. Notably, the recently reported full-length sequence of USP27X [8] has a very high degree of amino acid identity with USP22 and it is currently unclear whether the antibodies utilized in the reported IHC studies may also detect USP27X.
Surprisingly, our data on colorectal tumorigenesis in vivo strongly suggest that USP22 may exert tumor-promoting or -suppressive functions in carcinogenesis in a context-dependent manner. Therefore, it will be essential to further elucidate the mechanisms underlying the aggressive oncogenic growth in some biological contexts due to reduced USP22 levels. It is crucial to elucidate whether certain tumor subtypes are linked to low USP22 levels and, consequently, to determine which patient subgroups would benefit from USP22 inhibition. One of our central findings was the identification of increased activity of the mTOR pathway following USP22 depletion or deletion. We discovered that H3K9ac occupancy on the PRKAA2 gene was significantly reduced upon USP22 loss, thereby promoting mTOR activity. Thus, it is conceivable that USP22 expression levels could potentially help identify CRC patients (i.e., patients with tumors displaying low USP22 expression levels) which may particularly benefit from mTORi treatment. In fact, in our xenograft experiments we were able to illustrate that the deficiency in USP22 expression sensitized CRC cells towards everolimus treatment. Notably, given the fact that mTOR inhibitors are already approved for various indications and the promising results of these in combinatorial regimens in CRC patients [32] suggests that a more intensive investigation into the interconnection between USP22 and mTOR in CRC is warranted.
Interestingly, the potential tumor-suppressive effects that we observed for USP22 in this study are consistent with previous and recently published findings for the H2B ubiquitin ligase RNF40 in breast and CRC. While RNF40 appears to function as a tumor suppressor in ERα-positive breast cancer [25], its depletion significantly decreased the proliferation of HCT116 cells [24, 26]. Moreover, while intestinal-specific deletion of Rnf40 decreased inflammation following the induction of acute colitis in vivo, our results here suggest that Usp22 deletion may promote a proinflammatory tumor phenotype (as suggested by the increased H-score). Interestingly, the effects of both USP22 and RNF40 are likely to be independent of their function in modulating global H2B monoubiquitination since we could not observe any significant correlation between H2Bub1 levels and CRC patient survival nor could we observe correlations between global H2Bub1 and RNF40 levels in Apc1638N tumors in vivo [24]. Whether the apparently opposing roles of RNF40 and USP22 in CRC are linked remains to be determined, but it is conceivable that they may play competing roles in controlling the ubiquitination and deubiquitination of H2B or some other substrate, thereby promoting or blocking CRC progression, respectively.
Together, our results unexpectedly demonstrate that USP22 cannot strictly be considered as an oncogene, but it can, in fact, function as a tumor suppressor in some biological contexts. Further work will be necessary to determine which tumor types or subgroups will benefit most from inhibition of USP22 activity and, in other cases, whether decreased USP22 activity may be exploited for an individualized therapeutic approach to patient stratification for mTORi treatment.
References
Bosetti C, Levi F, Rosato V, Bertuccio P, Lucchini F, Negri E, et al. Recent trends in colorectal cancer mortality in Europe. Int J Cancer. 2011;129:180–91. https://doi.org/10.1002/ijc.25653
Vleugels JLA, van Lanschot MCJ, Dekker E. Colorectal cancer screening by colonoscopy: putting it into perspective. Dig Endosc. 2016;28:250–9. https://doi.org/10.1111/den.12533
Glinsky GV. Death-from-cancer signatures and stem cell contribution to metastatic cancer. Cell Cycle. 2005;4:1171–5. https://doi.org/10.4161/cc.4.9.2001
Glinsky GV, Berezovska O, Glinskii AB. Microarray analysis identifies a death-from-cancer signature predicting therapy failure in patients with multiple types of cancer. J Clin Investig. 2005;115:1503–21. https://doi.org/10.1172/JCI23412
Wicha MS, Liu S, Dontu G. Cancer stem cells: an old idea–a paradigm shift. Cancer Res. 2006;66:1883–90. https://doi.org/10.1158/0008-5472.CAN-05-3153. discussion 1895-6
Zhang X-Y, Varthi M, Sykes SM, Phillips C, Warzecha C, Zhu W, et al. The putative cancer stem cell marker USP22 is a subunit of the human SAGA complex required for activated transcription and cell-cycle progression. Mol Cell. 2008;29:102–11. https://doi.org/10.1016/j.molcel.2007.12.015
Zhang X-Y, Pfeiffer HK, Thorne AW, McMahon SB. USP22, an hSAGA subunit and potential cancer stem cell marker, reverses the polycomb-catalyzed ubiquitylation of histone H2A. Cell Cycle. 2008;7:1522–4. https://doi.org/10.4161/cc.7.11.5962
Atanassov BS, Mohan RD, Lan X, Kuang X, Lu Y, Lin K, et al. ATXN7L3 and ENY2 coordinate activity of multiple H2B deubiquitinases important for cellular proliferation and tumor growth. Mol Cell. 2016;62:558–71. https://doi.org/10.1016/j.molcel.2016.03.030
Li L, Osdal T, Ho Y, Chun S, McDonald T, Agarwal P, et al. SIRT1 activation by a c-MYC oncogenic network promotes the maintenance and drug resistance of human FLT3-ITD acute myeloid leukemia stem cells. Cell Stem Cell. 2014;15:431–46. https://doi.org/10.1016/j.stem.2014.08.001
Lin Z, Yang H, Kong Q, Li J, Lee S-M, Gao B, et al. USP22 antagonizes p53 transcriptional activation by deubiquitinating Sirt1 to suppress cell apoptosis and is required for mouse embryonic development. Mol Cell. 2012;46:484–94. https://doi.org/10.1016/j.molcel.2012.03.024
Ding F, Bao C, Tian Y, Xiao H, Wang M, Xie X, et al. USP22 promotes NSCLC tumorigenesis via MDMX up-regulation and subsequent p53 inhibition. Int J Mol Sci. 2014;16:307–20. https://doi.org/10.3390/ijms16010307
Gao Y, Lin F, Xu P, Nie J, Chen Z, Su J, et al. USP22 is a positive regulator of NFATc2 on promoting IL2 expression. FEBS Lett. 2014;588:878–83. https://doi.org/10.1016/j.febslet.2014.02.016
Melling N, Grimm N, Simon R, Stahl P, Bokemeyer C, Terracciano L, et al. Loss of H2Bub1 expression is linked to poor prognosis in nodal negative colorectal cancers. Pathol Oncol Res. 2016;22:95–102. https://doi.org/10.1007/s12253-015-9977-9
Liu YL, Yang YM, Xu H, Dong XS. Increased expression of ubiquitin-specific protease 22 can promote cancer progression and predict therapy failure in human colorectal cancer. J Gastroenterol Hepatol. 2010;25:1800–5. https://doi.org/10.1111/j.1440-1746.2010.06352.x
Liu Y, Yang Y, Xu H, Dong X. Implication of USP22 in the regulation of BMI-1, c-Myc, p16INK4a, p14ARF, and cyclin D2 expression in primary colorectal carcinomas. Diagn Mol Pathol. 2010;19:194–200. https://doi.org/10.1097/PDM.0b013e3181e202f2
Liu Y-L, Yang Y-M, Xu H, Dong X-S. Aberrant expression of USP22 is associated with liver metastasis and poor prognosis of colorectal cancer. J Surg Oncol. 2011;103:283–9. https://doi.org/10.1002/jso.21802
Jiang S, Song C, Gu X, Wang M, Miao D, Lv J, et al. Ubiquitin-specific peptidase 22 contributes to colorectal cancer stemness and chemoresistance via Wnt/β-catenin pathway. Cell Physiol Biochem. 2018;46:1412–22. https://doi.org/10.1159/000489156
Melo-Cardenas J, Zhang Y, Zhang DD, Fang D. Ubiquitin-specific peptidase 22 functions and its involvement in disease. Oncotarget. 2016;7:44848–56. https://doi.org/10.18632/oncotarget.8602
Koutelou E, Wang L, Schibler AC, Chao H-P, Kuang X, Lin K, et al. USP22 controls multiple signaling pathways that are essential for vasculature formation in the mouse placenta. Development. 2019. https://doi.org/10.1242/dev.174037.
Jeusset LM-P, McManus KJ. Ubiquitin specific peptidase 22 regulates histone H2B mono-ubiquitination and exhibits both oncogenic and tumor suppressor roles in cancer. Cancers. 2017. https://doi.org/10.3390/cancers9120167
Cremers N, Neeb A, Uhle T, Dimmler A, Rothley M, Allgayer H, et al. CD24 is not required for tumor initiation and growth in murine breast and prostate cancer models. PLoS ONE. 2016;11:e0151468. https://doi.org/10.1371/journal.pone.0151468
van der Bilt ARM, van Terwisscha Scheltinga AGT, Timmer-Bosscha H, Schröder CP, Pot L, Kosterink JGW, et al. Measurement of tumor VEGF-A levels with 89Zr-bevacizumab PET as an early biomarker for the antiangiogenic effect of everolimus treatment in an ovarian cancer xenograft model. Clin Cancer Res. 2012;18:6306–14. https://doi.org/10.1158/1078-0432.CCR-12-0406
Kosinsky RL, Wegwitz F, Hellbach N, Dobbelstein M, Mansouri A, Vogel T, et al. Usp22 deficiency impairs intestinal epithelial lineage specification in vivo. Oncotarget. 2015;6:37906–18. https://doi.org/10.18632/oncotarget.5412
Kosinsky RL, Chua RL, Qui M, Saul D, Mehlich D, Ströbel P, et al. Loss of RNF40 decreases NF-κB activity in colorectal cancer cells and reduces colitis burden in mice. J Crohns Colitis. 2018. https://doi.org/10.1093/ecco-jcc/jjy165.
Prenzel T, Begus-Nahrmann Y, Kramer F, Hennion M, Hsu C, Gorsler T, et al. Estrogen-dependent gene transcription in human breast cancer cells relies upon proteasome-dependent monoubiquitination of histone H2B. Cancer Res. 2011;71:5739–53. https://doi.org/10.1158/0008-5472.CAN-11-1896
Schneider D, Chua RL, Molitor N, Hamdan FH, Rettenmeier EM, Prokakis E, et al. The E3 ubiquitin ligase RNF40 suppresses apoptosis in colorectal cancer cells. Clin Epigenetics. 2019;11:98 https://doi.org/10.1186/s13148-019-0698-x
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–50. https://doi.org/10.1073/pnas.0506580102
Mootha VK, Lindgren CM, Eriksson K-F, Subramanian A, Sihag S, Lehar J, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–73. https://doi.org/10.1038/ng1180
Fan J-D, Lei P-J, Zheng J-Y, Wang X, Li S, Liu H, et al. The selective activation of p53 target genes regulated by SMYD2 in BIX-01294 induced autophagy-related cell death. PLoS ONE. 2015;10:e0116782. https://doi.org/10.1371/journal.pone.0116782
Uhlen M, Zhang C, Lee S, Sjöstedt E, Fagerberg L, Bidkhori G, et al. A pathology atlas of the human cancer transcriptome. Science. 2017. https://doi.org/10.1126/science.aan2507
Nordgren S, McPheeters G, Svaninger G, Oresland T, Hultén L. Small bowel length in inflammatory bowel disease. Int J Colorectal Dis. 1997;12:230–4.
Wang X-W, Zhang Y-J. Targeting mTOR network in colorectal cancer therapy. World J Gastroenterol. 2014;20:4178–88. https://doi.org/10.3748/wjg.v20.i15.4178
Melo-Cardenas J, Xu Y, Wei J, Tan C, Kong S, Gao B, et al. USP22 deficiency leads to myeloid leukemia upon oncogenic Kras activation through a PU.1-dependent mechanism. Blood. 2018;132:423–34. https://doi.org/10.1182/blood-2017-10-811760
Acknowledgements
The authors are grateful to N. Molitor and S. Beuermann for their technical assistance and the staff of the animal facility at the European Neuroscience Institute Göttingen (ENI-G). Furthermore, we thank S. Becker for performing FACS at the Cell Sorting Core Facility, Department of Hematology and Medical Oncology, University Medicine Göttingen (UMG), Göttingen, as well as G. Salinas and F. Ludewig for performing next-generation sequencing at the Transcriptome and Genome Analysis Laboratory (TAL) Göttingen. This work was supported by funding from the Roggenbuck Foundation (to RLK) and institutional funding provided to the Department of General, Visceral and Pediatric Surgery by the University Medical Center Göttingen (to SAJ). RLK is supported by the Dorothea Schlözer program (University of Göttingen).
Author information
Authors and Affiliations
Contributions
SAJ and RLK designed the study with input from YB-N. RLK performed all the cell culture experiments with the help of MZ and DS. Next-generation sequencing data were generated and analyzed by RLK and FW provided support with bioinformatic analyses. Mouse experiments were performed by RLK with the help of FW, DS, and MZ. Histological analyses were carried out by RLK and LW. ChIP was performed by RLK and MZ with the help of XW. SAJ and RLK wrote the manuscript. All authors have read and approved the final version of the article, including the authorship list.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by G. Melino
Supplementary information
Rights and permissions
About this article

Cite this article
Kosinsky, R.L., Zerche, M., Saul, D. et al. USP22 exerts tumor-suppressive functions in colorectal cancer by decreasing mTOR activity. Cell Death Differ 27, 1328–1340 (2020). https://doi.org/10.1038/s41418-019-0420-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-019-0420-8
This article is cited by
-
AMPKα2 promotes tumor immune escape by inducing CD8+ T-cell exhaustion and CD4+ Treg cell formation in liver hepatocellular carcinoma
BMC Cancer (2024)
-
Deubiquitinases in cancer
Nature Reviews Cancer (2023)
-
Phosphorylation of USP27X by GSK3β maintains the stability and oncogenic functions of CBX2
Cell Death & Disease (2023)
-
USP22 knockdown protects against cerebral ischemia/reperfusion injury via destabilizing PTEN protein and activating the mTOR/TFEB pathway
Naunyn-Schmiedeberg's Archives of Pharmacology (2023)
-
USP2 promotes cell proliferation and metastasis in choroidal melanoma via stabilizing Snail
Journal of Cancer Research and Clinical Oncology (2023)
