
Abstract
Chromosome translocation t(12;22)(p13;q12)/MN1-ETV6 and MN1 overexpression confer a subset of adverse prognostic AML but so far lack in-depth research. We focused on the clinical course and comprehensive genetic analysis of eight cases with t(12;22)(p13;q12) and one with t(12;17;22) (p13;q21;q13) to elucidate their molecular etiology and outcomes of allogeneic hemopoietic stem cell transplantation (allo-HSCT). The total incidence of t(12;22)(p13;q12) and related translocations was 0.32% in myeloid neoplasms. These patients were confirmed to have dismal prognosis when treated only with chemotherapy, and we firstly provided evidence that they can significantly benefit from timely allo-HSCT. Five cases were MN1-ETV6 positive, and a novel MN1-STAT3 fusion was identified in the patient with triadic translocation. Significant MN1 overexpression was observed in all three MN1-fusion-negative cases. Genetic analysis highlighted the evidence of an ectopic super-enhancer associated orchestrated mechanism of MN1 overexpression and ETV6 haploinsufficiency in t(12;22)(p13;q12) myeloid neoplasms, rather than the conventional thought of MN1-ETV6 fusion formation. We also disclosed the high concomitance of trisomy 8 and 531 Kbps focal 8q duplication in t(12;22)(p13;q12) cases. The new perspective about this entity of disease will enlighten further research to define the mechanism of tumorigenesis and discover effective treatments for MN1-driven malignancies.
Similar content being viewed by others


Introduction
Translocation of t(12;22)(p13;q12) is a rare but recurrent chromosomal abnormality in hematologic malignancies involving meningioma 1 (MN1) and ETS variant 6 (ETV6) genes [1]. Although more than 40 cases with t(12;22)(p13;q12) have been reported so far, MN1-ETV6 and the reciprocal ETV6-MN1 fusion transcripts were confirmed in only a dozen of them [1]. The translocation has been reported only in myeloid neoplasms, most of which are acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS) with poor responses to chemotherapy [1]. Also, MN1 overexpression has been reported in several subsets of AML and has been accepted as adverse prognosticators in AML with normal karyotype [1,2,3].
The pathogenic mechanism of t(12;22)(p13;q12) and the fact that a considerable proportion of cases with this translocation lack fusion transcripts remain mysterious. Whether the central pathogenesis lies in the fusion of MN1-ETV6 or ETV6-MN1 also remains elusive [4]. We focused here on AML cases with t(12;22)(p13;q12) to elucidate their molecular etiology and outcomes of allogeneic hemopoietic stem cell transplantation (allo-HSCT) treatment.
Subjects and methods
Patients
Inclusion criteria for cases in this study were the diagnosis of myeloid neoplasms from April 2012 to October 2018, and karyotyping analyses showing t(12;22)(p13;q12) or related translocations. Data included in the analysis were gender, age, diagnosis, clinical course, morphology test, immunophenotypic test, cytogenetics, fluorescence in situ hybridization (FISH), and molecular genetics analysis. The diagnosis was made according to the 2008 revision to the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia [5]. The follow-up cutoff date was May 22, 2019. This study was conducted in accordance with the Declaration of Helsinki and approved by the ethics committee of Hebei Yanda Lu Daopei Hospital. All patients or their legal guardians signed informed consent for sample collections and research.
Cytogenetic and molecular cytogenetic analysis
Cytogenetic analysis was performed on bone marrow (BM) mononuclear cells after a 24-h culture. Cultures were exposed overnight to 0.1 μg/mL colcemid (ThermoFisher Scientific Inc., Waltham, USA) and then harvested following standard procedures to obtain metaphases. The karyotype was interpreted according to the International System for Human Cytogenetic Nomenclature 2016 (ISCN 2016) after the analysis of at least 20 G-banded metaphases using the IKAROS software (MetaSystems Inc., Altlhusseim, Germany). FISH with an MN1 break-apart probe was performed with the help of professor Jinlan Pan according to the protocols previously reported [1].
High-throughput sequencing (HTS) mutation screening
Mutational hotspots or whole coding regions of 58 genes that are known to mutate frequently in hematologic malignancies were sequenced using a targeted, multiplexed, amplicon-based high-throughput sequencing protocol as previously reported [6].
Whole-genome sequencing (WGS) analysis
Standard 30 × WGS was performed on diagnostic BM samples from cases that were negative for MN1-ETV6. The libraries were constructed with NEBNext Ultra II DNA Library Prep Kit for Illumina (New England Biolabs Inc., Ipswich, USA) according to the manufacturer's instructions, followed by sequencing on Illumina HiSeq X Ten platform (Illumina, Inc., San Diego, USA) using HiSeq X Ten Reagent Kit v2.5 (Illumina, Inc., San Diego, USA) running on paired-end 150 bp mode. Reads were aligned to the Genome Reference Consortium Human Genome Build 37 (GRCh37/hg19) assembly of the human genome with bwa mem.
Structure variations, including translocations and copy number variations (CNVs), were analyzed according to the protocols we previously reported [7, 8]. The two super-enhancers (SEs) that have been annotated in the SEdb, with potential blood-cell specific functions within the genomic region of ETV6, were SE_00_00600344 (chr12:11898032-11928354), which spans ETV6 exon 2, and SE_02_25500737 (chr12:11986329-12016687), which spans ETV6 exon 3 [9].
Reverse-transcriptase polymerase chain reaction (RT-PCR) and PCR validation
MN1-ETV6, ETV6-MN1, and MN1 expression were analyzed by RT-PCR according to the protocols previously reported [1]. The putative breakpoints and transcripts were validated by PCR and Sanger sequencing.
Results
Patient characteristics
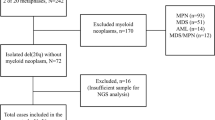
Eight cases with t(12;22)(p13;q12) and one case with t(12;17;22)(p13;q21;q13) were detected by G-banding karyotyping in 2782 newly-diagnosed AML and MDS patients (Table 1), with a relatively low incidence of 0.32%. There were six males, three females, and the median age of onset was 45 years (range 4–60 years).
The cases included three AML-M5, one AML-M2, one AML progressed from MDS (MDS-AML), one MDS with excess blasts 2 (MDS-EB-2), one chronic myelomonocytic leukemia (CMML), and two myeloid/T mixed-phenotype acute leukemia (MPAL) (Table 1).
Clinical outcome
All cases underwent standard or intensive chemotherapy for myeloid malignancies, mainly according to the National Comprehensive Cancer Network guidelines [10, 11]. The three cases treated only with chemotherapy died 3, 7, and 11 months after diagnosis, respectively. Six patients underwent haploidentical allo-HSCT [12], five of them were alive until the last follow-up, and the overall survival time was 11, 49, 47, 29, and 18 months, respectively. The other case (P1), who underwent salvage allo-HSCT after 2 years of chemotherapy and multiple relapses, relapsed 13 months after transplantation and died from a lung infection (Fig. 1).

allo-HSCT allogeneic hematopoietic stem cell transplantation.
Karyotyping and FISH results
Three cases had t(12;22)(p13;q12) as the only karyotype abnormality. Six patients harbored additional chromosomal abnormalities, five of whom had trisomy 8. Four cases had complex karyotypes (Table 1 and Fig. 2a). FISH was performed using the MN1 dual-color break-apart probe [1] on cases P4 and P9, who were MN1-ETV6 negative, and showed split signals in both cases, indicating a fracture within or adjacent to MN1 (Fig. 2b).

FISH fluorescence in situ hybridization. a One karyotype of case P8, the arrows indicate t(12;22(p13;q13), trisomy 8, and del(20) (q11.2). b MN1 gene break-apart probe FISH test result of case P1. The separation of the red and green signals in one of the cells indicate a fracture of the MN1 gene or its vicinity. c, d Sanger sequencing of MN1-ETV6 (c) and ETV6-MN1 (d) transcripts.
Fusion transcripts and MN1 expression
Five cases were positive for both MN1-ETV6 and ETV6-MN1 fusions (all type I, Table 1, Fig. 2c, d). The expression of MN1 was significantly upregulated in all three cases without MN1 fusion (P1, P3, and P4), but not in the six cases that carried MN1-ETV6 or MN1-STAT3 fusion (Table 1).
HTS mutation screening
The HTS mutation screening results of 58 genes were available in seven cases, but no significant feature was found related to the chromosome subtypes or MN1-ETV6 fusion (Table 1).
Genomic translocation analysis
The WGS analysis was performed on cases P1, P3, P4, and P9, who were negative for MN1-ETV6. In case P1 (Fig. 3a), WGS analysis showed the chr22:28117054 breakpoint located at 25 Kbps downstream of MN1, and the chr12:11942309 breakpoint located at ETV6 intron 2. Thus, the translocation resulted in the intact MN1 ectopia adjacent to the SE_02_25500737.

a Structural diagram of MN1 ecotopia in MN1-ETV6 negative cases. b, c Genomic sequence of MN1 intron 1- STAT3 intron 21 splicing (b) and Sanger sequencing of MN1-STAT3 transcripts (c). d Chromosome (Chr) 8 duplication analysis.
In case P3 (Fig. 3a), WGS analysis revealed tandem splicing, which sequentially involved telomeric chr22q with the breakpoint at chr22:28124526, a 5.3 Mbps chr16:51322289-56600706 fragment, a 5.3 Mbps chr5:14044903-19353114 fragment, and the chopped chr12 with the breakpoint at chr12:11574655. The breakpoint on chr22q was 18 Kbps downstream of MN1 and adhered to the chr16 gene-deserted fragment. The chr12:11574655 breakpoint was located at 226 Kbps upstream of ETV6. Hence, the intact MN1 translocated to a gene barren fragment in chr16, and there was no evidence of ETV6 disruption in this case.
In case P4 (Fig. 3a), the breakpoints of chr22:28047879 and chr12:11845460 were located at 94 Kbps downstream of MN1 and within ETV6 intron 1, respectively. Thus, it would result in the translocation of intact MN1 to ETV6 intron 1, adjacent to SE_00_00600344. So, the MN1 remained intact, but the ETV6 was disrupted in this case. The MN1 break-apart FISH probe used in this study was designed to label a ~710 Kbps region upstream and a ~620 Kbps region downstream of the 57 Kbps MN1 separately. Therefore, it makes sense that the FISH test showed split signals.
In case P9 (Fig. 3a), the triadic t(12;17;22)(p13;q21;q13) resulted in a chr22:28168851-chr17:40473489 splicing and a predicted MN1 exon 1-STAT3 exon 22 in-frame fusion transcript, which had never been reported (Fig. 3b). The RT-PCR, followed by Sanger sequencing, further confirmed the novel MN1-STAT3 transcript (Fig. 3c). Besides, a breakpoint in ETV6 intron 4 and a chr12:12013281-chr1:2980818 translocation was found, indicating the breakage of ETV6 and cryptic chr1 fragments. The assumed reciprocal STAT3 exon 21-MN1 exon 2 fusion transcript was not captured by RT-PCR, which was consistent with the triadic translocation trait.
Genomic CNV analysis
Notably, all five cases with MN1-ETV6 in this study were accompanied by trisomy 8 (Table 1). Then duplications >10 Kbps of chr8 were analyzed with WGS data in the other four fusion-negative cases, and three of them shared a 531 Kbps focal duplication (Fig. 3d). As a comparison, only one of the five randomly selected acute lymphoblastic leukemia cases (C1–C5 in Fig. 3d) had duplication involving this region. Therefore, we disclosed a high concomitance of trisomy 8 or this 531 Kbps focal chr8 amplification in this disease entity.
Discussion
In this study, we provided the analysis of t(12;22)(p13;q12) and related translocations in myeloid malignancies with the largest number of cases in a single center. We further performed MN1 expression and WGS analysis in cases with this translocation but negative for MN1-ETV6 fusion transcript. Our data showed a relatively low incidence of 0.32% of the translocation in myeloid malignancies. Clinical data also indicated that the translocation could occur at any age and with no predominance in AML morphological classifications, which were similar to the literature reports [1]. Nevertheless, the bi-phenotypic of P5 and P7 in this study further indicated that some cases might have mixed phenotypes of myeloid and T-cell lineages.
The clinical outcomes of our cases confirmed the dismal prognosis when treated only with chemotherapy, which is consistent with the literature reports [1]. Six cases underwent allo-HSCT in this study; five displayed notable efficacies, and the one who died from relapse suffered multiple relapses before the salvage allo-HSCT. Thus, we first provide evidence that these patients can benefit significantly from timely allo-HSCT.
It was reported that nearly all cases carrying t(12;22)(p13;q12) showed MN1 split signals in FISH testing, but only half of them were MN1-ETV6-positive [1]. Three out of eight t(12;22)(p13;q12) cases were negative for MN1-ETV6 and the reciprocal ETV6-MN1 fusion in this study. Moreover, both cases that went through FISH analysis showed MN1 split signals, which further confirmed that the form of fusion is unnecessary. We also identified a novel MN1-STAT3 fusion in one case with triadic t(12;17;22)(p13;q21;q13) translocation. This novel MN1 fusion also provides collateral evidence that it is the MN1-ETV6 but not ETV6-MN1 fusion that plays the essential pathological role in t(12;22)(p13;q12) leukemia.
We observed significantly upregulated MN1 expression in MN1-fusion-negative cases but not in fusion-positive cases. MN1 overexpression has been studied and accepted as an independent adverse prognostic marker in karyotype normal AML [13,14,15], and studies also indicated that the specific phenotype of MN1-leukemia depends on the cooperating mutations [16]. The MN1 transcript consists of a 3781 bp exon 1, which encodes the main functional domains of MN1 protein, and a 182 bp exon 2, which encodes a short C-terminal sequence without an explicitly annotated domain (Fig. 4). The breakpoints of MN1, in reported cases, were all located in intron 1, so MN1 retains its exon 1 in both the MN1-ETV6 and MN1-STAT3 transcripts. Thus, the RT-PCR primers designed to quantitatively detect MN1 expression, which spanned MN1 exon1 and exon 2 [1], could not amplify and reflect the abundance of the MN1 fusion transcripts.

Structural diagram of MN1, ETV6, MN1-ETV6, and MN1-STAT3 transcripts.
Overexpression of MN1 but not the MN1-ETV6 fusion has also been reported in the t(12;22)(p13;q12) positive AMU-AML1 cell line [17], and has been identified playing an essential synergistic role with CBFB-MYH11 in the leukemogenesis of inv(16) AML [18]. Kandilci A. et al. reported that CEBPA downregulation contributes to MN1-modulated leukemogenesis, and reintroduction of CEBPA in MN1-overexpressing hematopoietic cells prevents their hyperproliferation and restores myeloid differentiation [19]. Besides, MN1 has been reported significantly upregulated in ETV6-AML1-positive B-cell acute lymphoblastic patients [20]. MN1 has also been observed to fuse with another ETS family of transcription factors gene FLI1 in acute megakaryoblastic leukemia [21]. These reports together well-founded indicate that MN1 overexpression might closely cooperate with abnormalities of transcription-factor genes such as ETV6, CBFB, CEBPA, and FLI1 in leukemogenesis.
Over 30 ETV6 fusion partners have been characterized, the genomic breakpoint within ETV6 is scattered, and the contribution of the fused ETV6 fragment to the leukemogenesis remains elusive [22]. In most scenarios, the main pathogenic mechanism of ETV6 fusion is ectopic activation of its partner gene, together with the ETV6 insufficiency due to impaired integrity [22]. The newly reported ETV6-IGH translocation in primary central nervous system lymphoma, which lead to IGH overexpression together with ETV6 haploinsufficiency but without ETV6-IGH fusion transcripts, also further support this interpretation [23].
The MN1-STAT3 fusion protein contains only 69 amino acids of the STAT3 residual without annotated functional domains, and STAT3-MN1 was absent due to the triadic translocation. Then, the fusion protein reserves the primary functional domains of MN1 but not STAT3 in case P9 in this study. So, both type I/II MN1-ETV6 and the novel MN1-STAT3 in this study reserve the functional domains of MN1 (Fig. 4). Taken together, we suggest it is reasonable to speculate that the upregulated function of MN1 protein plays the major pathological role in MN1 overexpression, MN1-ETV6, and MN1-STAT3 cases.
Impaired integrity of ETV6 betides in all cases except one MDS-EB-2 case in this study, regardless of whether there is MN1-ETV6 fusion. ETV6 functions as a transcription repressor and plays a critical role in hematopoiesis [24]. Deletions and fusion-driven haploinsufficiency of ETV6 have been reported widely in various hematological malignancies, and it also acts as a tumor suppressor, with haploinsufficiency enough to manifest partial effects [25, 26]. Thus the highly concomitant ETV6 haploinsufficiency caused by gene truncation might play a synergistic or phenotypic determination role in t(12;22)(p13;q12) leukemia.
The mechanism of MN1 overexpression in leukemia is far from disclosed. SEs are a kind of DNA elements that function as distal regulators of gene expression and sometimes hijacked in cancers to drive the oncogene activities [9, 27]. One paradigm in hematology is the GATA2-MECOM aberration in inv(3)(q21q26) AML. The formerly identified RPN1-MECOM fusion was once considered as the primary pathogenic factor of inv(3)(q21q26) AML and had ever been accepted as a genetic classification marker in WHO 2008 criteria [5]. However, further researches have confirmed that the central pathogenesis of this translocation was the ectopia of a GATA2 SE that activated MECOM expression and conferred GATA2 haploinsufficiency simultaneously [27,28,29]. These findings have led to the rewriting of the corresponding content in the WHO 2016 classification [3].
There are two annotated ETV6 SEs with potential blood-cell specific functions spanning its exon 2 and exon 3, respectively [9]. The scenario of t(12;22)(p13;q12) portrays another analog of inv(3)(q21q26) in AML. The detectable or absent fusion transcript, oncogene activation, SE ectopia, and even haploinsufficiency caused by gene breakage are all similar. Nearly all MN1-ETV6 fusion transcripts reported are type I, which reserves ETV6 exon 3 together with the SE that spanning this exon. As to cases P1 and P4 who without MN1-ETV6 fusion in this study, the unabridged MN1 is adjacent to the truncated ETV6 and its SEs due to ectopia. Therefore, it is more likely that the ectopia hijacks the SEs within ETV6 and then activates the chimeric MN1-ETV6 or MN1 itself.
Trisomy 8 is often seen in the AML subgroup with transcription-factor-associated fusions CBFB-MYH11, RUNX1-RUNX1T1, and PML-RARA [30]. Notably, all five cases with MN1-ETV6 in this study were accompanied by trisomy 8, and genomic CNV analysis further revealed a shared 531 Kbps focal chr8 amplification in three of the other four cases. Since ETV6 is also a transcription regulator in hematogenesis, synergistic factors might lie in this genomic focal region in this disease entity.
Taken together, we provide the incidence, clinical course, and effectiveness of allo-HSCT of myeloid neoplasms with t(12;22)(p13;q12) in this study. We identify incomplete MN1-fusion penetrance, MN1 overexpression in fusion-negative cases, decapitated ETV6, a novel MN1-STAT3 fusion, and high concomitance of trisomy 8 or 8q focal duplication in t (12;22)(p13;q12) myeloid neoplasms by integrative genetic analysis. Our investigation highlights the evidence of an ectopia-associated orchestrated mechanism of MN1 aberrant activation and ETV6 haploinsufficiency in t(12;22)(p13;q12) myeloid neoplasms, rather than the conventional thought of MN1-ETV6 fusion formation. The incidence of MN1 overexpression is much higher than t(12;22)(p13;q12), and the mechanism remains to be explored [2]. Studies have reported that Mediator kinases can negatively regulate SE-associated gene expression and can be pharmacologically targeted as a therapeutic approach [29, 31]. The new perspective about this entity of disease will enlighten further research to define the mechanism of tumorigenesis and discover effective treatments for MN1-driven malignancies.
References
Shao H, Cen J, Chen S, Qiu H, Pan J. Myeloid neoplasms with t(12;22)(p13;q12)/MN1-EVT6: a systematic review of 12 cases. Ann Hematol. 2018;97:417–24.
Xiang L, Li M, Liu Y, Cen J, Chen Z, Zhen X, et al. The clinical characteristics and prognostic significance of MN1 gene and MN1-associated microRNA expression in adult patients with de novo acute myeloid leukemia. Ann Hematol. 2013;92:1063–9.
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–405.
Gong S, Guo M, Tang G, Yang J, Qiu H. Overexpression of TEL-MN1 fusion enhances resistance of HL-60 cells to idarubicin. Chemotherapy. 2018;63:308–14.
Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114:937–51.
Zhang Y, Wang F, Chen X, Zhang Y, Wang M, Liu H, et al. CSF3R Mutations are frequently associated with abnormalities of RUNX1, CBFB, CEBPA, and NPM1 genes in acute myeloid leukemia. Cancer. 2018;124:3329–38.
Chen X, Wang F, Zhang Y, Teng W, Cao P, Ma X, et al. A novel NPM1-RARG-NPM1 chimeric fusion in acute myeloid leukaemia resembling acute promyelocytic leukaemia but resistant to all-trans retinoic acid and arsenic trioxide. Br J Cancer. 2019;120:1023–5.
Nie D, Cao P, Wang F, Zhang J, Liu M, Zhang W, et al. Analysis of overlapping heterozygous novel submicroscopic CNVs and FANCA-VPS9D1 fusion transcripts in a Fanconi anemia patient. J Hum Genet. 2019;64:899–909.
Jiang Y, Qian F, Bai X, Liu Y, Wang Q, Ai B, et al. SEdb: a comprehensive human super-enhancer database. Nucleic Acids Res. 2019;47(D1):D235–D243.
O'Donnell MR, Tallman MS, Abboud CN, Altman JK, Appelbaum FR, Arber DA, et al. Acute Myeloid Leukemia, Version 3.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2017;15:926–57.
Greenberg PL, Stone RM, Al-Kali A, Barta SK, Bejar R, Bennett JM, et al. Myelodysplastic Syndromes, Version 2.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2017;15:60–87.
Lu DP, Dong L, Wu T, Huang XJ, Zhang MJ, Han W, et al. Conditioning including antithymocyte globulin followed by unmanipulated HLA-mismatched/haploidentical blood and marrow transplantation can achieve comparable outcomes with HLA-identical sibling transplantation. Blood. 2006;107:3065–73.
Grosveld GC. MN1, a novel player in human AML. Blood Cells Mol Dis. 2007;39:336–9.
Heuser M, Beutel G, Krauter J, Döhner K, von Neuhoff N, Schlegelberger B, et al. High meningioma 1 (MN1) expression as a predictor for poor outcome in acute myeloid leukemia with normal cytogenetics. Blood. 2006;108:3898–905.
Langer C, Marcucci G, Holland KB, Radmacher MD, Maharry K, Paschka P. Prognostic importance of MN1 transcript levels, and biologic insights from MN1-associated gene and microRNA expression signatures in cytogenetically normal acute myeloid leukemia: a cancer and leukemia group B study. J Clin Oncol. 2009;27:3198–204.
Pieters T, T'Sas S, Demoen L, Almeida A, Haenebalcke L, Matthijssens F, et al. Novel strategy for rapid functional in vivo validation of oncogenic drivers in haematological malignancies. Sci Rep. 2019;9:10577.
Gotou M, Hanamura I, Nagoshi H, Wakabayashi M, Sakamoto N, Tsunekawa N, et al. Establishment of a novel human myeloid leukemia cell line, AMU-AML1, carrying t(12;22)(p13;q11) without chimeric MN1-TEL and with high expression of MN1. Genes Chromosomes Cancer. 2012;51:42–53.
Carella C, Bonten J, Sirma S, Kranenburg TA, Terranova S, Klein-Geltink R, et al. MN1 overexpression is an important step in the development of inv(16) AML. Leukemia. 2007;21:1679–90.
Kandilci A, Grosveld GC. Reintroduction of CEBPA in MN1-overexpressing hematopoietic cells prevents their hyperproliferation and restores myeloid differentiation. Blood. 2009;114:1596–606.
Numata M, Yener MD, Ekmekçi SS, Aydın M, Grosveld G, Cardone M, et al. High MN1 expression increases the in vitro clonogenic activity of primary mouse B-cells. Leuk Res. 2015;39:906–12.
Wenge DV, Felipe-Fumero E, Angenendt L, Schliemann C, Schmidt E, Schmidt LH, et al. MN1-Fli1 oncofusion transforms murine hematopoietic progenitor cells into acute megakaryoblastic leukemia cells. Oncogenesis. 2015;4:e179.
De Braekeleer E, Douet-Guilbert N, Morel F, Le Bris MJ, Basinko A, De Braekeleer M. ETV6 fusion genes in hematological malignancies: a review. Leuk Res. 2012;36:945–61.
Bruno A, Labreche K, Daniau M, Boisselier B, Gauchotte G, Royer-Perron L, et al. Identification of novel recurrent ETV6-IgH fusions in primary central nervous system lymphoma. Neuro Oncol. 2018;20:1092–1100.
Songdej N, Rao AK. Hematopoietic transcription factor mutations: important players in inherited platelet defects. Blood. 2017;129:2873–81.
Hock H, Shimamura A. ETV6 in hematopoiesis and leukemia predisposition. Semin Hematol. 2017;54:98–104.
Rasighaemi P, Ward AC. ETV6 and ETV7: Siblings in hematopoiesis and its disruption in disease. Crit Rev Oncol Hematol. 2017;116:106–15.
Koche RP, Armstrong SA. Genomic dark matter sheds light on EVI1-driven leukemia. Cancer Cell. 2014;25:407–8.
Yamazaki H, Suzuki M, Otsuki A, Shimizu R, Bresnick EH, Engel JD, et al. A remote GATA2 hematopoietic enhancer drives leukemogenesis in inv(3)(q21;q26) by activating EVI1 expression. Cancer Cell. 2014;25:415–27.
Gröschel S, Sanders MA, Hoogenboezem R, de Wit E, Bouwman BAM, Erpelinck C, et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell. 2014;157:369–81.
Hemsing AL, Hovland R, Tsykunova G, Reikvam H. Trisomy 8 in acute myeloid leukemia. Expert Rev Hematol. 2019;12:947–58.
Pelish HE, Liau BB, Nitulescu II, Tangpeerachaikul A, Poss ZC, Da Silva DH, et al. Mediator kinase inhibition further activates super-enhancer-associated genes in AML. Nature. 2015;526:273–6.
Acknowledgements
We thank professor Jinlan Pan, director of the Key Laboratory of Thrombosis and Hemostasis of Ministry of Health, Jiangsu Institute of Hematology, for the help of MN1 FISH probe detection.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, T., Chen, X., Hui, S. et al. Ectopia associated MN1 fusions and aberrant activation in myeloid neoplasms with t(12;22)(p13;q12). Cancer Gene Ther 27, 810–818 (2020). https://doi.org/10.1038/s41417-019-0159-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41417-019-0159-x
