
Abstract
Caspases participate in regulated cell death mechanisms and are divided into apoptotic and proinflammatory caspases. The main problem in identifying the unique role of a particular caspase in the mechanisms of regulated cell death is their overlapping substrate specificity; caspases recognize and hydrolyze similar peptide substrates. Most studies focus on examining the non-prime sites of the caspases, yet there is a need for novel and more precise chemical tools to identify the molecular participants and mechanisms of programmed cell death pathways. Therefore, we developed an innovative chemical approach that examines the prime area of the caspase active sites. This method permits the agile parallel solid-phase synthesis of caspase inhibitors with a high yield and purity. Using synthesized compounds we have shown the similarities and differences in the prime area of the caspase active site and, as a proof of concept, we demonstrated the exclusive role of caspase-8 in necroptosis.
Similar content being viewed by others


Introduction
Caspases, the main proteolytic enzymes that mediate regulated cell death pathways, are a structurally and functionally related family of cysteine proteases that owe their name to the stringent requirement for aspartic acid in the P1 position of their peptide substrates [1]. Caspases are found in all metazoans and expressed as inactive zymogens that await activation in the cytoplasm. Of the 11 human caspases [2], members of the inflammatory group (caspase-1, caspase-4, caspase-5) are vital for the activation of specific cytokines and inflammatory cell death or pyroptosis, while members of the apoptotic group (caspase-3, caspase-6, caspase-7, caspase-8, caspase-9, and caspase-10) participate in the activation and progression of apoptosis.
The other type of allegedly programmed cell death is necroptosis, however, this process has been investigated much less extensively that above-mentioned apoptosis and pyroptosis. According to recent recommendation, necroptosis means “necrotic cell death dependent on receptor-interacting protein kinase-3 (RIPK3)” [3]. This process is caspases-independent, as their activity has to be silenced so that the cell could be destroyed in programmed fashion. Necroptosis was first recognized as a type of cell death that can be provoked by treatment with TNF (tumor necrosis factor), exclusively in the presence of caspase inhibitors such as zVAD-fmk which inhibits or disrupts the function of caspase-8 [4]. Apoptosis and necroptosis share several of the upstream signaling factors and the execution of either death pathway is coordinated by the array of regulatory molecules, including the FLIP protein [5], the cellular inhibitors of apoptosis [6], deubiquitinases A20 and CYLD [7]. So far the key caspase that needs to be blocked in order to trigger necroptosis is caspase-8, however, there is growing body of evidence, that the inhibition of caspase-10, another apical protease, can be important to this process [8].
There exists a substantial problem with defining the role of individual caspases in the control of cell death and survival—namely their cross-reactivity with available chemical tools [9]. Even with the development of highly selective, well-defined substrates, there is a need for tools that discriminate between the individual caspases. This difficulty is a consequence of the overlap in consensus sequences recognition by caspases [10,11,12], i.e., one caspase can efficiently hydrolyze a substrate designed for another caspase. To address this obstacle, chemical biology strategies in which peptide substrates and inhibitors are screened against individual caspases have been utilized [13]. While this delivers distinguishing characteristics, current methodologies are restricted to half of the available binding interactions. The substrate binding cleft of a protease is defined by the surfaces that interact with the substrate side chains. The cleft is divided into a non-prime region that binds substrate amino acids N-terminal to the scissile bond, and a prime region that binds amino acids C-terminal to the scissile bond [14]. Numerous studies on the kinds of caspase substrates that can be used to explore the prime and non-prime active sites have been published [13, 15]. However, in the case of inhibitors, the prime region is occupied by a reactive warhead [16] leaving the non-prime region to explore for inhibitor design.
There have been very few attempts to explore the prime region using inhibitors [17], and none have adopted wide-ranging library-based approaches. To provide an alternative caspase inhibitor design we first needed to employ a warhead that permitted amino acid extension into the prime region. We then crafted a simple, straightforward method based on solid-phase peptide synthesis and generated two libraries of peptidyl inhibitors intended to explore this region. We screened apoptotic caspases-3, -7, -8, -9, and -10 with the libraries, aiming specifically to generate diagnostic agents that could distinguish between the closely related caspase-8 and caspase-10. We selected the best hits to analyze paradigms of apoptosis, whose mechanisms depends on caspases [18,19,20], and necroptosis, which is negatively mediated by caspases [21]. Our compounds allowed us to differentiate the biological outcomes of caspase inhibition in cancer cells stimulated with various cell death inducers.
Materials and methods
Reagents
All chemicals and reagents were purchased from commercial suppliers and used without further purification. For the synthesis of the inhibitor libraries and individual substrates, we used Rink Amide AM resin (loading 0.74 mmol/g, Iris Biotech GmbH), Fmoc-protected amino acids (purity >98%, Iris Biotech GmbH, Bachem, Creosalus, P3 BioSystems, QM Bio), N-hydroxybenzotriazole (HOBt monohydrate purity >98%, Creosalus), diisopropylcarbodiimide (DICI, peptide grade, Iris Biotech GmbH), HATU and HBTU (peptide grade, ChemPep Inc.), 2,4,6-trimethylpyridine (2,4,6-collidine, peptide grade, Sigma-Aldrich), N,N-dimethylformamide (DMF, peptide grade, WITKO), acetonitrile (ACN, HPLC grade, WITKO), piperidine (PIP, peptide grade, Iris Biotech GmbH), trifluoroacetic acid (TFA, purity 99%, Iris Biotech GmbH), triisopropylsilane (TIPS, purity 99%, Sigma-Aldrich), methanol (MeOH, analytical grade, POCh), dichloromethane (DCM, analytical grade, POCh), diethyl ether (Et2O, analytical grade, POCh), acetic acid (AcOH, purity 98%, POCh) and phosphorus pentoxide (P2O5, purity 98%, POCh). 2-chlorotrityl chloride resin (100–200 mesh, 1.59 mmol/g) was purchased from Iris Biotech GmbH. N,N-diisopropylethylamine (DIPEA) was purchased from VWR International (Gdansk, Poland). 2,2,2-trifluoroethanol (TFE), HBr (30% wt. in AcOH), anhydrous tetrahydrofuran (THF), potassium fluoride (KF, purity >99,97%), 4-methylmorpholine (NMM), isobutylchloroformate (IBCF), tere-phthalic acid (TPA) and iso-phthalic acid (IPA) were purchased from Sigma-Aldrich. Diazomethane was generated according to the Aldrich Technical Bulletin (AL-180) protocol. For the apoptosome activation in the HEK293F cell-free system, horse cytochrome c and dATP were purchased from Sigma-Aldrich and the proteasome inhibitor MG-132 was purchased from ApexBio (Houston, USA). Methylated zVAD(O-Me)-fmk and non-methylated zVAD-fmk were purchased from Cayman Chemical (Michigan, USA). Recombinant human TNF-α was purchased from R&D Systems (cat. no. 210-TA, Minneapolis, USA). Necrostatin-1 (Nec-1) was obtained from Santa Cruz Biotechnology (Dallas, USA). The SMAC mimetic birinapant (TL32711) was purchased from Active Biochem (Maplewood, USA). Individual inhibitors and substrates were purified by HPLC on a Waters M600 solvent delivery module with a Waters M2489 detector system using a semi-preparative Waters Spherisorb S10ODS2 column and a semi-preparative Discovery® C8 HPLC column (particle size 10 µm). The solvent consisted of phase A (water/0.1% TFA) and phase B (acetonitrile/0.1% TFA). The purity of each compound was confirmed by an analytical HPLC system using a Waters Spherisorb S5ODS2 column and analytical Discovery® C8 column (particle size 5 µm). The molecular weight of each inhibitor and substrate was confirmed by high-resolution mass spectrometry on a High Resolution Mass Spectrometer WATERS LCT premier XE with Electrospray Ionization (ESI) and a Time of Flight (TOF) module.
Synthesis of the inhibitor libraries
(1) zVAD-bmk synthesis
The Cbz-Val-Ala-Asp(tBu)-OH peptide was synthesized on a 2-chlorotrityl chloride resin using Fmoc-protected amino acids. Five grams of 2-chlorotrityl chloride resin (7.95 mmol) were resuspended in 10 mL of anhydrous DCM and gently stirred once per 5 min for 30 min. Then, the DCM was filtered off and the resin was washed three times with fresh portions of DCM. Next, 3 eq of Fmoc-L-Asp(tBu)-OH amino acid in anhydrous DCM were pre-activated with 3 eq of DIPEA and added to the resin. The suspension was gently agitated for 4 h at room temperature. Subsequently, the mixture was filtered off and the resin was washed three times with DCM and three times with DMF to dispose of the excess reagent. The Fmoc-protecting group was removed using a 20% solution of piperidine in DMF (three cycles: 5 min, 5 min and 25 min). After the Fmoc group de-protection, a ninhydrin test was carried out. Next, the resin was washed six times with DMF and 3 eq of Fmoc-L-Ala-OH, pre-activated with 3 eq of HOBt and then, 3 eq of DICI in DMF was added to the reaction vessel. The reaction was carried out for 3 h at room temperature. After coupling the second amino acid, the Fmoc-protecting group was removed in the same manner as previously described and the last amino acid, Cbz-L-Val-OH, was coupled using HOBt and DICI. After solvent removal, the resin was washed three times with DMF, three times with DCM and three times with MeOH, then dried overnight over P2O5. The tripeptide was cleaved from the resin using the mild acidic condition of a mixture of TFE/AcOH/DCM (%, v/v/v 10:10:80), thus preserving the t-Bu protecting group on the side chain residue of aspartic acid. The liquid product was then mixed with hexane and the solvents were evaporated under reduced pressure. The resulting product was dissolved in an ACN/H2O mixture (%, v/v 75:25) and lyophilized. The purity of the resulting compound was confirmed by analytical HPLC and its molecular weight was determined by HR-MS. The crude tripeptide was then converted into a bromomethyl ketone (BMK) using a synthetic procedure described by Kato et al. [22]. Briefly, a 0.2 M solution of Cbz-VAD(tBu)-OH (5 mmol, 1 eq) in anhydrous THF was stirred in an ice/acetone bath at −10°C for 20 min. Then, 4- methylmorpholine (6.25 mmol, 1.25 eq) and isobutyl chloroformate (5.75 mmol, 1.15 eq) were added to the mixture. Immediately after adding the isobutyl chloroformate, a white precipitate formed that indicated the formation of an anhydride. The reaction was carried out at −10°C for 45 min. In a parallel experiment, an ethereal solution of diazomethane was generated according to the Aldrich Technical Bulletin protocol (AL-180). Next, the solution of mixed anhydrides was added dropwise to the ethereal diazomethane (16.6–21.4 mmol) at 0°C and stirred vigorously for 20 min. Then, the ice bath was removed and the reaction was carried out for another hour at room temperature. To generate Cbz-VAD(tBu)-CH2Br, 15 mL of a 1:2 solution of HBr (30% wt. in CH3COOH) and water were slowly added to the mixture over a period of 10 min. The reaction was monitored using analytical HPLC. After 30 min, the reaction was complete and the mixture was diluted with ethyl acetate, transferred to a separatory funnel and extracted three times with saturated aqueous NaHCO3, twice with brine and once with water. The organic fraction was dried over MgSO4 and evaporated under reduced pressure. The product was a pale yellow solid that was used for further synthesis without purification. The product purity, determined by HPLC, was > 90% and the overall yield was > 85%.
(2) zVAD-iso-Pht-X-NH2 and zVAD-tere-Pht-X-NH2 library synthesis
0.3 g of Rink Amide Resin (0.222 mmol, 1 eq) was suspended in 5 mL of DCM and gently stirred once every 5 min for 30 min. The DCM was removed by filtration and the resin was washed three times with DMF. Next, the Fmoc group was de-protected using 20% piperidine in DMF (three cycles of 5 min, 5 min and 25 min). The resin was washed six times with DMF, three times with DCM, and three times with MeOH then dried over P2O5. The resin was divided into 19 equal portions, each of which was added to 19 wells of a solid-phase peptide synthesis reactor. To swell the resin, 1 mL of DCM was added to each well and left there for 30 min, shaking gently once every 5 min. Afterward, the DCM was filtered and the resin was washed three times with DCM. Next, 3 eq of each of the 18 natural amino acids and norleucine were pre-activated with HATU and 2,4,6-collidine in a minimal amount of DMF and added to separate wells of the solid-phase peptide synthesis reactor (Asp amino acids were omitted due to synthesis issues). The reaction was carried out for 4 h, followed by three DMF washes of the solid-phase. The Fmoc-protecting group was removed and the resin was washed six times with DMF. Iso-phthalic acid was coupled to the free amine group of each of the 18 natural amino acids and norleucine with the use of the same coupling reagents (3 eq of HATU and 3 eq of 2,4,6-collidine). After 3 h of the coupling reaction, the resin was washed three times with DMF. In the subsequent step, 3 eq of Cbz-VAD(tBu)-BMK were dissolved in a minimal amount of DMF, pre-activated with 10 eq of KF and added to each well of the reactor. The reaction was carried out for 24 h with gentle agitation. The resin was then washed ten times with DMF, three times with DCM, and three times with MeOH then dried over P2O5. Finally, the inhibitors were cleaved from the resin using a mixture of TFA:TIPS:H2O (%, v/v/v 95:2.5:2.5) for 2 h at room temperature, shaking once per 10 min. Supernatants were collected in 15 mL Falcon conical-bottom centrifuge tubes and the resin was washed once again with TFA:TIPS:H2O (%, v/v/v 95:2.5:2.5). For each compound, both supernatant fractions were combined and precipitated in cold Et2O for 30 min at −20°C. Then, the samples were centrifuged (5 min, 4°C, 4400 rpm), the supernatants were decanted and the precipitates were washed with 10 mL of Et2O, then centrifuged (5 min, 4°C, 4400 rpm) and the supernatant was discarded. The precipitated compounds were dissolved in 0.8 mL of DMSO and purified using HPLC (Waters, column: Spherisorb, 5 µm particle size, L × I.D. 25 cm × 4.6 mm) with an H2O:ACN gradient and then lyophilized. The purified white/pale yellow products were dissolved in DMSO to a final concentration of 10 mM and stored at −80°C until used. The second library with tere-phthalic acid and the zVAD-2,6-dimethylterephthalic-X-NH2 inhibitors was synthesized in an analogous manner. The purity of each inhibitor was confirmed by analytical HPLC and analyzed using HRMS (Table S4, Fig. S5).
Individual substrate synthesis
To determine the kinetic parameters of the obtained inhibitors we synthesized broad-spectrum, ACC-labeled fluorogenic caspase substrates using a previously published method [23]. The Fmoc-ACC-OH fluorophore was synthesized according to Maly et al. [24]. Briefly, Rink Amide Resin was added to a glass reaction vessel containing DCM and was stirred gently every 10 min for 1 h then filtered and washed with DMF. The Fmoc-protecting group was removed using 20% piperidine in DMF and the resin was washed six times with DMF. Next, 2.5 eq of Fmoc-ACC-OH were pre-activated with HOBt monohydrate and DICI in DMF and the mixture was poured onto the resin. After 24 h of gentle agitation at room temperature, the Fmoc-ACC-OH coupling reaction was repeated to improve the amount of ACC coupling to the resin. When the reaction was complete, the resin was washed with DMF and the Fmoc-protecting group was removed with 20% piperidine in DMF. Next, 2.5 eq of Fmoc-L-Asp(tBu)-OH, 2.5 eq HATU and 2.5 eq of collidine in DMF were activated for 3 min and added to the reaction vessel containing H2N-ACC-resin and the reaction was carried out for 24 h. The resin was washed four times with DMF and the reaction was repeated using 1.5 eq each of Fmoc-L-Asp(tBu)-OH, HATU, and collidine in DMF. After the resin was washed with DMF, the Fmoc-protecting group was removed using 20% piperidine in DMF. The resin was subsequently washed three times with DMC and three times with MeOH and dried over P2O5. Next, the resin was divided and individual substrates were synthesized by solid-phase peptide synthesis. The 2.5 eq of Fmoc-P2-OH were pre-activated with 2.5 eq of HOBt and 2.5 eq of DICI in DMF and poured into the cartridge with 1 eq of NH2-Asp(tBu)-ACC-resin, followed by gentle agitation for 3 h at room temperature. Then, the reaction mixture was filtered and the resin was washed six times with DMF. The Fmoc-protecting group was removed using 20% piperidine in DMF. A ninhydrin test was carried out after each round of coupling and Fmoc de-protection. A solution of 2.5 eq of Fmoc-P3-OH, 2.5 eq of HOBt and 2.5 eq of DICI in DMF was added to the resin and the resin was agitated for 3 h. After removal of the solution, the resin was washed with DMF and coupling and de-protection of Fmoc-P4-OH was performed in the same manner as for the P2 position. The N-terminus was protected with an acetyl group using 5 eq of AcOH, 5 eq of HBTU and 5 eq of DIPEA in DMF. After solvent removal, the resin was washed six times with DMF, three times with DCM and three times with MeOH, dried over P2O5 and cleaved from the resin with a mixture of TFA/TIPS/H2O (%, v/v/v 95: 2.5: 2.5). The crude product was purified by HPLC and lyophilized. The purity of each substrate was confirmed by analytical HPLC and analyzed using HRMS. The substrates were then dissolved in peptide grade DMSO to a concentration of 20 mM and stored at −80°C until used.
Recombinant caspase assay conditions
Recombinant human apoptotic caspase-3, caspase-7, caspase-8, caspase-9, and caspase-10, recombinant human proinflammatory caspase-1, caspase-4, and caspase-5 and recombinant mouse proinflammatory caspase-1, and caspase-11 were expressed and purified as previously described [25, 26]. All the enzymes were active site-titrated using the zVAD(OH)-fmk inhibitor (cat. no. 14467, Cayman Chemical Company) as previously described [25]. The caspase buffer consisted of 20 mM Pipes, 10 mM NaCl, 1 mM EDTA, 10% sucrose (w/v) and 10 mM DTT (pH 7.2). To allow dimerization and to maintain the maximal activity, the buffer for the apical caspases (8, 9, and 10) and the proinflammatory caspases (1, 4, 5, and 11) was supplemented with 0.75 M sodium citrate.
Determination of the kinetic parameters for the inhibitors
We measured the Ki parameters for the reversible inhibitors using the Morrison equation [27]. For each enzyme, an appropriate fluorogenic substrate was used to monitor substrate hydrolysis (mCasp-1, Ac-LEHD-ACC KM = 36 μM; mCasp-11, Ac-LEHD-ACC KM = 165 μM; hCasp-1, Ac-LEHD-ACC KM = 49 μM; hCasp-3, Ac-DEVD-ACC KM = 21 μM; hCasp-4, Ac-LEHD-ACC KM = 112 μM; hCasp-5, Ac-LEHD-ACC KM = 124 μM; hCasp-7, Ac-DEVD-ACC KM = 57 μM; hCasp-8, Ac-LEHD-ACC KM = 15 μM; hCasp-9, Ac-LEHD-ACC KM = 106 μM; hCasp-10, Ac-LEHD-ACC KM = 24 μM). To meet the criteria for reversible inhibitor kinetics, the lowest inhibitor concentration was at least 4-fold higher than the caspase concentration used in the assay. The caspase was first pre-warmed in the assay buffer in the 96-well plate for 15 min at 37°C and then preincubated with the inhibitor for another 15 min at 37°C. Next, the substrate was added to the enzyme-inhibitor mixture and the progress of the reaction was monitored using CLARIOstar plate reader (BMG Labtech) operating in fluorescence kinetic mode (355 nm excitation and 460 nm emission). The kinetic parameters (kobs/I) for the irreversible inhibitors (tagged with an AOMK warhead) were determined under pseudo-first order kinetic conditions, where the lowest inhibitor concentration was at least 5 times higher than the caspase concentration. The inhibitor was diluted into the wells of the 96-well plate, mixed with the fluorescent substrate and pre-warmed for 15 min at 37°C. In parallel, the caspase was incubated at 37°C for 15 min, poured onto the plate and the reaction was started immediately under the same fluorescence setting as that of the reversible inhibitors. The reaction was monitored until the enzyme was completely inhibited. All the measurements for the reversible and irreversible inhibitors were performed at least three times and the data were analyzed using Graph Pad Prism 7.0 software (La Jolla, CA, USA).
Specificity in the prime region of the caspases
The Ki parameters of all 39 inhibitors for the individual recombinant caspases were used to create specificity profiles (heat maps) in the prime areas of the active sites of the caspases. For better data presentation, the Ki for the reference inhibitor (zVAD-Bzl) for each caspase was separately set at 1 and the Ki parameter for the other inhibitors was adjusted accordingly. The heat maps were created in Graph Pad Prism 7.0.
Correlation of inhibition profiles between caspases
To identify the differences between inhibitor specificities across caspases, we generated correlation matrices for the proinflammatory caspases and the apoptosis caspases, where we calculated the Pearson correlation coefficient (ρ) for each caspase pair. All calculations were performed using Graph Pad Prism 7.0 and presented either as a heat map or a scatter plot.
Caspase-8:FLIP complex formation and activity assays
FLIP protein was expressed and purified according to the procedure described by Boatright et al. [28]. The caspase-8:FLIP complex was assembled in regular caspase buffer supplemented with 0.75 M of sodium citrate. Caspase-8 monomers dimerize easily in citrate buffer, therefore, to avoid the proteolytic background from dimerized caspase-8, caspase-8 was preincubated at either low or high concentrations with an excess of FLIP protein and subjected to kinetic analysis using the Ac-LEHD-ACC substrate. Next, to optimize the kinetic assay conditions, caspase-8 was titrated with FLIP for 15 and 30 min and then reacted with Ac-LEHD-AFC.
Determination of the inhibition mode of selected inhibitors for caspase-8, caspase-10, and caspase-8:FLIP
To test whether the inhibitors were reversible or irreversible, we competitively labeled caspase-8, caspase-8:FLIP, and caspase-10. Briefly, 50 nM of active site-titrated caspase was preincubated for 30 min with 1 μM selected caspase inhibitor, followed by a 15 min incubation with a potent and irreversible caspase activity-based probe (100 nM, biotin-ahx-LEHD-AOMK, total reaction volume—100 μL). Next, the reaction products were precipitated using 100 μL of ice-cold 30% trichloroacetic acid (TCA) in water. TCA was added to each sample, incubated on ice for 30 min and centrifuged for 30 min (20,000 × g, 4°C). The supernatants were gently aspirated using a vacuum device, the pellet was resuspended in 200 μL of ice-cold 10% TCA and the samples were centrifuged for an additional 15 min (20,000 × g, 4°C). The supernatants were again removed and the pellet resuspended in 200 μL of acetone. The samples were centrifuged for 10 min (20,000 × g, 4°C) and the supernatant was removed. The samples were dried at 37°C for 15 min and then 80 μL of 1× SDS/DTT buffer was added to each sample, followed by denaturation at 95°C for 5 min. Then, 30 μL of each sample was subjected to SDS-PAGE (30 min, 200 V), followed by transfer to a nitrocellulose membrane (60 min, 10 V) and blocking with 2% BSA in TBS-T (60 min, RT). The biotin-labeled probe/caspase complex was detected by probing the membrane with fluorescently labeled streptavidin (30 min, RT, 1:10,000, IRDye® 800CW, LI-COR), followed by scanning in a LI-COR system (800 nm channel, Odyssey® CLx, Lincoln, NE, USA). The data were analyzed using Image Studio Lite software (Lincoln, NE, USA). The use of 1 μM (inhibitor-enzyme 20x excess) of zVAD-Bzl-X and zVAD-AOMK-X inhibitor ensured that all caspase activity was blocked before adding the LEHD activity-based probe. In the second experiment, caspase-8, caspase-8:FLIP and caspase-10 (100 nM) were preincubated with select inhibitors (1 µM) for 30 min and then the caspase-inhibitor complex was diluted 20× in assay buffer (final caspase conc. 5 nM, final inhibitor conc. 50 nM), the Ac-LEHD-AFC (100 μM) was added and the fluorescence was monitored immediately for 30 min. The dissociation of the caspase-inhibitor complex and the recovery of the enzyme activity indicated reversible inhibition. In a separate experiment, 5 nM of active site-titrated caspase was mixed with 50 nM of the selected inhibitor and 100 μM Ac-LEHD-AFC and the fluorescence of the reaction was measured.
Apoptosis and necroptosis assays
We used HT-29 human colon cancer cells, which respond well to apoptosis and necroptosis stimulants, to test whether we could use our inhibitors to identify caspases’ involvement in apoptosis vs. necroptosis. HT-29 cells were seeded on a 96-well cell culture plate (20,000 cells per well in 100 μL of phenol red-free McCoy’s media) and allowed to attach overnight. The next day, the cells were preincubated for 6 h with or without caspase inhibitors (25 μM) and with or without Nec-1 (50 μM). After this, the cells were stimulated with TRAIL (100 ng/mL) + CHX (5μg/mL) [29, 30] or TNF-α (100 μg/mL) + birinapant (2 μM) to induce apoptosis [31, 32]. After 24 h, the HT-29 cell viability was determined using MTS assay according to the manufacturer’s protocol (CellTiter 96® Aqueous One Solution Cell Proliferation Assay, G3582, Promega). The number of living cells was reported as a survival ratio calculated based on the number of un-stimulated control cells. Apoptotic cells (%) were calculated in the presence of 50 μM of Nec-1, and necroptotic cells (%) were calculated using the equation: [100% − (apoptotic cells + viable cells)] = necroptotic cells (in the absence of Nec-1) (Fig. S4).
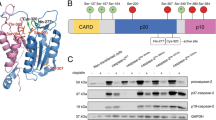
Caspases activation in TRAIL/CHX-treated HT29 cells
500,000 cells per well were seeded on 12-well plates and allowed to attach overnight. Cells were treated with TRAIL (100 ng/mL) and CHX (5 μg/mL) for various timepoints, harvested and resuspended in 100 μL of 1xSDS/DTT. After sample denaturation at 95°C cells were sonicated and subjected to western blot analysis in the same manner as described above (SDS-PAGE: 30 min at 200 V; transfer to nitrocellulose membrane: 60 min at 10 V; blocking-2% BSA in TBS-T, 60 min at room temperature). Membranes were separately incubated overnight with anti-caspase-8 mouse serum (1:250), anti-caspase-3 (1:1000, cat no. 9662, Cell Signaling) or mouse anti-caspase 10 antibody (1:500, MBL; cat. no M059-3) at 4°C, incubated with fluorescently labeled secondary antibody (30 min, RT, 1:10,000, goat anti-mouse IgG, AlexaFluor 680), followed by scanning in Azure system (Sapphire Biomolecular Imager). The data were analyzed using Azure Spot Analysis software.
Monitoring caspases (auto)cleavage in the presence of zVAD-fmk and zVAD-Bzl inhibitors
500,000 cells per well were seeded on 12-well plates and allowed to attach overnight. Next, 25 μM zVAD-fmk or zVAD-Bzl inhibitors were added to wells and preincubated for 2 h prior to TRAIL (100 ng/mL) and CHX (5μg/mL) addition. Cell were harvested at various timepoints, and subjected for SDS-PAGE and Western blotting. Caspase-8 processing was detected with an anti-caspase-8 mouse serum as described above.
Western Blot analysis of siRNA-induced knockdowns of caspase-8 and caspase-10
HT29 cells were seeded on 12-well cell culture plate (200,000 per well) and allowed to attach overnight. The next day caspase-8 and caspase-10 siRNA lyophilizate was dissolved in nuclease-free water to a final concentration of 26.7 μM, combined with Lipofectamine® 2000 Reagent in Optimem medium and incubated for 30 min. Next, the mixture was dissolved in McCoy’s medium (10%FBS, 5 mM glutamine, 5 mM penicillin/streptavidin) to a final concentration of 100 nM siRNA and 2 ug/mL lipofectamine. 1 mL of siRNA/lipofectamine solution was added to three wells and the medium to three control wells. Control and siRNA-treated cells were harvested after 24, 48, and 72 h of incubation, SDS-PAGE electroporated and subjected to Western blot analysis. Membranes were incubated overnight with anti-caspase-8 mouse serum (1:250) at 4°C or mouse anti-caspase 10 antibody (1:500, MBL; cat. no M059-3). Primary antibodies were detected with secondary goat anti-mouse antibody (30 min, RT, 1:10,000, Alexa Fluor 680) and membranes were scanned using Azure system (658 nm channel). The siRNA-induced knockdowns were analyzed using Image Studio Lite software (Lincoln, NE, USA) and the protein depletion was calculated based on the fluorescence intensity of detected bands.
Apoptosis and necroptosis assays using caspase-8 and caspase-10 knockdown cells
HT29 cells were seeded on two 96-well plates (10,000 cells/well) and subjected to siRNA-induced knockdown of caspase-8 or caspase-10. Lipofectamine® 2000 Reagent was incubated with caspase-8 or caspase-10 siRNA in Optimem medium for 30 min, then diluted in McCoy’s medium supplemented with 10%FBS, 5 mM penicillin/streptavidin and 5 mM glutamine to final concentration of 100 nM siRNA and 2 ug/mL lipofectamine. Next, 100 μL of prepared solution was added to each well and plates were incubated for 72 h at 37°C (5% CO2). After this time, siRNA solution was aspirated, and cells were preincubated for 2 h with various inhibitors in two different concentrations (5 and 25 μM). Next, extracellular death stimulant TRAIL (100 ng/mL) and protein synthesis inhibitor-CHX (5μg/mL) were added to induce apoptosis. To rescue cells from necroptosis, Nec-1 (50 μM) was added to each well. After 24 h, the HT-29 cell viability was determined using MTS assay according to the manufacturer’s protocol (CellTiter 96® Aqueous One Solution Cell Proliferation Assay, G3582, Promega).
Results and discussion
Inhibitor library design
We developed a comprehensive method for investigating the prime region of the active site in caspases through direct inhibitor library screening as this approach has not been previously taken. The concept of the library design was based on (1) the general caspase requirements for aspartic acid in the P1 position, (2) the broad-spectrum peptide sequence and (3) the electrophilic binding group that inhibits caspase activity and can be used to explore the prime region of the active site in caspases (Fig. 1a, b). zVAD-fmk is the most widely used pan-caspase inhibitor for determining the mechanisms by which the caspases are involved in apoptosis [33], therefore, we decided to use the zVAD (also called Cbz-VAD) caspase recognition motif as a scaffold for the addition of warheads that allow extension into the prime region. To determine the significance of the next building block position for compound activity, we used iso- and tere-isomers of phthalic acid (Pht) in the synthesis, since these isomers allow extension from the meta and para positions into prime region space. In the prime area of the inhibitor backbone, we embedded one of twenty natural amino acids to see whether they would enhance the potency and specificity of the inhibitor toward individual caspases. Next, caspase broad-spectrum peptide (zVAD) was transformed into reactive bromomethyl ketone (zVAD-BMK) which was subsequently coupled to HO-iso/tere-Pht-X-resin moiety (X stands for amino acid) on solid support, followed by HPLC purification (Fig. 1c). This solid-phase/solution phase parallel block chemical synthetic strategy allowed us to obtain a large number of highly pure caspase inhibitors in relatively short time (Fig. 1, Table S3).

A general outline of the solid-phase synthesis of the caspase inhibitor library. a The general architecture of caspase inhibitors for the investigation of the prime site specificity. b We selected the zVAD peptide motif as a general sequence recognized by all caspases and combined it with either iso-/tere-phthalic acid to allow for the generation of a library with various amino acids to explore the prime site. We used the same 20 amino acids in the iso-phthalic and tere-phthalic isomers. c A general scheme of the solid-phase library synthesis. Briefly, we transformed the z-VAD-OH peptide into a reactive bromomethyl ketone and coupled it to an electrophilic moiety (Pht) and one of twenty amino acids that were previously attached to the resin. Next, we cleaved all thirty-eight reaction products (19 iso-isomers and 19 tere-isomers) from the resin and purified them using HPLC
Determination of the kinetic parameters of the inhibitors
All the synthesized inhibitors containing the Pht binding group demonstrated a rapid reversible mode of inhibition, precluding the calculation of a kobs/I parameter, which is the rate of enzymes inactivation via covalent modification of their active site (Cys residue in caspases) by an irreversible inhibitor (Fig. S1). This may be related to the fact that Pht does not represent a good leaving group, because its negative charge is not stabilized adequately. Therefore, to test the activity and selectivity of the inhibitors for the caspases, we calculated the equilibrium constant (Ki, tight inhibition) across ten recombinant caspases (Fig. 2a). By measuring Ki parameter we directly determined the strength of the inhibitor-enzyme complex. (Fig. S1, Table S1, S2). The most relevant differences between the examined caspases are shown as a heat map (Fig. 2b). We used zVAD-Bzl as a control compound and all the calculated values were normalized to its Ki value (Fig. 2c). There were strong differences between the recognition model of the particular caspases, their preferences for the inhibitor isomers (iso- or tere-) and the arrangement of the amino acid in the prime region. For the proinflammatory caspases-4, -5, and -11 and apoptotic caspases-9 and -10, inhibitors containing the tere-isomer of phthalic acid were much more potent than the ones with the iso-isomer. On the other hand, caspases-1 (both human and mice) and human caspases-3, -7, and -8 displayed general kinetic preferences for iso-isomers. As the structures of obtained compounds are quite rigid, prime region of particular caspases has to be somehow adjusted to be pro- iso or pro- tere. Furthermore, individual amino acids situated in the prime area of the compound backbone were able to distinguish between individual enzymes, even the ones with strong, structural and functional similarities such as caspase-8 and caspase-10. We were able to determine the best and worst inhibitors for other caspases (Table S1, S2).

The inhibition parameters (Ki) of the individual compounds tested on individual, recombinant caspases. a Fluorogenic substrates (Ac-peptide-ACC) used for the Ki calculations. b The inhibition parameters (Ki) of all 39 inhibitors presented as a heat map where red indicates strong inhibitor binding (low Ki) and blue (and green) indicates weak inhibitor binding (high Ki). For better presentation, all kinetic values (Ki) across the individual caspases were presented as fold-change comparing to zVAD-bzl (value = 1, marked gray). c The Ki values of zVAD-Bzl for individual caspases
Table 1 shows the kinetic parameters (Ki) of selected reversible inhibitors for the proinflammatory and apoptotic caspases. In some cases, a change of only one amino acid in the prime area allowed for significant increases or decreases in the activity of the inhibitor for a particular enzyme, as shown for caspases-4, -5, and -11. The analysis of apoptotic caspases demonstrated that although the differences between Ki values for the opposite inhibitors are smaller than for proinflammatory caspases, we were still able to obtain good inhibitory discrimination with our approach. The best example here is caspase-8, for which the difference between the best (isoPht-Trp) and the worst (terePht-Phe) inhibitors was over 80-fold.
As apoptotic and proinflammatory caspases are involved in different processes we correlated the global features in their specificities to see whether we could extract a pattern showing similarities between closely related enzymes, even if they were from different organisms. In Fig. 3 we show the most interesting outcomes of the Ki correlation for proinflammatory caspases, which depicts the strong similarities in their prime site architecture. Moreover, by using a simple functional/activity approach, we demonstrated that human caspase-4 and caspase-5 are equivalent to mouse caspase-11, as the Pearson correlation coefficients for both iso- and tere-isomers were very high, 0.962 and 0.780 respectively (for caspase-4/-11 pair), and 0.829 and 0.833 (for caspase-5/-11 pair). This observation is in the line with a genetic evidence that in humans caspases-4 and -5 are both orthologues to murine caspase-11 (Fig. 3a) [34, 35]. Subsequently, we also made this analysis for five human apoptotic caspases, demonstrating that in general all these enzymes display much less conserved prime region architecture than inflammatory caspases (Fig. 3b). As an example, we investigated caspase-8 and caspase-9 in details, as they are the key players in two different pathways in apoptosis. There was no correlation between caspase-8/-9 with the iso-phthalic isomers of our compounds (ρ = 0.024), thus these inhibitors could be used to distinguish between the caspases by analyzing their prime area preferences (Fig. 3b). The analysis of tere-phthalic isomers showed that the inhibitor binding preferences of these caspases at this orientation are alike. Interestingly, we observed almost no correlation in inhibitors potency between caspase-8 and caspase-10, which might suggest that although these enzymes participate in the same pathway of apoptosis initiation, their substrate preferences are different, and could be possible to separate these proteases by selective inhibitors. The correlation analysis for the other caspases showed that our method can distinguish between closely-related pairs of caspases (i.e., caspase-4 and caspase-5; caspase-8 and caspase-10; and caspase-3 and caspase -7) (Figs. S1, S2).

The correlation of the Ki parameters of the zVAD iso- and tere-Pht-X inhibitors among select caspases. a A matrix of Ki correlations for the proinflammatory caspases (left) and the detailed correlation plots between human caspase-4 and mouse caspase-11 (right). These data demonstrate that there is a significant similarity in the prime site architecture of the proinflammatory caspases. b A matrix of the Ki correlation for human apoptotic caspases (left) and the detailed correlation plots between human caspase-8 and human caspase-9. These data demonstrate that the prime site architecture differs across the caspases. For example, there is a strong linear correlation between the tere-phthalic inhibitors for caspase-8 and -9 but no correlation for the iso-phthalic inhibitors. For each caspase pair, the Pearson correlation coefficient was calculated and the results were presented either as heat maps or scatter plots
Table 2 summarizes the kinetic parameters of the most selective inhibitors of the apoptotic and proinflammatory caspases. The presented values are the exact Ki values for each enzyme and the Ki compared with that of human caspase-3. The highest kinetic discrimination within apoptotic caspases we obtained was for caspase-8 (zVAD-isoPht-Trp), however for caspase-9 and -10 we also were able to develop selective inhibitors (zVAD-terePht-Ser and zVAD-terePht-Tyr). For the inflammatory caspases the best selectivity we obtained was for caspase-1 with zVAD-isoPht-Lys inhibitor which was 21- and 23-fold more potent over caspase-4 and -5 respectively. The most selective caspase-4 and -5 inhibitors displayed only poor discrimination between the enzymes. However, it must be stressed here, that the selectivity for each caspase can be significantly improved by applying the optimal peptide sequence in the non-prime position. Here, we analyzed only the caspases non-prime site, while the prime region was occupied by zVAD sequence that is known to be selective for caspase-1 and caspase-8. This explains why the absolute selectivity factors for these two enzymes were the most pronounced.
Determination of the inhibition mode of the selected inhibitors
Since the ultimate goal of our studies was to apply caspase inhibitors in cell-based assays, we decided to transform selected zVAD-Pht-X compounds into irreversible inhibitors, as the latter ones are generally more potent in cellulo. To do so we replaced the Pht group with a widely used acyloxymethyl ketone (AOMK; 2,6-dimethylterephthalic acid). We synthesized irreversible inhibitors with glycine, serine, and phenylalanine in the prime area to be able to distinguish between closely-related caspases such as caspase-8, caspase-9 and, caspase-10 and compared them with the reversible analogs (Fig. 4a). We examined the differences in the mechanism of action of Pht- and AOMK-based compounds on recombinant caspases-8 and -10, as both are involved in the extrinsic pathway of apoptosis (Fig. S5). As we expected, the zVAD-Pht-X inhibitors had reversible kinetics and the zVAD-AOMK-X inhibitors had irreversible kinetics with recombinant caspase-10 (Fig. 4b). Surprisingly, inhibitors with either warhead (Pht or AOMK) demonstrated classic reversible kinetics on recombinant caspase-8 within initial assay timeframe and tended to form an irreversible complex after prolonged incubation (Fig. 4b, Fig. S5). To explore this phenomenon, we incubated 50 nM of recombinant caspase-8 and caspase-10 with 1 μM of selected inhibitors for 30 min to fully inhibit caspase activity. Next, we added the pan-caspase irreversible biotin-LEHD-AOMK probe to detect enzymatic activity using fluorescence streptavidin (Fig. 4c). When caspases were inhibited with irreversible inhibitor (for example zVAD-fmk) no enzymatic activity was detected with biotin-labeled probe. However, when enzyme was inhibited in reversible fashion, biotin-LEHD-AOMK probe competed for the active site with such inhibitor, which resulted in the covalent enzyme modification (Fig. 4c). With this experiment, we showed that even compounds considered to be irreversible inhibitors of caspase-8, such as zVAD-AOMK-Gly/Ser/Phe, turned out to form reversible complexes under tested conditions. However, on the other hand, non-substituted zVAD-Bzl and zVAD-AOMK appeared to display bimodal inhibition. To further explore this, we preincubated caspase-8 with selected inhibitors for 60 min, allowing the enzyme-inhibitor complex to mature, then diluted it, and measured the recovery of enzyme activity in the presence of fluorogenic substrate (Ac-LEHD-ACC). The rate of substrate cleavage was lower compared to the one where the inhibitor and caspase-8 were mixed right before assay (no pre-incubation step), indicating that some portion of the enzyme was irreversibly inhibited (Fig. 4d). Caspase-10, which has a strong structural and functional likeness to caspase-8, was irreversibly inhibited by both fmk and AOMK-based warheads and displayed reversible inhibition toward Pht-inhibitors (Fig. 4b, d).

Dissecting the mechanisms of the inhibition of caspases by zVAD-tere-Bzl-X and zVAD-iso-Bzl-X inhibitors. Panel a The structures of zVAD inhibitors with tere-phthalic (Bzl) or 2,6-dimethylterephthalic (AOMK) warheads. Panel b The inhibition curves of caspase-8 and caspase-10 by zVAD-Bzl and zVAD-AOMK inhibitors indicated that only caspase-10 vs. zVAD-AOMK had classic irreversible inhibitor kinetics. Panel c Reversible/irreversible inactivation of caspases-8 and -10 using selected inhibitors followed by the use of irreversible pan-caspase biotin-LEHD-AOMK probe. Panel d zVAD-Bzl and zVAD-AOMK inhibitors were shown to display reversible inhibition kinetics towards caspase-8 in the initial timeframes of the assay. In brief, caspase-8 was either preincubated with inhibitors (green line) or the inhibitor was added right before the assay (orange line). Next, the caspase-8-inhibitor complex was diluted, Ac-LEHD-AFC substrate was added to the reaction mixture and the fluorescence was read immediately. The recovery of enzyme activity demonstrated reversible inhibitor kinetics. The blue line serves as a control (enzyme plus substrate, no inhibitor)
Dissecting cell death pathways in human colon cancer cells
To explore the utility of our inhibitors we dissected the roles of caspase-8 and caspase-10, which are involved in the interplay of apoptosis and necroptosis (Fig. 5a). If the activity of caspase-8 is blocked or disrupted, the cell triggers necroptosis, which can be blocked by necrostatin-1 (Nec-1), an inhibitor of RIPK-1, [36]. Another important protein is FLIPL which forms a heterodimer with caspase-8 that blocks the necroptosis pathway [37, 38]. Therefore, we assembled a catalytically active caspase-8:FLIPL complex (Fig. S6) and found that although the Ki values were higher than the ones for caspase-8 alone, the specificity remained unchanged (Fig. 5d, e). Next, we checked whether the outcomes of apoptosis and necroptosis (cell viability) correlated with the activity of caspase-8, caspase-10 or both enzymes. For example, a recent study reported that caspase-10 can actually mediate cell death in HT-29 cells, but only if caspase-8 is ablated [8]. To investigate this, we first performed western blotting analysis, to confirm the presence and the activation of caspase-8 and caspase-10 in response to TRAIL + cycloheximide (CHX) stimulation at various timepoints (Fig. 5b). The data showed that indeed, both caspases are processed, leading to caspase-3 activation, which in turn triggered cell death. Since caspase-8, when inhibited, promotes necroptotic cell death, we also dissected the influence of the most potent caspase-8 inhibitors on the TRAIL/CHX-induced cell death (Fig. 5c). The results demonstrated that zVAD-fmk fully inhibits caspase-8 activation, even after prolonged incubation, while reversible zVAD-Bzl only partially attenuates caspase-8 autoprocessing. To test the influence of various inhibitors on the outcomes of cell death stimulation, HT-29 cell were preincubated with the inhibitors; TRAIL + cycloheximide (CHX) was added to induce cell death (apoptosis and necroptosis) and TRAIL + CHX + Nec-1 was added to induce apoptosis. The potent broad-spectrum irreversible caspase inhibitor (zVAD-fmk) blocked apoptosis and induced necroptosis in the HT29 cell line (Fig. 5d, e). However, cells pretreated with zVAD-fmk and Nec-1 survived, as both pathways were inhibited (Fig. S4). Next, we pretreated HT-29 cells with selective inhibitors for caspase-8 (Pht warhead) and caspase-10 (AOMK warhead) and induced either apoptosis or necroptosis. There was a good correlation between the potency of caspase-8 inhibitors and the inhibition of apoptosis (and the promotion of necroptosis) (Fig. 5d). The most potent caspase-8 inhibitor (zVAD-Bzl, Ki = 2.55 nM) switched HT-29 cells from apoptosis to necroptosis, whereas the least potent caspase-8 inhibitor (zVAD-Bzl-Phe, Ki = 137 nM) did not. Similar results were obtained for other caspase-8 selective compounds (-Bzl-Gly and -Bzl-Ser), which were also able to induce programmed necrosis in these cells. On the other hand, the potent and irreversible caspase-10 inhibitor (zVAD-AOMK-Phe, kobs/I = 135,000 M−1 s−1) did not affect any of the cell death pathways, demonstrating that the inhibition of caspase-10 is not relevant to any of the types of cell death in the wild-type HT-29 cell line (Fig. 5e). To further demonstrate the usability of our inhibitors to study cell death mechanisms, we induced the apoptosis in HT-29 using TNF-α/birinapant stimulants, and triggered the necroptosis switch by pre-incubation the cells with selected inhibitors. By doing this we demonstrated that in this system caspase-8, but not caspase-10 inhibition is the main driver to induce programmed necrosis (Fig. S4).

Necroptosis in HT29 cells was mediated by caspase-8 and not by caspase-10. a The extrinsic pathway of apoptosis leads to the activation of the death-inducing signaling complex (DISC) containing active caspase-8 and caspase-10, which further leads to the activation of downstream caspase-3 and caspase-7. When caspase-8 activity is blocked, an alternative pathway is triggered, leading to a type of cell death called necroptosis. Cells can be rescued from necroptosis when treated with necrostatin-1 (Nec-1), a potent RIPK inhibitor. b Time-course of the activation of caspases-8, -10, and -3 in TRAIL/CHX-stimulated HT29 cells via Western blotting analysis. c The inhibition of caspase-8 activation in apoptotic HT29 cells using irreversible zVAD-fmk and reversible zVAD-Bzl inhibitors. zVAD-fmk was shown to completely prevent caspase-8 activation, while zVAD-Bzl only partially attenuates its auto-cleavage (top panel). Nevertheless, both inhibitors were able to block apoptosis, and induce necroptosis (bottom panel) Panel d and e. HT-29 cells were preincubated with selected inhibitors, followed by induction of apoptosis (TRAIL+CHX) or necroptosis (TRAIL+CHX+inhibitors). To inhibit necroptosis, Nec-1 inhibitor was used. zVAD-fmk, a potent, broad-spectrum caspase inhibitor rescued cells from apoptosis but not necroptosis. When the cells were pretreated with both zVAD-fmk and Nec-1, both processes were inhibited. The use of selective inhibitors for caspase-8 and caspase-8:FLIPL (zVAD-Bzl-X) or caspase-10 (zVAD-AOMK-X) demonstrated that caspase-8 but not caspase-10 was involved in necroptosis in HT-29 cells. zVAD-AOMK-Phe, a potent and selective caspase-10 inhibitor had no effect on either apoptosis or necroptosis, while zVAD-Bzl (a selective and potent caspase-8 inhibitor) partially rescued HT29 cells from apoptosis and induced necroptosis
Next, we aimed to additionally challenge our conclusions that caspase-8, but not caspase-10 is mainly responsible for necroptosis in HT29 cells. To do so, we performed caspase-8 and caspase-10 siRNA-mediated knockdowns in HT29 cells, followed by the induction of cell death (Fig. 6a). Western blotting analysis revealed that indeed, transfecting cells with caspase-8 or caspase-10 siRNA resulted in a significant decrease of the expression of these enzymes, however, we could not completely deplete caspase-8 (Fig. 6b, c). In the first step, caspase-8 or caspase-10 genes were silenced by administering siRNA/lipofectamine solution for 72 h prior to experiment. Afterward, wild type and knockdown cells were preincubated with the panel of inhibitors and stimulated with TRAIL/CHX to induce cell death. The data demonstrated that caspase-8 depletion resulted in the significant increase of cell survival when compared to wild type and caspase-10 depleted cells, highlighting its prominent role in TRAIL/CHX-mediated cell death (Fig. 6d). In a follow-up experiment, all HT29 cell variants (WT, Casp-8 siRNA and Casp-10 siRNA) were preincubated with selected zVAD inhibitors containing various binding groups (-fmk, -bzl, and -aomk) and +/− necrostatin-1, followed by TRAIL/CHX treatment. The differential use of Nec-1 allowed to directly measure the level of necroptosis and apoptosis in HT29 cells. The data show that in caspase-8 depleted cells, the necroptosis is significantly reduced, whereas depletion of caspase-10 had no effect on cell death, neither apoptosis, nor necroptosis (Fig. 6e). Moreover, the reduced necroptosis in casp-8 siRNA cells is in line with the model where inhibited caspase-8 is required for necrosome assembly [39,40,41].

Dissecting the role of caspases in necroptosis using wild type and caspase-8/caspase-10 depleted HT29 cells. a The schematic representation of three cell models to study necroptosis: wild type, caspase-8 depleted and caspase-10 depleted cells. b, c Time-course of the depletion of caspase-8 and caspase-10 in HT29 cells using siRNAs. The efficiency of caspase knockdowns was calculated based on the fluorescence intensity of detected bands. d The effect of caspase-8/caspase-10 depletion on TRAIL/CHX-induced cell death. Wild type cells and caspase siRNA variants were preincubated with panel of caspase inhibitors and stimulated with TRAIL/CHX for 24 h. Cells with reduced caspase-8 expression were less susceptible for cell death (apoptosis+necroptosis), while cells lacking caspase-10 displayed similar “death pattern” as wild type HT29. The “−” in the x-axis indicates that inhibitor is lacking amino acid in the prime position. e Necroptotic pattern in three HT29 variants. All cell types were preincubated with inhibitors and with/without Nec-1 followed by TRAIL/CHX treatment. Next, the level of apoptosis and necroptosis were calculated. Results showed that caspase-8 siRNA cells displayed significantly lower necroptosis comparing to both, wild type and caspase-10 siRNA cells
Conclusions
The development of selective tools that enable the distinction between particular caspases is of great importance since they are of exceptional use in biochemical and biological studies. Although there have been many attempts to find selective compounds that can be used to suppress individual caspases and reveal their involvement in various programmed cell death pathways, none have been sufficient enough to distinguish between closely related cysteine proteases that exhibit overlapping substrate specificity. To address this issue we used a novel, chemical strategy to examine the prime region in caspases, as its analysis is informative. The advantage of our approach is the ease, the speed and yield of the solid-state synthesis of bifunctional, prime site-exploring compounds. Using our inhibitors we showed the differences and similarities in the binding strengths of particular caspases (expressed as Ki values) and proved the importance of the prime region in their active sites. Our detailed kinetic analysis demonstrated that although all these inhibitor contain the same peptide motif (zVAD) motif in the non-prime region, they can be fairly distinguished using only one amino acid in the prime region of active site groove. In addition, the selectivity toward particular caspase can be largely improved by applying its optimal non-prime peptide sequence.
In order to test our compounds in cell-based experiments, we transformed our reversible inhibitors into irreversible AOMK-based analogs, since these compounds are more potent in in cellulo studies. Surprisingly, we discovered that these compounds exhibited only reversible inhibition kinetics for caspase-8 and irreversible inhibition kinetics for the other caspases, i.e., caspase-3, caspase-7, and caspase-10. We then we used selected Bzl and AOMK inhibitors to dissect cell death mechanisms in the HT-29 human colon cancer cell line. Necroptosis is a type of cell death that can be initiated exclusively by treatment with tumor necrosis factor (TNF) in the presence of caspase inhibitors such as zVAD-fmk, which inhibits caspase-8 function [20, 42]. Using small inhibitors, we demonstrated that necroptosis in HT-29 human colon cancer cells was mediated by caspase-8 (and caspase-8:FLIPL), as its selective inhibition blocked apoptosis and promoted necroptosis. The inhibition of caspase-10 did not affect cell viability and we concluded that it had no measurable impact on apoptosis or necroptosis. In addition, we performed caspase-8 and caspase-10 knockdown experiments to further confirm the negligible role of caspase-10 in TRAIL/CHX/inhibitors-mediated cell death. Indeed, depletion of caspase-10 from HT-29 cells did not affect necroptosis, whereas silencing of caspase-8 resulted in increased cell survival. We postulate that our methodology may be used to determine which caspases are involved in other types of cell death, i.e., pyroptosis, as we have already demonstrated that by exploring the prime site specificity, a significant inhibitory potency and selectivity can be achieved.
References
Grutter MG. Caspases: key players in programmed cell death. Curr Opin Struct Biol. 2000;10:649–55.
Rawlings ND, Waller M, Barrett AJ, Bateman A. MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic acids Res. 2014;42(Database issue):D503–9.
Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–27.
Vercammen D, Beyaert R, Denecker G, Goossens V, Van Loo G, Declercq W, et al. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J Exp Med. 1998;187:1477–85.
Silke J, Strasser A. The FLIP Side of Life. Sci Signal. 2013;6:pe2.
Silke J, Meier P. Inhibitor of apoptosis (IAP) proteins-modulators of cell death and inflammation. Cold Spring Harb Perspect Biol 2013;5: pii: a008730.
Lork M, Verhelst K, Beyaert R. CYLD, A20 and OTULIN deubiquitinases in NF-kappaB signaling and cell death: so similar, yet so different. Cell Death Differ. 2017;24:1172–83.
Tanzer MC, Khan N, Rickard JA, Etemadi N, Lalaoui N, Spall SK, et al. Combination of IAP antagonist and IFNgamma activates novel caspase-10- and RIPK1-dependent cell death pathways. Cell Death Differ. 2017;24:481–91.
Berger AB, Sexton KB, Bogyo M. Commonly used caspase inhibitors designed based on substrate specificity profiles lack selectivity. Cell Res. 2006;16:961–3.
Benkova B, Lozanov V, Ivanov IP, Mitev V. Evaluation of recombinant caspase specificity by competitive substrates. Anal Biochem. 2009;394:68–74.
Pereira NA, Song Z. Some commonly used caspase substrates and inhibitors lack the specificity required to monitor individual caspase activity. Biochem Biophys Res Commun. 2008;377:873–7.
McStay GP, Salvesen GS, Green DR. Overlapping cleavage motif selectivity of caspases: implications for analysis of apoptotic pathways. Cell Death Differ. 2008;15:322–31.
Poreba M, Szalek A, Kasperkiewicz P, Rut W, Salvesen GS, Drag M. Small molecule active site directed tools for studying human caspases. Chem Rev. 2015;115:12546–629.
Schechter I, Berger A. On the size of the active site in proteases. I. Papain. Biochem Biophys Res Commun. 1967;27:157–62.
Poreba M, Szalek A, Rut W, Kasperkiewicz P, Rutkowska-Wlodarczyk I, Snipas SJ, et al. Highly sensitive and adaptable fluorescence-quenched pair discloses the substrate specificity profiles in diverse protease families. Sci Rep. 2017;7:43135.
Powers JC, Asgian JL, Ekici OD, James KE. Irreversible inhibitors of serine, cysteine, and threonine proteases. Chem Rev. 2002;102:4639–750.
Ganesan R, Jelakovic S, Campbell AJ, Li ZZ, Asgian JL, Powers JC, et al. Exploring the S4 and S1 prime subsite specificities in caspase-3 with aza-peptide epoxide inhibitors. Biochemistry. 2006;45:9059–67.
Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495–516.
McStay GP, Green DR. Measuring apoptosis: caspase inhibitors and activity assays. Cold Spring Harb Protoc. 2014;2014:799–806.
Riedl SJ, Shi Y. Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol cell Biol. 2004;5:897–907.
Weinlich R, Oberst A, Beere HM, Green DR. Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol. 2017;18:127–36.
Kato D, Boatright KM, Berger AB, Nazif T, Blum G, Ryan C, et al. Activity-based probes that target diverse cysteine protease families. Nat Chem Biol. 2005;1:33–8.
Poreba M, Kasperkiewicz P, Snipas SJ, Fasci D, Salvesen GS, Drag M. Unnatural amino acids increase sensitivity and provide for the design of highly selective caspase substrates. Cell Death Differ. 2014;21:1482–92.
Maly DJ, Leonetti F, Backes BJ, Dauber DS, Harris JL, Craik CS, et al. Expedient solid-phase synthesis of fluorogenic protease substrates using the 7-amino-4-carbamoylmethylcoumarin (ACC) fluorophore. J Org Chem. 2002;67:910–5.
Stennicke HR, Salvesen GS. Caspases: preparation and characterization. Methods. 1999;17:313–9.
Gonzalez ML, Poreba M, Snipas SJ, Groborz K, Drag M, Salvesen GS. Extensive peptide and natural protein substrate screens reveal that mouse caspase-11 has much narrower substrate specificity than caspase-1. J Biol Chem. 2018;293:7058–67.
Copeland RA. Enzymes: a practical introduction to structure, mechanism, and data analysis. 2nd edn. New York: Wiley; 2000.
Boatright KM, Deis C, Denault JB, Sutherlin DP, Salvesen GS. Activation of caspases-8 and -10 by FLIP(L). Biochem J. 2004;382(Pt 2):651–7.
Sosna J, Philipp S, Fuchslocher Chico J, Saggau C, Fritsch J, Foll A, et al. Differences and similarities in TRAIL- and tumor necrosis factor-mediated necroptotic signaling in cancer cells. Mol Cell Biol. 2016;36:2626–44.
Voigt S, Philipp S, Davarnia P, Winoto-Morbach S, Roder C, Arenz C, et al. TRAIL-induced programmed necrosis as a novel approach to eliminate tumor cells. BMC Cancer. 2014;14:74.
Fauster A, Rebsamen M, Huber KV, Bigenzahn JW, Stukalov A, Lardeau CH, et al. A cellular screen identifies ponatinib and pazopanib as inhibitors of necroptosis. Cell Death Dis. 2015;6:e1767.
Vince JE, Wong WW, Khan N, Feltham R, Chau D, Ahmed AU, et al. IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell. 2007;131:682–93.
Van Noorden CJF. The history of Z-VAD-FMK, a tool for understanding the significance of caspase inhibition. Acta Histochem. 2001;103:241–51.
Kamens J, Paskind M, Hugunin M, Talanian RV, Allen H, Banach D, et al. Identification and characterization of ICH-2, a novel member of the interleukin-1 beta-converting enzyme family of cysteine proteases. J Biol Chem. 1995;270:15250–6.
Munday NA, Vaillancourt JP, Ali A, Casano FJ, Miller DK, Molineaux SM, et al. Molecular cloning and pro-apoptotic activity of ICErelII and ICErelIII, members of the ICE/CED-3 family of cysteine proteases. J Biol Chem. 1995;270:15870–6.
Declercq W, Vanden Berghe T, Vandenabeele P. RIP kinases at the crossroads of cell death and survival. Cell. 2009;138:229–32.
Pop C, Oberst A, Drag M, Van Raam BJ, Riedl SJ, Green DR, et al. FLIP(L) induces caspase 8 activity in the absence of interdomain caspase 8 cleavage and alters substrate specificity. Biochem J. 2011;433:447–57.
Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, et al. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature. 2011;471:363–7.
Moriwaki K, Chan FK. RIP3: a molecular switch for necrosis and inflammation. Genes Dev. 2013;27:1640–9.
Vandenabeele P, Declercq W, Van Herreweghe F, Vanden Berghe T. The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Sci Signal. 2010;3:re4.
Tummers B, Green DR. Caspase-8: regulating life and death. Immunol Rev. 2017;277:76–89.
Zhou W, Yuan J. Necroptosis in health and diseases. Semin Cell Dev Biol. 2014;35:14–23.
Acknowledgements
This project was funded by the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No. 661187 (to MP) and by National Science Centre in Poland (grant OPUS, UMO-2018/29/B/NZ102249 to MP). The Salvesen laboratory is supported by Sanford Burnham Prebys NCI Cancer Center Support Grant P30CA030199 and NIH grant (GM99040). The Drag laboratory is supported by the Foundation for Polish Science and National Science Centre in Poland.
Author information
Authors and Affiliations
Contributions
MP and MD conceived the project, KG, GS, MD, and MP designed and KG and MP performed the experiments, MLG and SJS contributed new reagents and tools, KG, GS, MD, and MP drafted and edited the paper. All authors reviewed the results and approved the final version of the paper.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by A. Ashkenazi
Supplementary information
Rights and permissions
About this article

Cite this article
Groborz, K., Gonzalez Ramirez, M.L., Snipas, S.J. et al. Exploring the prime site in caspases as a novel chemical strategy for understanding the mechanisms of cell death: a proof of concept study on necroptosis in cancer cells. Cell Death Differ 27, 451–465 (2020). https://doi.org/10.1038/s41418-019-0364-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-019-0364-z
