
Abstract
We have previously reported that homologous recombination (HR) is dysregulated in multiple myeloma (MM) and contributes to genomic instability and development of drug resistance. We now demonstrate that base excision repair (BER) associated apurinic/apyrimidinic (AP) nucleases (APEX1 and APEX2) contribute to regulation of HR in MM cells. Transgenic as well as chemical inhibition of APEX1 and/or APEX2 inhibits HR activity in MM cells, whereas the overexpression of either nuclease in normal human cells, increases HR activity. Regulation of HR by AP nucleases could be attributed, at least in part, to their ability to regulate recombinase (RAD51) expression. We also show that both nucleases interact with major HR regulators and that APEX1 is involved in P73-mediated regulation of RAD51 expression in MM cells. Consistent with the role in HR, we also show that AP-knockdown or treatment with inhibitor of AP nuclease activity increases sensitivity of MM cells to melphalan and PARP inhibitor. Importantly, although inhibition of AP nuclease activity increases cytotoxicity, it reduces genomic instability caused by melphalan. In summary, we show that APEX1 and APEX2, major BER proteins, also contribute to regulation of HR in MM. These data provide basis for potential use of AP nuclease inhibitors in combination with chemotherapeutics such as melphalan for synergistic cytotoxicity in MM.
Similar content being viewed by others


Introduction
Most cancers display a variety of genomic alterations at diagnosis1,2,3,4,5,6 and acquire new changes with progression of disease to advanced stages7,8,9,10 indicating an instability of genome1,11,12,13,14,15,16,17. We have observed a similar pattern of change in multiple myeloma (MM), both at diagnosis and in relapse samples1. The ability to constantly evolve not only enables the MM cells to acquire new characteristics for growth, survival, and progression of disease, but also presents a great obstacle to treatment. Understanding mechanisms underlying genomic instability is therefore essential to develop effective strategies for prevention and treatment of cancer.
Normal cellular DNA is subject to a large number of chemical and physical modifications18,19 which may include missing or altered bases, deletions/insertions, DNA strand breaks and cross-linking etc. Therefore, multiple DNA repair mechanisms exist and ensure that these alterations are constantly and efficiently repaired in normal cells. Homologous recombination (HR), which depends on sequence homology/identity, is one of the most precise DNA repair mechanism20. HR is involved in the repair of a variety of DNA damage types including DNA double-stranded breaks, DNA strand crosslinks and single-stranded DNA gaps, and plays a vital role in the maintenance of genomic integrity21. However, dysregulation of HR can also lead to genomic instability22. Consistent with this, we have previously demonstrated that elevated HR mediates genomic instability and progression in MM23. Using another cancer model system (Barrett’s adenocarcinoma), we demonstrated that dysregulated HR disrupts genomic integrity24 and stability25, and transgenic as well as chemical suppression of HR, significantly reduced both the genomic instability as well as growth of cancer cells in mice26. Identification of mechanisms underlying dysregulation of HR thus may help prevent genomic evolution and provide novel targets for cancer prevention.
Investigating mechanisms underlying dysregulation of HR, we identified significant role for apurinic/apyrimidinic (AP) nucleases (APEX1 and APEX2). APEX1 and APEX2 are the key base excision repair (BER) pathway proteins involved in the recognition and repair of AP (or abasic) sites27,28,29,30 and play a significant role in genome maintenance. Development of abasic site, one of the most common DNA damage, can occur either spontaneously or by removal of a damaged DNA base by specific enzymes, DNA glycosylases. The repair of AP sites is extremely efficient. APEX1 and APEX2 are major BER proteins involved in its initiation. APEX1 is the abasic endonuclease implicated in both the BER and 3′-end processing in mammalian cells29,31,32. It is a multifunctional protein and is also responsible for repair of alkylation and oxidative DNA damage in a cell. The protein also functions as a reduction–oxidation (redox) factor and hence maintains many transcription factors in an active reduced state33,34. APEX1 is a strong endonuclease but also has a weak 3′-5′ exonuclease activity for proofreading function35,36. APEX2 on the other end has a weak endonuclease but a strong exonuclease activity35. Elevated levels of APEX1 have been found in cervical37, ovarian38,39, prostate40, germ cell tumors41, and correlate with the radiosensitivity of cervical cancers42. Here we have explored the role of APEX1 and APEX2 in dysregulation of HR in MM.
Materials and methods
Cell lines and patient samples
MM cell lines RPMI8226 and U266 were purchased from the American Type Tissue Culture Collection (Rockville MD), ARP was a gift from Dr. J. Epstein (University of Arkansas for Medical Sciences) and MM.1S obtained from Dr. Steven Rosen (Northwestern University, Chicago, IL). Cell lines, confirmed to be negative for mycoplasma, were cultured in RPMI1640 medium containing 10% fetal bovine serum (HyClone, South Logan, UT) as described previously43,44,45,46,47,48,49 and maintained in logarithmic growth.
Bone marrow aspirate samples from normal donors and patients were obtained following informed consent under protocol approved by Institutional Review Board, Dana Farber Cancer Institute. Plasma cells were isolated by Magnet Assisted Cell Sorting (MACS, Miltenyi Biotech) according to the manufacturer’s protocol, and their purity (>95%) confirmed by monitoring cell surface expression of CD45 and CD3850.
Reagents
Anti-APEX1 antibody, catalogue # NB100-101 was purchased from Novus Biologicals, Inc., (Littleton, CO). Anti-APEX2 antibody is developed by us. Anti-RAD51 antibody, catalogue # SC-8349 was purchased from Santa Cruz, Dallas, TX. Antibodies against GAPDH (catalogue # mAb2118), HP1 (catalogue # 261), and γH2A.X (Ser139, catalogue #2577) were from Cell Signaling Technology, Inc., Danvers, MA. For immunoprecipitation studies, anti-APEX1 antibody (Cat. # ab194, Abcam) and anti-p73 antibody (Cat. # NBP2-24737, Novus Biologicals) were used.
Modulation of AP nuclease expression
AP nuclease expression was modulated using chemical as well as transgenic manipulations. AP nuclease activity was suppressed using APEX1 inhibitor (APE1 Inhibitor III; API3) (Axon Medchem LLC, Reston, VA), methoxyamine (MX) which inhibits AP nuclease activity by binding to aldehyde group of abasic site (Sigma Aldrich, Saint Louis, MO) and lentivirus based shRNAs targeting APEX1 and APEX2 (Sigma Aldrich, Saint Louis, MO). For overexpression, the plasmids carrying these genes under CMV promoter were used.
AP nuclease activity assay
AP nuclease activity was assessed using a fluorescence based assay reported previously51. Briefly, the oligonucleotides carrying AP site mimic (Tetrahydrofuran; THF) and a fluorescein label on one strand and a quenching moiety (Dabcyl-Q) on other strand are synthesized commercially (Eurogentec Ltd., San Diego, CA). The AP nuclease activity cleaves AP site releasing the short fluorescein-labeled fragment, thus resulting in the increase in fluorescence.
Recombination assays
Three different functional assays were used to evaluate HR in this study. A plasmid based HR activity assay has been reported previously25,26. Here, we also used a modification of this assay in which HR substrate plasmid is mixed with MM cell lysate under in vitro HR conditions52, and product of HR measured by Q-PCR. This modified version of HR assay was validated and was consistent with the assay conducted within intact cells (Supplementary Figure 1). We also used a fluorescence based homologous strand exchange (SE) assay to measure HR, as reported by53.
Detection of DNA breaks
DNA breaks were estimated by evaluating MM cells for levels of γ-H2AX. Expression of γ-H2AX was measured by Western blotting using anti-H2AX (Ser139, antibody # 2577; Cell Signaling Technology, Inc., Danvers, MA) as reported by us previously24.
Western blotting and immunocytochemical detection of proteins
For western blotting, the proteins fractionated on gradient SDS-acrylamide gel were electroblotted onto nitrocellulose paper and sequentially treated with indicated primary antibodies and either anti-rabbit or anti-mouse horseradish peroxidase (HRPO) conjugates. Blots were then washed and specific proteins detected using an enhanced chemiluminescence, according to the instructions provided by manufacturer (Amersham Life Sciences Inc., Arlington Heights, IL). For immunocytochemical detection of APEX1, cytospins of normal plasma cells and MM cells were fixed in methanol/acetone (1:1, v/v) for 10 min at –20 °C. Fixed cells were rinsed, rehydrated in PBS, and incubated with antibody to APEX1. Vectastain Elite ABC kit (Vector Laboratories, Burlingame, CA) was used for immunostaining as described by the manufacturer.
Chromatin immunoprecipitation and co-immunoprecipitation
Chromatin immunoprecipitations were performed using EZ-Magna ChIP™ A/G Chromatin Immunoprecipitation Kit (Millipore). Cells were fixed in formaldehyde (1%) for 10 min at room temperature. Glycine was then added to a final concentration of 0.125 M to quench the reaction. Cells were washed twice in cold PBS and once with PBS containing protease inhibitors, incubated in lysis buffer with protease inhibitors on ice for 15 min, and centrifuged at 800 × g for 5 min at 4 °C. The supernatant was removed and the nuclear pellets re-suspended in nuclear lysis buffer with protease inhibitors and incubated on ice for 10 min before sonication. Sonication was performed using a probe sonicator in 1.5 ml tubes. Sonication conditions involved 11 cycles at 75% amplitude, 30 s pulse, 1 min on ice, followed by 1 cycle at 80% amplitude. The lysates were centrifuged at 12,000 × g for 10 min at 4 °C and the chromatin supernatants were used for immunoprecipitation, using 4 µg of APEX1 antibody (catalogue # ab194, Abcam) or p73 antibody (catalogue # NBP2-24737, Novus Biologicals) or IgG controls. Real-time PCR reactions, using primers for amplification of a region of RAD51 promoter, were conducted to assess the fraction of RAD51 promoter DNA associated with proteins under investigation. Primers specific to GAPDH promoter region were used as negative control.
Investigation of protein–protein interactions, expression, and phosphorylation levels
For investigation of direct protein–protein interactions, a cell cycle antibody array (Hypromatrix, Worcester, MA) containing 60 antibodies, and/or a custom antibody array having 40 antibodies immobilized on a nitrocellulose membrane, were sequentially incubated with MM cell lysates and then HRP-conjugated anti-APEX1 or APEX2 antibodies. Interacting partners were then identified by their address on the array.
Evaluating impact on micronuclei, a marker of genomic instability
To evaluate impact on genomic instability, the cells were cultured in the presence or absence of inhibitor of AP nuclease activity (API3), melphalan or combination of both, and cells evaluated for micronuclei, a marker of unstable genome54,55, using Micronucleus Assay MicroFlow kit (Litron Laboratories, New York, USA).
Statistical analyses
For evaluation of APEX1 and APEX2 expression in MM datasets, expression dataset (GSE2113; Fig. 1c) and (GSE6477; Fig. 1d) were downloaded from GEO (https://www.ncbi.nlm.nih.gov/geo/) database and expression profiles pre-processed with R using affy and limma packages for Affymetrix Human Genome U133A Array. RMA normalization was applied to both datasets. 7 MGUS and 39 MM samples from GSE2113 were compared using modified t test in limma. Expression levels in GSE6477 dataset (N = 16, MGUS = 22, SMM = 24, MM = 73, and Relapse = 28) was plotted using R and normal and MM samples were compared with limma. For gene array (Supplementary Figure 2), RNA derived from untreated and methoxyamine treated ARP cells was evaluated using HG-U133 array (Affymetrix); Arrays were normalized and expression values calculated, using DNA-Chip Analyzer, as described previously 23,44,45.

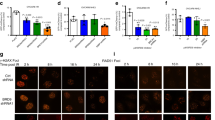
a-b. Protein levels of APEX1 and APEX2 are elevated in MM cells; a. Immunocytochemistry showing APEX1 in normal samples (Donors 1–3) and MM cell lines; b. Western blot showing APEX2 expression in normal and MM cell lines; c. Relative expression (Log2) of APEX1 and APEX2 in 7 MGUS and 39 MM samples from GSE2113 dataset were computed using modified t test in limma. d. Relative expression (Log2) of APEX1 and APEX2 in GSE6477 dataset (N = 16, MGUS = 22, SMM = 24, MM = 73 and Relapse = 28) plotted using R and normal and MM samples compared with limma
Results
APEX1 and APEX2 are overexpressed in MM
In our previous study, evaluation of gene expression by microarray indicated that expression of APEX1 and/or APEX2 in MM cell lines and a subset of MM patient samples was upregulated relative to normal plasma cell samples23. To further investigate AP nucleases in MM, here we evaluated their expression at protein level using immunocytochemistry (for APEX1) and western blotting (for APEX2) and confirmed upregulation in MM relative to normal plasma cells (Fig. 1a, b). To further investigate AP nuclease expression in other MM datasets, expression datasets (GSE2113, GSE6477) were downloaded from GEO (https://www.ncbi.nlm.nih.gov/geo/) database and expression profiles pre-processed with R using affy and limma packages for Affymetrix Human Genome U133A Array. RMA normalization was applied to both datasets. 7 MGUS and 39 MM samples from GSE2113 were compared using modified t test in limma. APEX1 was 1.8 fold upregulated in MM compared to MGUS (p value < 1e-4) and APEX2 was 1.2 fold upregulated in MM (p value = 0.047) (Fig. 1c). Expression levels in GSE6477 dataset (N = 16, MGUS = 22, SMM = 24, MM = 73, and Relapse = 28) were plotted using R and normal and MM samples compared with limma. Both APEX1 and APEX2 were upregulated with p values 0.02 and 1e-3, respectively (Fig. 1d).
Elevated AP nuclease expression dysregulates HR in MM cells
To investigate the functional role of elevated APEX 1/2 on various cellular processes, we evaluated the impact of chemical inhibition of AP nuclease activity with mithoxyamine (MX) and identified prominent perturbation of HR-related genes (Supplementary Figure 2; full microarray data submitted as Supplementary Table 1). We also observed that the expression of APEX1 and APEX2 in MM patient dataset (gse26863) correlated with the expression of a panel of HR genes (Supplementary Figure 3) identified in a functional screen56. Therefore, we next investigated the impact of transgenic and chemical modulations of APEX1 and/or APEX2 on HR activity, using three different functional HR assays; measuring HR activity by transfecting a plasmid substrate into cell25,26; utilizing the same plasmid under in vitro HR assay conditions52; and by measuring homologous strand exchange (SE) activity53, an essential step in the initiation of HR. APEX1-knockdown inhibited HR activity in ARP and RPMI8226 MM cells as measured by in vitro HR assay (Fig. 2a) as well as by SE assay in ARP (Fig. 2b) and RPMI8226 (data not shown) cells. Similarly, shRNA-mediated knockdown of APEX2 also inhibited HR activity in ARP cells (Fig. 2c). Treatment of MM cell lines (RPMI8226 and U266) with a small molecule inhibitor of APEX1 (API3) also inhibited HR activity in these cells (Fig. 2d, panel I). Additionally, the treatment of ARP MM cells with methoxyamine (MX) an inhibitor of overall AP activity by binding to aldehyde group of abasic sites led to a dose-dependent inhibition of HR activity, as measured by SE assay (Fig. 2d, panel II). Consistent with these data, the upregulation of AP nuclease in normal human (fibroblast) cells, separately and in combination with each other, increased HR activity by ~1.8-fold and ~3-fold (p > 0.05), respectively (Fig. 2e). Taken together, these data confirm that APEX1 and APEX 2 are involved in regulation of HR, and their elevated expression may at least in part contribute to dysregulation of HR activity in MM cells.

MM cells were subjected to various transgenic or chemical manipulations to inhibit APEX1 and/or APEX2, and cells evaluated for HR activity. a. MM cell lines (ARP and RPMI8226), transduced with lentivirus particles carrying control shRNA (CS) or APEX1-shRNA (KD), were selected in puromycin and evaluated for APEX1 expression by Western blotting (bottom panels) and HR activity using a plasmid based in vitro HR assay (top panels); error bars represent SDs of triplicate assays. b. Control and APEX1-knockdown ARP cells shown in panel A were also evaluated for HR using a different assay which measures homologous strand exchange (SE), a critical step in HR. c. MM cells (ARP), transduced with lentivirus particles carrying control shRNA (CS) or APEX2-shRNA (KD), were selected in puromycin and evaluated for APEX2 expression by western blotting (bottom panel) and HR activity using a plasmid based in vitro HR assay (top panel); error bars represent SDs of triplicate assays. d. (panel I) MM cell lines (RPMI and U266) were treated with DMSO (C) or APEX1 inhibitor (API3; 5 μM) for 24 h and evaluated for impact on HR; (panel II) ARP cells were treated with various concentrations of AP nuclease inhibitor methoxyamine (MX) for 48 h and impact on HR evaluated. e. Normal fibroblast cells transfected with control plasmid (C) or those for overexpression of APEX1 (APEX1-OE), APEX2 (APEX2-OE) or both AP nucleases (AP-OE) were evaluated for HR activity in intact cells using plasmid based assay25,26; error bars represent SEMs of triplicate assays
AP nucleases regulate RAD51 expression in MM cells.
To further investigate how AP nucleases impact HR activity, we evaluated AP-knockdown cells for their effect on RAD51 recombinase expression as well as RAD51 foci formation. As seen in Fig. 3a, APEX1-knockdown (KD) inhibits RAD51 expression in ARP and RPMI8226 MM cell lines (panel a). Consistent with these observations, the knockdown of APEX1, APEX2 or both inhibited RAD51 focus formation in ARP cells (Fig. 3b). Similarly, the small molecule inhibitor of AP activity (API3) also inhibited RAD51 expression in RPMI8226 and ARP (Fig. 3c) cells associated with reduction in RAD51 transcript (Fig. 3d), suggesting a transcription level regulation. As expected, the transgenic upregulation of APEX1 in normal human fibroblast cells increased RAD51 expression as well as RAD51 foci (Fig. 3e). These data demonstrate that APEX1 and APEX2 in MM cells are involved in the regulation of RAD51, the protein which mediates homologous pairing, a key step in HR.

a. Suppression of APEX1 inhibits RAD51 protein. Control and APEX1-knockdown ARP and RPMI8226 cells (for which APEX1 knockdown is shown in Fig. 2a, bottom) were evaluated for impact on RAD51 expression using western blotting. CS, control shRNA; KD, APEX1-knockdown; b. RAD51 foci in control (CS) ARP cells or those with knockdown (KD) of APEX1, APEX2 or both these genes (APEX1/2-KD); c. RPMI8226 and ARP cells, control and treated with AP inhibitor A PI3 (5 μM for 24 h) were evaluated for RAD51 by western blotting; d. Inhibition of APEX1 inhibits transcript levels of RAD51. RPMI8226 cells were treated with API3 (5 μM) for 24 h and transcript levels of RAD51 evaluated by quantitative RT-PCR; error bars indicate SDs of triplicate assays. e. Western blot showing RAD51 (I) and RAD51 foci in normal fibroblast cells, control (C) and APEX1-overexpressing (APEX1-OE)
APEX1 is involved in p73-mediated transcriptional regulation of RAD51 in MM cells
To further investigate how AP nuclease regulates RAD51, we investigated the interacting partners of APEX1 and APEX2 using standard as well as custom-synthesized antibody arrays (Supplementary Figure 4). We observed that both APEX1 (Fig. 4a-I; Supplementary Figure 4A) and APEX2 (Supplementary Figure 4B) interact with a number of HR-related proteins. Interestingly, both antibody arrays (Fig. 4a-I; Supplementary Figure 4A) and co-immnoprecipitation experiment (co-IP; Fig. 4a-II) identified APEX1 interaction with P73, a known transcriptional regulator of RAD51. Furthermore, a chromatin immunoprecipitation (ChIP) assay showed that both APEX1 and P73 bind to RAD51 promoter in MM cells; and inhibition of APEX1 by small molecule inhibitor or shRNA-mediated APEX1 knockdown, inhibits the association of P73 with RAD51 promoter (Fig. 4b I-IV). To further investigate the role of APEX1 in transcriptional regulation of RAD51, we transfected MM cells with a plasmid carrying luciferase gene under RAD51 promoter. APEX1 in these cells was then inhibited either by API3 or APEX1-shRNA, and luciferase activity assessed. APEX1 inhibition, by either method was able to inhibit RAD51 promoter activity in MM cells (Fig. 4c I-II). Taken together, these data suggest that APEX1 is involved in P73-mediated regulation of RAD51 expression in MM cells.

a (I) Antibody array: Cell Cycle Antibody Array (Hypomatrix) was sequentially treated with ARP cell lysate and HRP-conjugated anti-APEX1 antibody. Interacting partners were then identified by their location on the array. (II) Immunoprecipitation: Interaction between endogenous APEX1 and p73 was detected in the ARP cells, either control (DMSO) or those treated with 5 µM API3 for 24 h. Nuclear lysates were immunoprecipitated using anti-P73 antibody or IgG (control) and protein complexes resolved on Western blot which was probed with anti-APEX1 antibody; Input, lysate before immunoprecipitation. b (I-III): APEX1 and P73 bind to RAD51 promoter in MM cells. Protein-DNA complexes from ARP cells treated as below were immunoprecipitated using anti-P73 antibody (I and III) or anti-APEX1 antibody (II). DNA was then extracted and occupancy of RAD51 promoter evaluated by Q-PCR; Treatments: C, control (DMSO); API3, cells treated with APEX1 inhibitor 5 µM for 24 h; CS, control shRNA; APEX1-KD, APEX1-knockdown cells; (IV) Western blot showing APEX1 in CS and APEX1-KD cells. c. Inhibition of APEX1 inhibits RAD51 promoter activity in MM cells. (I) RAD51 promoter was cloned upstream of firefly luciferase gene in a plasmid, which was then introduced into ARP cells. APEX1 in these cells was inhibited by treating them with small molecule (API3, 5 µM) for 24 h, and cells evaluated for luciferase activity; (II) ARP cells transduced with lentivirus particles carrying control (CS) or APEX1-shRNA (APEX1-KD) were selected in puromycin and following confirmation of knockdown, cells were co-transfected with RAD51 promoter plasmid (described in panel I) and a plasmid carrying Gaussia luciferase as control for transfection efficiency. RAD51 promoter activity was assessed from ratio of two luciferase activities; error bars represent SDs of triplicate assays
Elevated AP expression contributes to melphalan-resistance in MM
We have previously shown that increased HR activity contributes to development of drug resistance in MM23. Since AP nuclease expression regulates HR activity, we investigated its role in melphalan resistance in MM cells. RPMI8226 and melphalan-resistant MM cells (LR5) were treated with API3 (2.5 μM) and impact on AP and HR activities monitored as described. Both AP and HR activities, although higher in resistant LR5 relative to melphalan-sensitive RPMI8226 cells, were inhibited ∼50% by API3 in both cell lines (Fig. 5a, b). Moreover, AP nuclease inhibitor sensitized both sensitive and resistant MM cell lines to melphalan treatment (Fig. 5c). Importantly, the dose of melphalan (20 μM) which had almost no effect on viability of resistant LR5 cells, induced >50% cell death in the presence of AP nuclease inhibitor (Fig. 5c, panel I). To further investigate if inhibition of AP nuclease expression increases efficacy of melphalan in MM, we suppressed both APEX1 and APEX2 in ARP cells and observed sensitization of MM cells to both melphalan and a PARP inhibitor PJ34 (Fig. 5d).

a. AP nuclease activity was measured in parental RPMI8226 and melaphalan-resistant LR5 cell lines in the presence or absence of AP nuclease inhibitor API3 (2.5 µM) as described in 'Methods'; time-dependent change in AP activity is shown. b. HR activity measured in parental RPMI8226 and melaphalan-resistant LR5 cell lines in the presence or absence of AP nuclease inhibitor API3 (2.5 µM) as described in 'Methods'; error bars represent SDs of triplicate assays. c. LR5 (I) and RPMI8226 (II) cells were treated with different concentrations of melphalan (MEL) alone or in combination with 2.5 µM of APEX1 inhibitor (API3) for 48 h and cell viability measured. d. AP (APEX1/2-KD) knockdown increases sensitivity of MM cells to melphalan and PARP inhibitor: d. Control (CS) ARP cells or those in which both APEX1 and APEX2 were suppressed (APEX1/2-KD; same cells as shown in Fig. 3b) were treated with different concentrations of melphalan (I) or PARP inhibitor, PJ34 (II) and cell viability measured by Cell Titer Glow for 7 days
Inhibition of AP activity increases melphalan-induced cytotoxicity while reducing DNA damage and instability
To investigate impact of AP nuclease inhibitor on melphalan-induced cytototoxicity, we evaluated their effect on DNA damage and genomic instability. RPMI8226 cells were treated with API3 (at a lower dose of 1 μM), melphalan or combination of both for 48 h and evaluated for cell viability and genomic instability. As shown in Fig. 6, treatment with melphalan increased cell death and genomic instability in MM cells, whereas AP nuclease inhibitor, although increased melphalan-induced cell death (panel A), was able to reverse melphalan-induced DNA-damage (γ-H2AX) (panel B) and genomic instability (assessed by micronucleus assay) (panels C) in MM cells.

MM (RPMI) cells, control (C) or those treated with melphalan (M), AP nuclease inhibitor (API3; 1 μM) or combination (API3-M) were evaluated for cell viability (a), DNA breaks by evaluating γH2AX expression (a DNA break marker) by western blotting (b) and genomic instability as assessed from micronucleus assay (c). Error bars in panel A represent SDs of triplicate assays
Discussion
HR is a DNA repair mechanism that utilizes sequence homology in the participating DNA strands to copy missing information either from a sister chromatid (in the G2 phase) or from homologous chromosome (in G1 phase of cell cycle)57. Since availability and utilization of sister chromatid as template in G2 phase of cell cycle makes the process of HR much more accurate (or error free), the regulatory mechanisms ensure that HR in G1 is suppressed and mitotic recombination machinery uses sister chromatid instead of a homolog chromosome as a template to repair the damage58. Thus HR, especially associated with G2 phase of cell cycle, has a unique role in the maintenance of genomic integrity and stability by accurately repairing double-strand breaks in DNA. Moreover, if DNA breaks escape the repair system and persist from G2 into mitosis, they can recombine in G1 to produce gene rearrangements. In normal cellular environment, the process of HR is tightly regulated and precise. However, the process of HR which involves incision and recombination of genomic DNA fragments, can also be deleterious, if dysregulated or altered. Consistent with this, we have previously shown that elevated HR activity mediates genomic instability and development of drug resistance in MM23. Investigating the mechanisms underlying dysregulation of HR, here we have identified AP nucleases (APEX1 and APEX2) as novel regulators of HR in MM. Using multiple cell types and various approaches and evaluating various features of HR, we show that transgenic as well as chemical suppression of AP nuclease activity inhibits HR in MM cells, whereas transgenic upregulation of AP nucleases in normal cells increases HR activity. These data suggest that besides important roles in BER, AP nucleases also regulate HR in MM. These data are consistent with the observations in glioblastoma indicating the role of APEX1 in the regulation of HR59. However, our study further demonstrates that ability of both APEX1 and APEX2 to regulate HR can be partly attributed to their ability to regulate RAD51 expression in MM cells. We show that transgenic as well as chemical suppression of AP nucleases inhibits RAD51 expression in MM as well as esophageal cancer (not shown) cells, whereas transgenic upregulation of APEX1 as well as APEX2 induces RAD51 expression. An important finding here is that APEX1 is involved in the regulation of HR, through P73-mediated transcriptional regulation of RAD51. However, we do not rule out the possibility that APEX1 may also be involved in regulation of RAD51/HR by other mechanisms. Consistent with this view, we observe that in addition to P73, APEX1 also interacts with other HR regulators including BRCA1, P53, and probably RAD51 itself (Supplementary Figure 4A). Although association of APEX1 with P73, a transcriptional regulator of RAD5160, was confirmed, APEX2 did not show interaction with P73 but with several other HR regulators including BRCA1, BRCA2, BARD1, RAD52, and P53. These data suggest that although APEX1 and APEX2 have some unique interacting partners, in general, both nucleases interact with major HR regulators and interconnect HR with BER.
Consistent with the role of AP nuclease expression in HR activity, both AP and HR activities were higher in melphalan-resistant, relative to sensitive cells. Moreover, the inhibition of AP nuclease activity increased the melphalan cytotoxicity even overcoming melphalan-resistance in MM cells. These data further support the evidence that AP nuclease inhibitors can be combined with melphalan and possibly other chemotherapeutic agents to increase their cytotoxicity. In MM cells, melphalan increased cell death as well as genomic instability. However, addition of AP inhibitor, although increased melphalan-induced cytotoxicity, was able to reverse the genomic instability caused by melphalan. Since melphalan causes DNA breaks, the cells with extensive breaks undergo apoptosis. However, melphalan-treated cells which have limited breaks and survive apoptosis, may undergo HR-mediated repair which may expose them to genomic rearrangements and instability. This is evident by increase in micronuclei (Fig. 6) and also by observation that AP nuclease inhibitor increases melphalan-induced cytotoxicity but reverses associated genomic instability. Commensurate with these data, we have also observed that just like AP nuclease inhibitor, inhibitors of RAD51 also increase melphalan-induced cytotoxicity but reverse genomic instability associated with it (unpublished data from our laboratory). Our results therefore show that AP nucleases play an important role in HR through regulation of RAD51 expression, and their inhibitors have potential to inhibit growth as well as ability to inhibit/delay evolution of MM cells. These inhibitors also have ability to increase cytotoxicity of chemotherapeutic agents and potential to reduce their harmful genomic impact.
References
Bolli, N. et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat. Commun. 5, 2997 (2014).
Grady, W. M. Genomic instability and colon cancer. Cancer Metastas-. Rev. 23, 11–27 (2004).
Kawano, M. M. Genomic instability in multiple myeloma. Nihon Rinsho. 65(Suppl 1), 215–219 (2007).
Sawyer, J. R. et al. Genomic instability in multiple myeloma: evidence for jumping segmental duplications of chromosome arm 1q. Genes Chromosomes Cancer 42, 95–106 (2005).
Sieber, O. M., Heinimann, K. & Tomlinson, I. P. Genomic instability--the engine of tumorigenesis? Nat. Rev. Cancer 3, 701–708 (2003).
Timuragaoglu, A., Demircin, S., Dizlek, S., Alanoglu, G. & Kiris, E. Microsatellite instability is a common finding in multiple myeloma. Clin. Lymphoma Myeloma 9, 371–374 (2009).
Croft, J. et al. Analysis of the premalignant stages of Barrett’s oesophagus through to adenocarcinoma by comparative genomic hybridization. Eur. J. Gastroenterol. Hepatol. 14, 1179–1186 (2002).
Paulson, T. G. et al. Chromosomal instability and copy number alterations in Barrett’s esophagus and esophageal adenocarcinoma. Clin. Cancer Res. 15, 3305–3314 (2009).
Rabinovitch, P. S., Reid, B. J., Haggitt, R. C., Norwood, T. H. & Rubin, C. E. Progression to cancer in Barrett’s esophagus is associated with genomic instability. Lab. Invest. 60, 65–71 (1989).
Usmani, B. A. Genomic instability and metastatic progression. Pathobiology 61, 109–116 (1993).
Biswas, S. et al. Mutational inactivation of TGFBR2 in microsatellite unstable colon cancer arises from the cooperation of genomic instability and the clonal outgrowth of transforming growth factor beta resistant cells. Genes Chromosomes Cancer 47, 95–106 (2008).
Lengauer, C., Kinzler, K. W. & Vogelstein, B. Genetic instabilities in human cancers. Nature 396, 643–649 (1998).
Stilgenbauer, S. et al. Clonal evolution in chronic lymphocytic leukemia: acquisition of high-risk genomic aberrations associated with unmutated VH, resistance to therapy, and short survival. Haematologica 92, 1242–1245 (2007).
Stoler, D. L. et al. Genomic instability in invasive breast carcinoma measured by inter-Simple Sequence Repeat PCR. Breast Cancer Res. Treat. 97, 107–110 (2006).
Stoler, D. L. et al. The onset and extent of genomic instability in sporadic colorectal tumor progression. Proc. Natl Acad. Sci. USA 96, 15121–15126 (1999).
Stoler, D. L. et al. Genomic instability measurement in the diagnosis of thyroid neoplasms. Head. Neck 24, 290–295 (2002).
van Tilborg, A. A. et al. Molecular evolution of multiple recurrent cancers of the bladder. Hum. Mol. Genet. 9, 2973–2980 (2000).
Jackson, S. P. & Bartek, J. The DNA-damage response in human biology and disease. Nature 461, 1071–1078 (2009).
Solomon, E., Borrow, J. & Goddard, A. D. Chromosome aberrations and cancer. Science 254, 1153–1160 (1991).
Heyer, W. D., Ehmsen, K. T. & Liu, J. Regulation of homologous recombination in eukaryotes. Annu. Rev. Genet. 44, 113–139 (2010).
Moynahan, M. E. & Jasin, M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Biol. 11, 196–207 (2010).
Guirouilh-Barbat, J., Lambert, S., Bertrand, P. & Lopez, B. S. Is homologous recombination really an error-free process? Front. Genet. 5, 175 (2014).
Shammas, M. A. et al. Dysfunctional homologous recombination mediates genomic instability and progression in myeloma. Blood 113, 2290–2297 (2009).
Pal, J. et al. Impact of RAD51C-mediated homologous recombination on genomic integrity in Barrett’s adenocarcinoma cells. J. Gastroenterol. Hepatol. Res. 6, 2286–2295 (2017).
Pal, J. et al. Genomic evolution in Barrett’s adenocarcinoma cells: critical roles of elevated hsRAD51, homologous recombination and Alu sequences in the genome. Oncogene 30, 3585–3598 (2011).
Lu, R. et al. Targeting homologous recombination and telomerase in Barrett’s adenocarcinoma: impact on telomere maintenance, genomic instability and tumor growth. Oncogene 33, 1495–1505 (2014).
Barzilay, G. & Hickson, I. D. Structure and function of apurinic/apyrimidinic endonucleases. Bioessays 17, 713–719 (1995).
Barzilay, G., Walker, L. J., Rothwell, D. G. & Hickson, I. D. Role of the HAP1 protein in repair of oxidative DNA damage and regulation of transcription factors. Br. J. Cancer Suppl. 27, S145–S150 (1996).
Mol, C. D., Hosfield, D. J. & Tainer, J. A. Abasic site recognition by two apurinic/apyrimidinic endonuclease families in DNA base excision repair: the 3’ ends justify the means. Mutat. Res. 460, 211–229 (2000).
Rothwell, D. G. et al. The structure and functions of the HAP1/Ref-1 protein. Oncol. Res. 9, 275–280 (1997).
Abbotts, R. & Madhusudan, S. Human AP endonuclease 1 (APE1): from mechanistic insights to druggable target in cancer. Cancer Treat. Rev. 36, 425–435 (2010).
Parsons, J. L., Dianova, I. I. & Dianov, G. L. APE1 is the major 3’-phosphoglycolate activity in human cell extracts. Nucleic Acids Res. 32, 3531–3536 (2004).
Fung, H. & Demple, B. A vital role for ape1/ref1 protein in repairing spontaneous DNA damage in human cells. Mol. Cell 17, 463–470 (2005).
Kelley, M. R. & Parsons, S. H. Redox regulation of the DNA repair function of the human AP endonuclease Ape1/ref-1. Antioxid. Redox Signal. 3, 671–683 (2001).
Burkovics, P., Szukacsov, V., Unk, I. & Haracska, L. Human Ape2 protein has a 3’-5’ exonuclease activity that acts preferentially on mismatched base pairs. Nucleic Acids Res. 34, 2508–2515 (2006).
Hadi, M. Z., Ginalski, K., Nguyen, L. H. & Wilson, D. M. 3rd Determinants in nuclease specificity of Ape1 and Ape2, human homologues of Escherichia coli exonuclease III. J. Mol. Biol. 316, 853–866 (2002).
Schindl, M. et al. DNA repair-redox enzyme apurinic endonuclease in cervical cancer: evaluation of redox control of HIF-1alpha and prognostic significance. Int. J. Oncol. 19, 799–802 (2001).
Freitas, S., Moore, D. H., Michael, H. & Kelley, M. R. Studies of apurinic/apyrimidinic endonuclease/ref-1 expression in epithelial ovarian cancer: correlations with tumor progression and platinum resistance. Clin. Cancer Res. 9, 4689–4694 (2003).
Tanner, B. et al. Nuclear expression of apurinic/apyrimidinic endonuclease increases with progression of ovarian carcinomas. Gynecol. Oncol. 92, 568–577 (2004).
Kelley, M. R. et al. Elevated and altered expression of the multifunctional DNA base excision repair and redox enzyme Ape1/ref-1 in prostate cancer. Clin. Cancer Res. 7, 824–830 (2001).
Robertson, K. A. et al. Altered expression of Ape1/ref-1 in germ cell tumors and overexpression in NT2 cells confers resistance to bleomycin and radiation. Cancer Res. 61, 2220–2225 (2001).
Herring, C. J. et al. Levels of the DNA repair enzyme human apurinic/apyrimidinic endonuclease (APE1, APEX, Ref-1) are associated with the intrinsic radiosensitivity of cervical cancers. Br. J. Cancer 78, 1128–1133 (1998).
Shammas, M. A. et al. Telomerase inhibition and cell growth arrest following telomestatin treatment in multiple myeloma. Clin. Cancer Res 10, 770–776 (2004).
Shammas, M. A. et al. Telomerase inhibitor GRN163L inhibits myeloma cell growth in vitro and in vivo. Leukemia 22, 1410–1418 (2008).
Shammas, M. A. et al. Specific killing of multiple myeloma cells by (-)-epigallocatechin-3-gallate extracted from green tea: biologic activity and therapeutic implications. Blood 108, 2804–2810 (2006).
Shammas, M. A. et al. Telomerase inhibition and cell growth arrest after telomestatin treatment in multiple myeloma. Clin. Cancer Res. 10, 770–776 (2004).
Tassone, P. et al. Cytotoxic activity of the maytansinoid immunoconjugate B-B4-DM1 against CD138+multiple myeloma cells. Blood 104, 3688–3696 (2004).
Tassone, P. et al. In vitro and in vivo activity of the maytansinoid immunoconjugate huN901-N2’-deacetyl-N2’-(3-mercapto-1-oxopropyl)-maytansine against CD56+ multiple myeloma cells. Cancer Res. 64, 4629–4636 (2004).
Tassone, P. et al. Combination therapy with interleukin-6 receptor superantagonist Sant7 and dexamethasone induces antitumor effects in a novel SCID-hu In vivo model of human multiple myeloma. Clin. Cancer Res. 11, 4251–4258 (2005).
Davies, F. E. et al. Insights into the multistep transformation of MGUS to myeloma using microarray expression analysis. Blood 102, 4504–4511 (2003).
Dorjsuren, D. et al. Diverse small molecule inhibitors of human apurinic/apyrimidinic endonuclease APE1 identified from a screen of a large public collection. PLoS ONE 7, e47974 (2012).
Liu, J., Sneeden, J. & Heyer, W. D. In vitro assays for DNA pairing and recombination-associated DNA synthesis. Methods Mol. Biol. 745, 363–383 (2011).
Huang, F. et al. Identification of specific inhibitors of human RAD51 recombinase using high-throughput screening. ACS Chem. Biol. 6, 628–635 (2011).
Balmus, G. et al. A high-throughput in vivo micronucleus assay for genome instability screening in mice. Nat. Protoc. 10, 205–215 (2015).
Terradas, M., Martin, M. & Genesca, A. Impaired nuclear functions in micronuclei results in genome instability and chromothripsis. Arch. Toxicol. 90, 2657–2667 (2016).
Peng, G. et al. Genome-wide transcriptome profiling of homologous recombination DNA repair. Nat. Commun. 5, 3361 (2014).
Shammas, M. A. & Shmookler Reis, R. J. Recombination and its roles in DNA repair, cellular immortalization and cancer. Age 22, 71–88 (1999).
Orthwein, A. et al. A mechanism for the suppression of homologous recombination in G1 cells. Nature 528, 422–426 (2015).
Strobel, T. et al. Ape1 guides DNA repair pathway choice that is associated with drug tolerance in glioblastoma. Sci. Rep. 7, 9674 (2017).
Lin, Y. L. et al. p63 and p73 transcriptionally regulate genes involved in DNA repair. PLoS. Genet. 5, e1000680 (2009).
Acknowledgements
This work was supported by Department of Veterans Affairs Merit Review Award I01BX001584-01 (NCM), NIH grants P01-155258 and 5P50 CA100707 (MAS, MKS, NCM) and Leukemia and Lymphoma Society translational research grant (NCM).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kumar, S., Talluri, S., Pal, J. et al. Role of apurinic/apyrimidinic nucleases in the regulation of homologous recombination in myeloma: mechanisms and translational significance. Blood Cancer Journal 8, 92 (2018). https://doi.org/10.1038/s41408-018-0129-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41408-018-0129-9
This article is cited by
-
Preclinical validation and phase I trial of 4-hydroxysalicylanilide, targeting ribonucleotide reductase mediated dNTP synthesis in multiple myeloma
Journal of Biomedical Science (2022)
-
Integrated genomics and comprehensive validation reveal drivers of genomic evolution in esophageal adenocarcinoma
Communications Biology (2021)
-
Exploiting synthetic lethality to target BRCA1/2-deficient tumors: where we stand
Oncogene (2021)
-
Dysregulated APOBEC3G causes DNA damage and promotes genomic instability in multiple myeloma
Blood Cancer Journal (2021)
-
Genomic alterations and abnormal expression of APE2 in multiple cancers
Scientific Reports (2020)
