
Abstract
Foxp3-expressing regulatory T (Treg) cells are essential for averting autoimmune diseases and maintaining immune homeostasis. However, the molecular mechanisms underlying the development and maintenance of Treg cells are still unclear. Here, we found that T cell-specific deletion of the gene encoding the deubiquitinase POH1 compromised the development of mature T cells, especially CD4+Foxp3+ Treg cells. Moreover, POH1 deficiency significantly attenuated the transition of CD25+ Treg cell precursors into Foxp3+ Treg cells accompanied by downregulation of interleukin 2 (IL-2)-STAT5 signaling. Deletion of POH1 in generated CD4+Foxp3+ Treg cells led to an early onset of fetal autoimmune disorders and a decrease in the pool size of peripheral Treg cells in mice, which were mostly due to decreased expansion of these cells. Thus, these results revealed that POH1 has a pivotal role in the development and maintenance of CD4+Foxp3+ Treg cells and contributes to immune tolerance.
Similar content being viewed by others


Introduction
CD4+ Foxp3-expressing regulatory T (Treg) cells play a central role in preserving immune tolerance. Impaired generation and maintenance of Foxp3+ Treg cells lead to severe autoimmune disorders [1,2,3,4]. Natural Treg (nTreg) cells originate from the thymus and are characterized as a distinct T cell lineage that represents 5−10% of peripheral CD4+ T cells. The thymic development of nTreg cells can be divided into two sequential stages based on the dependence of TCR signaling [5, 6]. The high affinity interaction of T cell antigen receptor (TCR) with self-antigens and costimulatory molecule CD28 generates the CD25+ Treg cell precursors and remodels the chromatin at the Foxp3 locus via the NF-κB-c-Rel transcription factors [7,8,9,10,11]. A subsequent increased responsiveness to interleukin 2 (IL-2) signals results in the transition of Treg cell precursors into Foxp3+ Treg cells dependent on the STAT5 transcription factor and finalizes the development of Treg cells [5, 6, 12, 13]. In addition, it has been reported that expression of the tumor-necrosis factor (TNF) receptor superfamily (TNFRSF) on Treg cell progenitors enhances their sensitivity to IL-2, thereby supporting the conversion of Treg cell precursors into Foxp3+ Treg cells [14]. After egress to the periphery, Treg cells proliferate at a high rate and differentiate into effector Treg cells to maintain host homeostasis [15]. The proliferation of Treg cells is regulated by cytokines, including IL-2 and IL-7, and the CD28 coreceptor [16,17,18]. It has been demonstrated that TCR signaling also contributes to the expansion and function of peripheral Treg cells, especially for effector Treg cells [19]. Impaired TCR signaling in mature Treg cells leads to decreased proliferation and suppressive function. Although great advances have been made for understanding the mechanisms underlying the differentiation and expansion of Treg cells, the precise molecular events responsible for regulating these processes remain unclear and need to be fully elucidated.
POH1/rpn11/PSMD14 is a deubiquitinase in the 19S regulatory particle of the proteasome and regulates various biological processes, including protein stability, double-strand DNA break repairs and drug resistance [20,21,22]. Our previous study has demonstrated the role of POH1 in the promotion of tumor formation via deubiquitinating and stabilizing E2F1 [23]. Recently, it has been found that POH1 positively regulates TRIM21-mediated NF-κB activation and TNF-α production in AdV/IgG-stimulated murine embryonic fibroblasts [24]. However, there is much less information about the effect of POH1 on T cells.
To investigate the function of POH1 in the development of Treg cells, we generated mice with a T cell-specific deletion of the gene encoding POH1. Deletion of POH1 in T cells resulted in downregulation of CD25 and defective proliferation in CD4Single+ thymocytes in response to TCR stimulation. Moreover, POH1 deficiency led to lower frequency of CD25+ Treg cell precursors and defective transition of precursors into Foxp3+ Treg cells accompanied by impaired activation of IL-2-STAT5 signaling. Deletion of POH1 in T cells resulted in a profound defect in Treg cell proliferation and decreased pool size of Treg cells. Furthermore, conditional deletion of POH1 in generated Foxp3+ Treg cells impaired cell-cycle progression of Foxp3+ Treg cells and resulted in fatal autoimmune disorders. Overall, our findings indicated that POH1 has a pivotal role in the development and maintenance of CD4+Foxp3+ Treg cells.
Methods and materials
Mice
The generation of POH1 flox/flox (poh1fl/fl) mice has previously been described [23]. Cd4Cre, Foxp3gfp, and Foxp3YFPCre mice (B6 background) were purchased from the Jackson Laboratory (Jackson Laboratory, Bar Harbor, Maine). Pathogen-free mice were housed under specific pathogen-free conditions and fed regular food and water. All of the experiments involving mice were performed in accordance with the guidelines approved by Shanghai JiaoTong University Animal Care commission. Genotyping was performed by PCR with the standard protocol and the proper primers.
Flow cytometry and cell sorting
Single-cell suspensions were prepared from the thymus, spleen, and lymph nodes. Nonspecific staining was blocked with monoclonal antibody to mouse CD16/CD32 (BD Biosciences Pharmingen, San Joes, CA, USA) and cells were stained with Peridinin chlorophyll protein (Percp)-conjugated anti-CD4 (100432, GK1.5, from biolegend, Santiago, USA), fluorescein isothiocyanate (FITC)-conjugated anti-GITR (11-5874-82, DTA-1), anti-Ki67 (11-5698-82, SolA15), and anti-CD44 (11-0441-81, ZM7), PE-conjugated anti-CD8 (12-0081-85, 53–6.7), anti-CD103 (12-1031-81, 2E7), anti-IL-17 (12-7177-81, eBio17B7), anti-OX40 (12-1341-81, OX-86), anti-CD80 (12-0801-82, 16-10A1) and anti-CD86 (12-0862-82, GL1), phycoerythrin Cy7 (Pecy7)-conjugated anti-CD25 (25−0251-81, PC61.5) and anti-B220 (25−0452-82, RA3-6B2), allophycocyanin (APC)-conjugated anti-CD25 (17-0251-42, PC61.5), anti-CD8 (17-0081-82, 53–6.7), anti-CD45.1 (17-0453-82, A20), anti-CXCR3 (17-1831-82, CXCR3-173), anti-IFN-γ (17-7311-82, XMG1.2), anti-CTLA4 (17-1522-80, UC10-4B9) and anti-CD62L (17-0621-81, MEL-14) (all from eBioscience, Santiago, USA). FITC-, PE-, or APC-conjugated anti-Foxp3 (FJK-16s) staining set (eBioscience, Santiago, USA) was used for Foxp3 staining according to the manufacturer’s recommendations. For intracellular staining of p-STAT5, cells were stimulated with or without IL-2 (50 U/ml) for 15 min, and then fixed in 2% formaldehyde, permeabilized in 90% methanol and labeled with FITC-conjugated anti-p-STAT5 monoclonal antibody (612598, 47/Stat5(pY694), BD Biosciences Pharmingen). For sorting of different subsets of thymocytes, thymocytes from poh1fl/flCd4creFoxp3gfp or control mice were depleted of CD8+ T cells with anti-CD8 microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany) and the remaining cells were stained with anti-CD4 and anti-CD25 (all identified above). The CD4Single+CD25−Foxp3-GFP−, CD4Single+CD25+Foxp3-GFP− Treg cell precursors, CD4Single+CD25+Foxp3-GFP+ Treg cells were sorted. Flow cytometry was done with Calibur or LSR Fortessa (BD Biosciences). Cell sorting was done with MoFlo XDP (Beckman Coulter). Results were analyzed using FlowJo software (TreeStar).
BrdU incorporation assay in vivo
Mice were injected intraperitoneally with 1.5 mg of bromodeoxyuridine (BrdU; BD Biosciences Pharmingen) 16 h before analysis. For detection of BrdU, cells were first stained with anti-Foxp3 followed by intracellular staining of BrdU with an anti-BrdU antibody and 7AAD using a BrdU flow kit according to the manufacturers’ instructions (BD Biosciences Pharmingen). Samples were analyzed by flow cytometry.
CFSE labeling assay
Isolated CD4Single+CD25−Foxp3-GFP− T cells from thymocytes were resuspended in PBS containing 0.1% BSA at 107 cells/mL. Carboxyfluorescein diacetate succinimidyl ester (CFSE; Invitrogen, Carlsbad, CA) was added at a final concentration of 5 μM, and the cells were incubated for 10 min at 37 °C in 5% CO2. The stain was terminated using ice-cold complete RPMI1640 medium at 4 °C for 5 min. The cells were then washed three times with PBS containing 0.1% BSA, resuspended in complete RPMI 1640 medium and stimulated with plate-bound anti-CD3 and anti-CD28 (BD Biosciences Pharmingen) for 4 days. Division of cells was determined with a FACS Calibur and analyzed using FlowJo software.
Cell culture conditions
Cells were resuspended in complete RPMI 1640 medium supplemented with 10% FCS, 100 U/mL penicillin, 100 μg/mL streptomycin, 50 μM 2-mercaptoethanol and 2 mM L-glutamine (Gibco, Grand Island, NY, USA).
CFSE-labeled CD4Single+CD25−Foxp3-GFP− T cells were stimulated with plate-bound anti-CD3 (5 μg/mL) and anti-CD28 (3 μg/mL) for 4 days (all from BD Biosciences Pharmingen). Cells were then collected, and the expression of CD25 and division were analyzed. For the inducible Treg cell assay, CD4Single+CD25−Foxp3-GFP−T cells were stimulated with plate-bound anti-CD3 (5 μg/mL) and anti-CD28 (3 μg/mL) in the presence or absence of TGF-β (2 ng/mL; R&D systems, Minneapolis, MN, USA) and IL-2 (20 U/mL; Peprotech NJ, USA) for 4 days. The expression of Foxp3 was detected by flow cytometry.
To examine cytokine production, cells were stimulated with PMA (20 ng/mL; Sigma-Aldrich, St Louis, MO) and ionomycin (1 μg/mL; Sigma-Aldrich) for 4 h and analyzed via flow cytometry.
RNA sequencing
Splenic CD4+Foxp3+ (YFP+) cells were sorted from 2-week-old poh1fl/flFoxp3Cre (n = 10) or poh1+/+Foxp3Cre (n = 5) mice using the MoFlo XDP. Total RNA was extracted by TRIzol and was subjected to RNA-seq analysis for each group. Total RNA was used for Illumina TruSeq paired-end sequencing library construction following manufacturer’s protocols at the Shanghai Biotechnology Corporation. Samples were sequenced on the Illumina HiSeq 2500. Approximately 50 million clean reads were obtained and then mapped to the mouse genome (mm10). The mapped fragments of genes were calculated using HTSeq followed by trimmed mean of M value normalization to evaluate gene expression by normalized Reads Per Kilobase of exon per Megabase of library size (RPKM). Significant differential expression genes between POH1-sufficient and POH1-deficient Treg cells were identified as those with fold-change ≥ 2 and a false discovery rate ≤ 0.05 using edgeR software. RNA sequencing data were uploaded to the GEO database (accession code GSE110084).
Bioinformatics analysis of RNA-seq
‘TCR-upregulated’ (i.e., TCR-dependent) genes [19] were defined as genes upregulated (a change in expression of at least 1.75-fold, Padj ≤ 0.05) in TCR-sufficient CD44high Treg cells relative to their expression in TCR-deficient CD44high Treg cells with a FDR-adjusted P value of ≤0.05 (GSE61077).
Statistical analysis
Comparison between two groups was performed by an unpaired Student’s t test. A value of P < 0.05 was considered significant.
Results
POH1 deletion impairs thymic foxp3+ treg cell development and T cell homeostasis
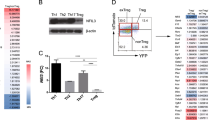
POH1 expression was upregulated in mouse CD4+ T cells upon stimulation of anti-CD3 and anti-CD28 antibodies (Fig. 1a), suggesting that POH1 might have a physiological function in T cells. To this end, we crossed mice carrying loxp-flanked POH1 (poh1fl/fl) with Cd4cre transgenic mice to generate mice with a T cell-specific ablation of POH1 (poh1fl/flCd4cre). We confirmed the efficient deletion of poh1 in CD4Single+ and CD8Single+ thymocytes by DNA detection (Fig. S1a) and real-time PCR (Fig. S1b). poh1fl/flCd4cre mice exhibited lower frequencies of CD4Single+ and CD8Single+ thymocytes compared to their control littermates (Fig. 1b, c). The percentages of CD4+ and CD8+ T cells in the periphery were profoundly reduced in poh1fl/flCd4cre mice (Fig. S1c). We further generated mixed bone marrow (BM) chimeras to corroborate the effects of POH1 deletion on thymocyte development and peripheral homeostasis. Briefly, donor BM cells from poh1fl/flCd4cre (CD45.2+) or poh1+/+Cd4cre (CD45.2+) mice mixed with wild-type (WT) (CD45.1+) BM cells were transferred into lethally irradiated CD45.1+ host mice. Eight weeks after reconstitution, the frequencies of CD4Single+ and CD8Single+ T cells among thymocytes derived from poh1fl/flCd4cre mice were lower than those from CD45.1 WT mice in the mixed chimeras (Fig. 1d). Moreover, a substantial decrease in the proportions of CD4+ and CD8+ T cells was observed in splenocytes and lymph nodes from POH1-deficient BM chimeras compared to control chimeras (Fig. S1d). The peripheral CD4+ T cells in poh1fl/flCd4cre mice contained a higher proportion of activated CD44highCD62Llow cells than that in poh1+/+Cd4cre mice (Fig. S2a and b). Accordingly, the production of pro-inflammatory cytokines, including interferon (IFN)-γ and IL-17, in POH1-deletion peripheral CD4+ T cells was higher than that in control CD4+ T cells (Fig. S2c).

Effects of POH1 ablation on thymic Foxp3+ Treg cell generation. a Western blot analyzing the expression of POH1 and β-actin with or without stimulation of anti-CD3 and anti-CD28 in splenic CD4+ T cells. Data are representative of three independent experiments with similar results. b Flow cytometry analyzing the percentage of CD4Single+ and CD8Single+ T cells in thymocytes derived from 4-week-old poh1fl/flCd4cre and poh1+/+Cd4cre control littermates. c Percentages of various thymocyte subsets derived from 4-week-old poh1fl/flCd4cre and control littermates are presented (n = 9 per genotype). The results from four separate experiments were pooled. d BM cells from poh1fl/flCd4cre (CD45.2+) or poh1+/+Cd4cre mice (CD45.2+) were mixed 1:1 with BM from WT (CD45.1+) C57BL/6 mice, and these cells were injected into lethally irradiated CD45.1+ mice to generate bone marrow chimeras. Eight weeks later, the proportion of CD4Single+ and CD8Single+ T cells among the thymocytes from the same origin is shown. The results from three separate experiments were pooled. e Flow cytometry analyzing the percentage of Foxp3+ Treg cells among CD4Single+ thymocytes from 4-week-old poh1fl/flCd4cre and control mice. f Pooled data from e representing four separate experiments (n = 8 per genotype). g BM cells from poh1fl/flCd4cre (CD45.2+) or poh1+/+Cd4cre mice (CD45.2+) were mixed 1:1 with BM from WT (CD45.1+) C57BL/6 mice, and these cells were injected into lethally irradiated CD45.1+ mice to generate bone marrow chimeras. Eight weeks later, the proportion of Foxp3+ Treg cells among the CD4Single+ thymocytes from the same origin is shown. The results from three separate experiments were pooled. Numbers adjacent to outlined areas show the percent of cells in each. Each symbol represents a single mouse, and horizontal bars represent the mean. *P < 0.05; ***P < 0.001, two-tailed unpaired Student’s t test
While a considerable decrease was observed in the frequencies of thymic CD4Single+ T cells in poh1fl/flCd4cre mice, a more apparent reduction in the percentages of Foxp3+ Treg cells, which reached an approximate 10-fold decrease, occurred in the CD4Single+ thymocytes (Fig. 1e, f). More importantly, the mixed bone marrow experiments showed that POH1 deficiency in T cells resulted in a drastic decrease in the proportion of Foxp3+ Treg cells in T cells, indicating that the decline in the number of Foxp3+ Treg cells in poh1fl/flCd4cre mice was due to the cell-intrinsic effects of POH1 deletion (Fig. 1g and S3a). Therefore, these results demonstrated that POH1 is essential for T cell homeostasis and the generation of nTreg cells.
POH1 deficiency inhibits the differentiating ability of thymic treg precursors
We next focused on the role of POH1 in the development of nTreg cells. We examined if POH1 functions in the development of nTreg cell precursors. POH1 deficiency resulted in a decline in the frequency of CD25+Foxp3− Treg cell precursors and a more pronounced decrease in the proportions of Foxp3-expressing cells (Fig. 2a, b), suggesting a defect in the transition of Treg precursors to Foxp3+ Treg cells in the context of POH1 deficiency. We bred poh1fl/+Cd4cre mice to mice containing the Foxp3 knock-in allele, Foxp3gfp (expressing Foxp3-tagged with green fluorescent protein (GFP)), and generated poh1fl/flCd4creFoxp3gfp mice and control mice were used for purification of CD4Single+CD25−Foxp3-GFP− conventional T (Tcon) cells. Consistently, upon stimulation with anti-CD3 and anti-CD28 antibodies, thymic Tcon cells in the absence of POH1 were unable to induce the expression of CD25 (Fig. 2c) and failed to proliferate (Fig. 2d). We then further evaluated if POH1 has an effect on the transition of Treg cell precursors into Foxp3+ Treg cells. We purified thymic CD4Single+CD25+Foxp3-GFP− Treg cell precursors from poh1fl/flCd4creFoxp3gfp mice and their littermates, and we modeled the second step of nTreg cell differentiation with IL-2 stimulation and then determined the expression of Foxp3. Twenty-four hours after stimulation with IL-2, only approximate 4% of cells from cultured poh1fl/flCd4cre Treg cell precursors acquired the expression of Foxp3, which was in contrast to 16% of those from control cells (Fig. 2e). The IL-2-STAT5 signaling is suggested to be critical for the transition of thymic Treg precursors into Foxp3+Treg cells. More specifically, phosphorylation of STAT5 (p-STAT5) binds to the first intron and promoter of the gene encoding Foxp3 and is critical for initiating Foxp3 expression [12, 13]. In response to IL-2, POH1-deficient Treg cell precursors showed a considerable decline in p-STAT5 levels compared to POH1-sufficient Treg cell precursors (Fig. 2f). Given that the members of the TNFRSF on Treg progenitors are also involved in the differentiation of Treg cells [14], we examined the expression of the TNFRSF members, GITR and OX40, on the CD4Single+CD25+Foxp3− subset. The results showed a pronounced downregulation in the expression of the TNFRSF members on POH1-deficient Treg cell precursors compared to that on control cells (Fig. 2g). Therefore, these results indicated that POH1 deficiency compromises the expression of GITR and OX40 on the precursors of Treg cells as well as IL-2-STAT5 signaling, resulting in decreased efficiency in the transition of precursors into Foxp3+ Treg cells.

POH1-deficiency impairs the transition of precursors into Treg cells. a Flow cytometry analyzing the percentage of CD25+Foxp3−, CD25−Foxp3+ and CD25+Foxp3+ cells among CD4Single+ thymocytes derived from 4-week-old poh1fl/flCd4cre or poh1+/+Cd4cre mice. b Pooled data from a representing three separate experiments. Each symbol represents a single mouse, and horizontal bars represent the mean. c, d Flow cytometry analyzing the expression of CD25 (c) and CFSE labeling (d) in sorted CD4Single+CD25−Foxp3-GFP−thymocytes from poh1fl/flCd4cre or poh1+/+Cd4cre mice. Cells were stimulated with plated bound anti-CD3 and anti-CD28 for 4 days. The numbers above bracketed lines indicate the percentage of proliferated cells. Data are representative of two independent experiments with similar results. e Flow cytometry analyzing the Foxp3 expression in purified thymic CD4Single+CD25+Foxp3-GFP−Treg cell precursors from poh1fl/flCd4cre mice and control littermates after stimulation with or without IL-2 for 24 h. Data are representative of three independent experiments with similar results. f Flow cytometry analyzing the p-STAT5 in purified thymic CD4Single+CD25+Foxp3-GFP− Treg cell precursors from poh1fl/flCd4cre mice and control littermates after stimulation with or without IL-2 for 15 min. Data are representative of two independent experiments with similar results. g Flow cytometry analyzing the expression of GITR and OX40 in CD4Single+CD25+Foxp3-GFP−Treg cell precursors from the thymus of 4-week-old poh1fl/flCd4cre or poh1+/+Cd4cre mice. Data are representative of four independent experiments with similar results. Numbers adjacent to outlined areas show the percent of cells in each. **P < 0.01; ***P < 0.001, two-tailed unpaired Student’s t test
POH1 deletion impairs thymic treg cell expansion
We next examined the phenotype of POH1-deficient thymic CD4+Foxp3+ Treg cells. While the expression of several signature molecules of Treg cells, including GITR, OX40, and CTLA4, was unaffected in POH1-deficient thymic Treg (tTreg) cells (Fig. 3a), the frequency of CD44highCD62Llow cells in these Treg cells was approximately 50% of that in POH1-sufficient Treg cells (Fig. 3b). Because proliferating Treg cells were restricted to the CD44high subset [25], we next determined if POH1 deficiency affects the proliferation of tTreg cells. tTreg cells in the poh1fl/flCd4cre background showed drastically decreased expression of the proliferation marker Ki67 in comparison to that in their littermates (Fig. 3c, d). Accordantly, these POH1-deleted tTreg cells did not incorporate the thymidine analog BrdU (Fig. 3e, f). These results suggested that the reduction of tTreg cells in poh1fl/flCd4cre mice is, in part, caused by impaired cell proliferation. Given the pivotal role of POH1 in the differentiation and proliferation of tTreg cells, we next investigated if POH1 is essential for the generation of induced Treg (iTreg) cells. We sorted CD4Single+CD25−Foxp3−GFP− cells from poh1fl/flCd4creFoxp3gfp mice and their littermates, and we cultured the cells with or without TGF-β and IL-2 in the presence of anti-CD3 and anti-CD28 antibodies. POH1 deficiency substantially inhibited the generation of iTreg cells (Fig. 3g). Thus, these findings suggest that POH1 is indispensable for the generation of both tTreg and iTreg cells.

POH1 is required for Treg cell expansion. a, b Flow cytometry analyzing the expression of GITR, OX40, CTLA4 (a), CD44 and CD62L (b) in CD4Single+Foxp3+ Treg cells from the thymus of poh1fl/flCd4cre mice and control littermates. Data are representative of three independent experiments with similar results. c Flow cytometry analyzing the expression of Ki67 in CD4Single+Foxp3+ Treg cells from the thymus of poh1fl/flCd4cre mice and control littermates. d Pooled data from c representing three separate experiments. Each symbol represents a single mouse, and horizontal bars represent the mean. e Flow cytometry analyzing the incorporation of BrdU in CD4Single+Foxp3+ Treg cells from the thymus of poh1fl/flCd4cre mice and control littermates. f The percentage of BrdU+ cells in thymic Treg cells from poh1fl/flCd4cre mice and control littermates is shown. Data are shown as the mean + SD (n = 3 per genotype) and are representative of two independent experiments. g Flow cytometry analyzing Foxp3 expression in CD4Single+CD25−Foxp3-GFP− thymocytes from poh1fl/flCd4cre or oh1+/+Cd4cre mice. Cells were stimulated with or without TGF-β and IL-2 in the presence of plated bound anti-CD3 and anti-CD28 for 4 days. Data are representative of three independent experiments with similar results. **P < 0.01; ***P < 0.001, two-tailed unpaired Student’s t test
We further assessed the characteristics of peripheral CD4+Foxp3+ Treg (pTreg) cells in poh1fl/flCd4cre mice and found that the levels of the Treg cell signature molecules, GITR, OX40, and CTLA4, in these cells were comparable to those in poh1+/+Cd4cre pTreg cells (Fig. S3b). The proportion of CD44highCD62Llow effector/memory Treg cells was significantly higher in the absence of POH1 than in the presence of POH1 (Fig. S3c). In addition, surface expression of the CD103 and CXCR3 activation markers was higher in POH1-deficient Treg cells than in POH1-sufficient Treg cells (Fig. S3d). The activated phenotype of POH1-ablated peripheral Foxp3+ Treg cells might be a consequence of the lymphopenic environment in these mice.
Mice with treg-specific deficiency of POH1 develop lethal autoimmunity
To further corroborate the requirement of POH1 in the maintenance of differentiated Foxp3+ Treg cells, we generated mice with Treg-specific deficiency of POH1 (poh1fl/flFoxp3Cre) by crossing poh1fl/fl mice with mice expressing the YFP-Cre fusion recombinase from the Foxp3 locus (Foxp3YFPCre). In the absence of POH1 in Treg cells, mice became sick as indicated by a hunched posture, reduced body size and decreased mobility as well as crusted ears, eyelids and tails (Fig. 4a left). The highly aggressive and overwhelming autoimmunity manifested by shrinking of the thymus, lymphadenopathy, and splenomegaly led to the death of mice at 3−4 weeks of age (Fig. 4a right and Fig. 4b). poh1fl/flFoxp3Cre mice showed inflammatory cell infiltration in peripheral organs, including the kidneys, lungs, and livers (Fig. 4c). Of note, poh1fl/flFoxp3Cre mice contained a larger proportion of the CD44highCD62Llow effector/memory population in peripheral CD4+ T cells compared to control littermates (Fig. 4d). Furthermore, deletion of POH1 in Treg cells resulted in higher production of the pro-inflammatory cytokine, IFN-γ, but not IL-17 in CD4+ T cells from the spleen and lymph nodes (Fig. 4e). Accordingly, the expression levels of the CD80 and CD86 costimulatory molecules on B cells were increased in the poh1fl/flFoxp3Cre mice (Fig. 4f). These results indicated a critical role for POH1 in generated Treg cells.

Treg-specific deficiency of POH1 leads to lethal autoimmune inflammation. a Typical littermate and poh1fl/flFoxp3Cre mice (left). Thymi, spleens and lymph nodes from 19-day-old poh1fl/flFoxp3Cre mice and poh1+/+Foxp3Cre control littermates (right). The images are representative of 20 independent mice with similar observations. b Survival of poh1fl/flFoxp3Cre and control mice monitored for 50 days (n = 9 mice per genotype). c Hematoxylin and eosin staining of kidney, lung and liver sections from 19-day-old poh1fl/flFoxp3Cre mice and control littermates. Data are representative of four independent experiments with similar results. d Flow cytometry analyzing the expression of CD44 and CD62L in CD4+ T cells from the spleens and lymph nodes of 19-day-old poh1fl/flFoxp3Cre mice and control littermates. Data are representative of five independent experiments with similar results. e Flow cytometry profiles of spleens and lymph nodes from poh1fl/flFoxp3Cre mice and poh1+/+Foxp3Cre control littermates showing expression of IFN-γ and IL-17 in CD4+ T cells stimulated for 4 h with PMA and ionomycin in the presence of brefeldin A. Data are representative of four independent experiments with similar results. f Flow cytometry analyzing the CD80 and CD86 expression in B cells from spleens of 19-day-old poh1fl/flFoxp3Cre mice and control littermates. Data were pooled from two independent experiments. Each symbol represents a single mouse, and horizontal bars represent the mean. *P < 0.05; ***P < 0.001, two-tailed unpaired Student’s t test
The frequency of tTreg cells in CD4Single+ T cells from poh1fl/flFoxp3Cre mice was comparable to that from poh1+/+Foxp3Cre mice, which may have resulted from a ‘carryover’ effect. In contrast, the expression of Foxp3 among CD4+ T cells in the spleen and lymph nodes was profoundly diminished in poh1fl/flFoxp3Cre mice compared to its expression in poh1+/+Foxp3Cre mice (Fig. 5a). CD25 expression on pTreg cells was not affected by POH1 deficiency (Fig. 5b). To directly evaluate if POH1 deletion affects the responsiveness of Foxp3+ Treg cells to IL-2, we isolated splenic Treg cells based on YFP expression from poh1fl/flFoxp3cre and poh1+/+Foxp3cre control mice. The cells were cultured with or without IL-2 for 15 min, and p-STAT5 levels were then determined. POH1-deficient and POH1-sufficient Treg cells had a comparable level of p-STAT5 after IL-2 stimulation (Fig. 5c). The increased frequency of CD44lowCD62Lhigh naive-like Foxp3+ cells in pTreg cells was found in the absence of POH1 (Fig. 5d). Several characteristic factors of Treg cells, including GITR, OX40, CTLA4, and PD1, were also assessed. The expression of GITR, CTLA4, and PD1 was lower in POH1-deficient Treg cells than in POH1-sufficient Treg cells (Fig. 5e). We then speculated if POH1 ablation affects the proliferation of pTreg cells. A significantly lower frequency of Ki67+ proliferative Treg cells in poh1fl/flFoxp3Cre mice was found compared to that in their control littermates (Fig. 5f). In addition, POH1 deficiency substantially decreased proliferation of Treg cells resulting from cell-cycle arrest at the G1-S transition (Fig. 5g, h).

POH1 maintains the pool size of generated Treg cells. a Flow cytometry analyzing the expression of Foxp3 among CD4+ T cells from the thymi, spleens and lymph nodes of 19-day-old poh1fl/flFoxp3Cre mice and control littermates. Data were pooled from three independent experiments. b Flow cytometry analyzing the expression of CD25 in Foxp3+ Treg cells from spleens of poh1fl/flFoxp3Cre mice and control littermates. Data are representative of four independent experiments with similar results. c Flow cytometry analyzing p-STAT5 in purified splenic Foxp3+ Treg cells from spleens of poh1fl/flFoxp3Cre mice and control littermates after stimulation with or without IL-2 for 15 min. Data are representative of two independent experiments with similar results. d–f Flow cytometry analyzing the expression of CD44, CD62L (d), GITR, CTLA4, OX40, PD1 (e), and Ki67 (f) in peripheral Foxp3+ Treg cells from poh1fl/flFoxp3Cre mice and control littermates. Data are representative of three independent experiments with similar results. g Flow cytometry analyzing the cell cycle of splenic Foxp3+ Treg cells from poh1fl/flFoxp3Cre mice and control littermates. h Cell cycle analysis of splenic Foxp3+ Treg cells from poh1fl/flFoxp3Cre mice and control littermates. Each symbol represents a single mouse, and horizontal bars represent the mean
Alterations in the transcriptional profile of POH1-deficient foxp3+ treg cells
To investigate the potential mechanisms underlying the defective proliferation conferred by POH1 deficiency, we sorted pTreg cells from poh1fl/flFoxp3cre and poh1+/+Foxp3cre control mice and analyzed gene expression by high-throughput RNA sequencing (RNA-seq). The gene expression profile of POH1-deficient Treg cells (poh1−/− Foxp3+) was markedly different than that in POH1-sufficient Treg cells (poh1+/+ Foxp3+) (Fig. 6a). Comparison to a previously defined TCR signature [19] demonstrated that a set of TCR-activated genes was transcriptionally downregulated by POH1 ablation in Treg cells (Fig. 6b–e). Accordingly, POH1 deletion in Treg cells also caused altered expression of the genes related to cell-cycle progression. p21, encoded by the Cdkn1a gene and responsible for cell-cycle arrest at the G1-S transition, was transcriptionally upregulated in poh1fl/flFoxp3cre Treg cells relative to poh1+/+Foxp3cre Treg cells (Fig. 6d, e). The RNA-seq data were validated by real-time PCR (Fig. 6e). In addition, the gene sets downregulated in POH1-deficient Treg cells included those encoding certain cell surface receptors and intracellular molecules involved in migration, suppressive function, and properties of Treg cells [26] (Fig. S4).

RNA-seq analysis of POH1-deficient and POH1-sufficient Treg cells. a Gene expression (log2 normalized) in Treg cells from poh1fl/flFoxp3Cre mice (poh1−/− Foxp3+) or poh1+/+Foxp3Cre control littermates (poh1+/+ Foxp3+). b Empirical cumulative distribution function for the change in expression (log2 values) of all genes expressed in POH1-deficient Treg cells and for the subsets of genes upregulated (TCR up) in a TCR-dependent manner. Numbers in parentheses (key) indicate total genes in each group. c Expression of selected genes upregulated in a TCR-dependent manner in POH1-deficient or control Treg cells. d Expression of selected genes associated with cell cycle. e Validation of RNA-seq data. Genes were selected from the RNA-seq data, and the mRNA expression was analyzed by real-time PCR in Treg cells from 2-week-old poh1fl/flFoxp3Cre mice or poh1+/+Foxp3Cre control littermates. mRNA levels were normalized with GAPDH, and mRNA levels in control Treg cells were arbitrarily set to 1. Data are shown as the mean + SD and are representative of two independent experiments
Discussion
The development of Foxp3+ Treg cells is a tightly regulated process that is essential for immune homeostasis. The ubiquitin-dependent pathway has an important role in mediating the differentiation and maintenance of Foxp3+ Treg cells. Our present study demonstrated that the deubiquitinating enzyme, POH1, is an important factor that regulates the differentiation of tTreg cells. Deletion of POH1 in T cells leads to a less efficient transition from Treg cell precursors to Treg cells accompanied by decreased activation of IL-2-STAT5 signaling. Furthermore, POH1 is required for the proliferation of generated Treg cells.
POH1 deficiency in T cells resulted in a lower frequency of CD4Single+ and CD8Single+ T thymocytes, indicating that POH1 has a significant role in the development of T cells. Moreover, POH1 ablation caused a more substantial decrease in Treg generation than that of Tcon cells in the thymus, suggesting that POH1-mediated regulation is particularly critical for the differentiation of nTreg cells. Accumulated evidence has demonstrated that TCR and IL-2 signals are required for the differentiation of nTreg cells. TCR signaling is essential for generation of CD25+ Treg cell progenitors, while IL-2 signaling is required for the transition of Treg cell precursors into Treg cells [5, 6]. We observed a less efficient transition from progenitors to Treg cells in response to IL-2 in the absence of POH1, which may have reflected a direct inhibition of signaling via IL-2R because downstream p-STAT5 was also reduced. Given that activated STAT5 directly binds to the first intron and promoter of Foxp3 and initiates its expression [12, 13], we hypothesize that POH1 regulation of the transition of Treg cell precursors to Treg cells is at least partially through modulating IL-2-STAT5 signaling. A previous study has demonstrated that Treg cell progenitors have high expression of the members of TNFRSF, including GITR, OX40, and TNFR. Co-stimulation with GITRL, OX40L, and TNF increases the sensitivity of progenitor cells to IL-2, thereby enhancing IL-2-dependent activation of STAT5 [14]. Accordingly, downregulation of GITR and OX40 caused by POH1 deficiency might contribute to a decreased response of Treg cell progenitors to IL-2 and low efficiency in the conversion of progenitors to Treg cells. Further studies are needed to clarify if other events underlie POH1 regulation of the transition.
We generated poh1fl/flFoxp3cre mice to study the role of POH1 in generated Foxp3+ Treg cells. A large number of Treg cells in poh1fl/flFoxp3cre mice had a naive-like phenotype and defective proliferation, suggesting that effector differentiation and expansion of Treg cells are dependent on the presence of POH1. Indeed, our results showed that TCR-driven transcription was weakened in POH1-defieient Treg cells, suggesting that TCR signaling is impaired in peripheral POH1-ablated Treg cells. Although the IL-2 activation of STAT5 was significantly decreased in POH1-deleted Treg cell precursors upon IL-2 stimulation, the strength of IL-2-STAT5 signaling in generated Treg cells (poh1fl/flFoxp3cre) was not remarkably changed in the absence of POH1, indicating the complexity of POH1-mediated regulation at different stages of Treg cell development. Moreover, this phenomenon is reminiscent of generated Treg cells lacking TCR signaling that display impairment in cell proliferation but not in the activation of IL-2-STAT5 signaling [19]. Nevertheless, our data suggest that the compromised proliferation of POH1-deficient Treg cells and their naive-like phenotypes are at least partly due to defective TCR signaling.
The impaired proliferative activity of Treg cells caused by POH1 ablation was associated with defective cell-cycle progression. p21, the p53-regulated cyclin-dependent kinase inhibitor, causes cell-cycle arrest at the G1-S transition and has an important role in controlling the proliferation of many cell types, including colonic Treg cells [27]. It has been reported that POH1 negatively regulates the expression of p21 in H1299 cancer cells [28]. In our study, we observed that POH1 ablation in Treg cell resulted in upregulation of p21 expression. It is also noteworthy that the other major regulators of cell proliferation, including cyclin E-CDK2 and cyclin D1-CDK4/6 genes, were downregulated in POH1-ablated Treg cells. These data suggested that POH1 in Treg cells has a role in promoting cell-cycle progression.
Currently, several reports have demonstrated that aberrantly high expression of POH1 in tumor cells contributes to the development and progression of certain human cancers [23, 29, 30]. However, the clinical relevance of POH1 function in the maintenance of immune homeostasis remains uncertain. More specifically, although we reveal that POH1 expression is required for the generation and maintenance of Treg cells in mice, it is unclear whether POH1 expression is dysregulated and thereby causally related to inappropriate function of Treg cells in clinical patients. Intriguingly, a previous study showed that the transcript abundance of POH1 in PBMCs of patients with the autoimmune diseases is decreased in comparison with that of healthy individuals [31]. However, more detailed delineations of the expression pattern and biological function of POH1 in different types of human immune cells are certainly required for addressing the importance of POH1 in regulating immune responses and pathogenic processes. Therefore, the clinical significance of POH1 in immune cells warrants further investigation.
References
Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–61.
Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–6.
Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol. 2012;30:531–64.
Dawson NAJ, Vent-Schmidt J, Levings MK. Engineered tolerance: tailoring development, function, and antigen-specificity of regulatory T cells. Front Immunol. 2017;8:1460.
Lio CW, Hsieh CS. A two-step process for thymic regulatory T cell development. Immunity. 2008;28:100–11.
Burchill MA, Yang J, Vang KB, Moon JJ, Chu HH, Lio CW, et al. Linked T cell receptor and cytokine signaling govern the development of the regulatory T cell repertoire. Immunity. 2008;28:112–21.
Jordan MS, Boesteanu A, Reed AJ, Petrone AL, Holenbeck AE, Lerman MA, et al. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat Immunol. 2001;2:301–6.
Kieback E, Hilgenberg E, Stervbo U, Lampropoulou V, Shen P, Bunse M, et al. Thymus-derived regulatory T cells are positively selected on natural self-antigen through cognate interactions of high functional avidity. Immunity. 2016;44:1114–26.
Ruan Q, Kameswaran V, Tone Y, Li L, Liou H-C, Greene M-I, et al. Development of Foxp3+ regulatory T cells is driven by the c-Rel enhanceosome. Immunity. 2009;31:932–40.
Long M, Park SG, Strickland I, Hayden MS, Ghosh S. Nuclear factor-κB modulates regulatory T cell development by directly regulating expression of Foxp3 transcription factor. Immunity. 2009;31:921–31.
Vang KB, Yang J, Pagán AJ, Li LX, Wang J, Green JM, et al. Cutting edge: CD28 and c-Rel-dependent pathways initiate regulatory T cell development. J Immunol. 2010;84:4074–7.
Burchill MA, Yang J, Vogtenhuber C, Blazar BR, Farrar MA. IL-2 receptor β-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J Immunol. 2007;178:280–90.
Yao Z, Kanno Y, Kerenyi M, Stephens G, Durant L, Watford WT, et al. Nonredundant roles for Stat5a/b in directly regulating Foxp3. Blood. 2007;109:4368–75.
Mahmud SA, Manlove LS, Schmitz HM, Xing Y, Wang Y, Owen DL, et al. Costimulation via the tumor-necrosis factor receptor superfamily couples TCR signal strength to the thymic differentiation of regulatory T cells. Nat Immunol. 2014;15:473–81.
Pierson W, Cauwe B, Policheni A, Schlenner SM, Franckaert D, Berges J, et al. Antiapoptotic Mcl-1 is critical for the survival and niche-filling capacity of Foxp3+ regulatory T cells. Nat Immunol. 2013;14:959–65.
Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol. 2005;6:1142–51.
Simonetta F, Gestermann N, Martinet KZ, Boniotto M, Tissières P, Seddon B, et al. Interleukin-7 influences FOXP3+CD4+ regulatory T cells peripheral homeostasis. PLoS ONE. 2012;7:e36596.
Appleman LJ, Berezovskaya A, Grass I, Boussiotis VA. CD28 costimulation mediates T cell expansion via IL-2-independent and IL-2-dependent regulation of cell cycle progression. J Immunol. 2000;164:144–51.
Levine AG, Arvey A, Jin W, Rudensky AY. Continuous requirement for the TCR in regulatory T cell function. Nat Immunol. 2014;15:1070–8.
Verma R, Aravind L, Oania R, McDonald WH, Yates JR 3rd, Koonin EV, et al. Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science. 2002;298:611–5.
Butler LR, Densham RM, Jia J, Garvin AJ, Stone HR, Shah V, et al. The proteasomal de-ubiquitinating enzyme POH1 promotes the double-strand DNA break response. EMBO J. 2012;31:3918–34.
Spataro V, Simmen K, Realini CA. The essential 26S proteasome subunit Rpn11 confers multidrug resistance to mammalian cells. Anticancer Res. 2002;22:3905–9.
Wang B, Ma A, Zhang L, Jin WL, Qian Y, Xu G, et al. POH1 deubiquitylates and stabilizes E2F1 to promote tumour formation. Nat Commun. 2015;6:8704.
Fletcher AJ, Mallery DL, Watkinson RE, Dickson CF, James LC. Sequential ubiquitination and deubiquitination enzymes synchronize the dual sensor and effector functions of TRIM21. Proc Natl Acad Sci USA. 2015;112:10014–9.
Huehn J, Siegmund K, Lehmann JC, Siewert C, Haubold U, Feuerer M, et al. Developmental stage, phenotype, and migration distinguish naive- and effector/memory-like CD4+ regulatory T cells. J Exp Med. 2004;199:303–13.
Campbell DJ, Koch MA. Phenotypical and functional specialization of FOXP3+ regulatory T cells. Nat Rev Immunol. 2011;11:119–30.
Obata Y, Furusawa Y, Endo TA, Sharif J, Takahashi D, Atarashi K, et al. The epigenetic regulator Uhrf1 facilitates the proliferation and maturation of colonic regulatory T cells. Nat Immunol. 2014;15:571–9.
Byrne A, McLaren RP, Mason P, Chai L, Dufault MR, Huang Y, et al. Knockdown of human deubiquitinase PSMD14 induces cell cycle arrest and senescence. Exp Cell Res. 2010;316:258–71.
Wang CH, Lu SX, Liu LL, Li Y, Yang X, He YF, et al. POH1 knockdown induces cancer cell apoptosis via p53 and Bim. Neoplasia. 2018;20:411–24.
Zhu R, Liu Y, Zhou H, Li L, Li Y, Ding F, et al. Deubiquitinating enzyme PSMD14 promotes tumor metastasis through stabilizing SNAIL in human esophageal squamous cell carcinoma. Cancer Lett. 2018;418:125–134.
Maas K, Chan S, Parker J, Slater A, Moore J, Olsen N, et al. Cutting edge: molecular portrait of human autoimmune disease. J Immunol. 2002;169:5–9.
Acknowledgements
This work was supported by grants from National Natural Science Foundation of China (31770976, 81572293, and 81402348), the State Key Laboratory of Oncogenes and Related Genes (91–1705), the Shanghai Rising-Star Program (17QA1403700).
Author contributions
YL and YZL designed research; YL, LZ, BSW, ZJY, GQX, AHM, MT, TTJ, LW, and XLX performed research; YL, LZ, and YZL analyzed data; and YL, LZ, and YZL wrote the paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Edited by M. Piacentini.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Liu, Y., Zhang, L., Wang, B. et al. Requirement for POH1 in differentiation and maintenance of regulatory T cells. Cell Death Differ 26, 751–762 (2019). https://doi.org/10.1038/s41418-018-0162-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-018-0162-z
This article is cited by
-
Six immune-related promising biomarkers may promote hepatocellular carcinoma prognosis: a bioinformatics analysis and experimental validation
Cancer Cell International (2023)
-
Deubiquitinating enzymes (DUBs): DoUBle-edged swords in CNS autoimmunity
Journal of Neuroinflammation (2020)
-
Regulatory T cells in tumor microenvironment: new mechanisms, potential therapeutic strategies and future prospects
Molecular Cancer (2020)
