
Abstract
Mitsugumin 53 (MG53) is a tripartite motif family protein that has been reported to attenuate injury via membrane repair in different organs. Contrast-induced acute kidney injury (CI-AKI) is a common complication caused by the administration of iodinated contrast media (CM). While the cytotoxicity induced by CM leading to tubular cell death may be initiated by cell membrane damage, we wondered whether MG53 alleviates CI-AKI. This study was designed to investigate the effect of MG53 on CI-AKI and the underlying mechanism. A rat model of CI-AKI was established, and CI-AKI induced the translocation of MG53 from serum to injury sites on the renal proximal tubular (RPT) epithelia, as illustrated by immunoblot analysis and immunohistochemical staining. Moreover, pretreatment of rats with recombinant human MG53 protein (rhMG53, 2 mg/mL) alleviated iopromide-induced injury in the kidney, which was determined by measuring serum creatinine, blood urea nitrogen and renal histological changes. In vitro studies demonstrated that exposure of RPT cells to iopromide (20, 40, and 80 mg/mL) caused cell membrane injury and cell death, which were attenuated by rhMG53 (10 and 50 μg/mL). Mechanistically, MG53 translocated to the injury site on RPT cells and bound to phosphatidylserine to protect RPT cells from iopromide-induced injury. In conclusion, MG53 protects against CI-AKI through cell membrane repair and reducing cell apoptosis; therefore, rhMG53 might be a potential effective means to treat or prevent CI-AKI.
Similar content being viewed by others


Introduction
Contrast-induced acute kidney injury (CI-AKI) [1] is a common complication of invasive cardiovascular procedures. It occurs in up to 30% of patients who receive iodinated contrast media (CM), which is considered the third most common cause of hospital-acquired AKI [2,3,4]. CI-AKI is associated with increased morbidity, extended length of hospital stays, and increased medical care costs [5,6,7]. While the precise mechanism underlying CI-AKI is not fully understood, the toxic effects of CM on renal proximal tubular (RPT) cells likely play a role in CI-AKI [8,9,10,11]. There are few effective treatments available for CI-AKI, but adequate hydration is the basis of all preventative strategies [6, 12, 13]. Interventions, including N-acetylcysteine, vitamin C, statins, furosemide and other drugs, have yielded controversial results [3, 14,15,16,17,18]. Thus, it is necessary to identify effective preventative measures and post-CM therapies to combat CI-AKI and improve clinical outcomes.
The cell membrane is a barrier that prevents extracellular substances from freely entering the cell and is vital for homeostatic maintenance in mammalian cells [19]. However, membrane damage can result from numerous threats, including physical, chemical and biochemical factors [20], leading to intracellular dyshomeostasis and eventually cell death. In studies of CM cytotoxicity, attention has rarely been drawn to cell membrane injury, but it may be the initial factor in CM-induced cytotoxicity that leads to apoptosis of endothelial and tubular cells [21].
Mitsugumin 53 (MG53) is a cell membrane repair protein that is predominantly expressed in striated muscles [22,23,24]. Recombinant human MG53 protein (rhMG53) has demonstrated efficacy in the prevention or treatment of injuries in a variety of organs, such as the heart, muscle, lung, and cornea [25–28]. Our previous study reported that MG53 is also present in the kidney, particularly in RPT cells, and MG53 knockout mice show renal insufficiency under stress conditions [29]. However, endogenous protein expression of MG53 in RPT cells is low, ~1/50 of the level present in skeletal muscle [29], indicating a possible benefit of exogenous MG53 in protecting the kidney tubules. We hypothesized that MG53 alleviates CI-AKI through cell membrane repair. In this study, we investigated endogenous MG53 in a rat model of CI-AKI and examined the additional therapeutic potential of exogenous rhMG53.
Materials and methods
Animals
Adult Sprague Dawley (SD) rats (males and females, weighing 225 ± 15 g) were obtained from the Laboratory Animal Center of Daping Hospital (Chongqing, China). All procedures were in accordance with institutional regulations and with the approval of the Experimental Animals Committee of Daping Hospital.
CI-AKI model
The rat model of CI-AKI was induced according to a previously described protocol [30,31,32]. Briefly, the rats were deprived of food with free access to water for 12 h before the operation, anesthetized by intraperitoneal injection of sodium pentobarbital (60 mg/kg) and placed on a heating pad to maintain body temperature at 37 °C during surgery. Jugular vein cannulation was performed in the CI-AKI group, and indomethacin (10 mg/kg, dissolved in dimethyl sulfoxide) was first administered. Then, L-nitro-arginine methyl ester (L-NAME, 10 mg/kg, dissolved in saline) was administered 15 min later, and iopromide (a low-osmolality, nonionic iodinated contrast media, 1600 mg I/kg) was injected 15 min after L-NAME administration. Saline was used instead of iopromide in the control group. In addition, rhMG53 or rhodamine-labeled rhMG53 (2 mg/kg) was administered before the indomethacin in the rhMG53 and rhMG53 + CI-AKI groups. The incisions were sutured, and the rats were then allowed to recover with free access to food and water. Twenty-four hours after iopromide administration, the rats were re-anaesthetized, and then a laparotomy and nephrectomy were performed. Blood samples were obtained from the left ventricle. Kidneys were harvested, rinsed with oxygenated saline and stored for further analysis.
Renal function
The renal function of the rats was assayed by measuring serum creatinine (SCr) and blood urea nitrogen (BUN). Serum samples were analyzed on an automated Beckman Analyzer (Beckman Instruments GmbH, Munich, Germany).
Renal histopathology and immunohistochemical staining
Kidney samples were fixed in 4% paraformaldehyde and then dehydrated in increasing concentrations of ethanol. The samples were cleared in xylene and embedded in paraffin. The samples were cut into 4 μm thick sections, followed by staining with hematoxylin and eosin (H&E). Each sample section was examined by a pathologist in a blind manner. Morphological changes in the tubules, including tubular epithelial cell swelling, vacuolization, cast formation, and desquamation, were scored to assess the degree of renal damage as previously described [33]. Histologic scoring ranged from 0 to 5 points based on the area of injury involvement.
Immunohistochemical (IHC) staining was performed using a rabbit anti-MG53 antibody (1:100) [28, 29, 34], and reactions were detected using horseradish peroxidase-conjugated goat anti-rabbit IgG (Zsbio, Beijing, China). The color was developed using 3,3'-diaminobenzidine tetrahydrochloride (DAB, Solarbio, Beijing, China) and stopped by rinsing in deionized water, followed by microscopy.
To observe the localization of exogenous rhodamine-labeled rhMG53, the kidneys were harvested, embedded in optimal cutting temperature (OCT) compound, and snap-frozen. Samples were cut into 7 μm thick cryosections and fixed in freshly prepared 4% paraformaldehyde in phosphate-buffered saline (PBS) at 4 °C. 4’,6-diamidino-2-phenylindole (DAPI) was used to stain the nuclei.
Cell culture
Immortalized RPT cells (Taconic, Germantown, NY, USA) from Wistar-Kyoto (WKY) rats were cultured at 37 °C in 95% air and 5% CO2 in DMEM/F-12 (HyClone, South Logan, UT, USA) with transferrin (5 μg/mL), insulin (5 μg/mL), epidermal growth factor (10 ng/mL), dexamethasone (4 μg/mL), 1% v/v penicillin/streptomycin (Invitrogen Life Technologies, Karlsruhe, Germany), and 10% (v/v) fetal bovine serum (FBS, Invitrogen Life Technologies, Karlsruhe, Germany). Subculture was performed by washing the cell monolayers twice with PBS, followed by the addition of 0.25% trypsin-ethylenediamine tetraacetic acid (EDTA) solution (Invitrogen Life Technologies, Karlsruhe, Germany) and incubation at 37 °C until the cells detached. Trypsin was inactivated by the addition of growth medium before seeding into fresh T25 flasks at densities of 2 × 104–4 × 104 cells/cm2. The medium was changed every 2 to 4 days.
Immunoblotting
Proteins were extracted from kidney tissues or RPT cells with tissue extraction reagent (Thermo Scientific, Waltham, MA, USA) containing a protease inhibitor cocktail (Roche, Indianapolis, IN, USA). The homogenate was sonicated and stored on ice for 1 h. The lysate was centrifuged at 12,000 rpm for 30 min. The supernatant was collected, and the protein concentration was measured using a BCA kit (Solarbio, Beijing, China). Protein samples (50 μg) were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis with 10% polyacrylamide gels and electrotransferred to nitrocellulose membranes. After the non-specific binding sites were blocked in Tris-buffered saline (TBS) containing 5% non-fat dry milk for 1 h, the membranes were incubated at 4 °C overnight with rabbit anti-MG53 antibody. Mouse anti-GAPDH antibody (1:5000, Proteintech, Wuhan, China) was used as an internal control. The membranes were washed and then incubated with IRDye 680-labeled goat anti-rabbit or goat anti-mouse secondary antibodies (1:15,000, Li-Cor Biosciences, Lincoln, NE, USA) at room temperature for 1 h. The membranes were washed three times in TBS-Tween 20, and the bands were detected by an Odyssey infrared imaging system (Li-Cor Biosciences, Lincoln, NE, USA). The images were analyzed using Odyssey Application Software to obtain the integrated intensities.
Real-time quantitative polymerase chain reaction
MG53 messenger RNA (mRNA) levels were quantified by real-time quantitative polymerase chain reaction (RT-qPCR). Total RNA was extracted using RNAiso Plus reagent (TaKaRa, Tokyo, Japan), and complementary DNA synthesis was performed using PrimeScript™ RT master mix (TaKaRa, Tokyo, Japan) according to the manufacturer’s instructions. For RT-qPCR, the mRNA level of MG53 was measured using TB Green™ Premix Ex Taq™ (TaKaRa, Tokyo, Japan) and a Thermal Cycler Dice™ Real-Time System (Bio-Rad, Hercules, CA, USA). The following PCR conditions were applied: 95 °C for 3 min, 40 cycles at 95 °C for 10 s and 62 °C for 30 s, and 62 °C for 10 s. The specific primers used were listed below: MG53, forward primer (5′-CCACAGTTTCTGCCGTGCCT-3′) and reverse primer (5′-TAGTGCTTAGCGCCTGTGGC-3′); β-actin, forward primer (5′-TCACTGTCCACCTTCCAGCAGA-3′) and reverse primer (5′-AGGGTGTAAAACGCAGCTCAGTAA-3′). The relative mRNA expression levels were evaluated using the 2−ΔΔCt method and normalized to β-actin. The PCR products were also analyzed by agarose gel electrophoresis.
TUNEL assay
TUNEL assays were performed with the DeadEnd™ Fluorometric TUNEL System (Promega, Madison, WI, USA) according to the manufacturer’s instructions. Briefly, slides were immersed in 4% paraformaldehyde for 25 min at 4 °C, followed by permeabilization with 0.2% Triton® X-100 in PBS for 5 min. The slides were equilibrated for 5 min in the manufacturer’s buffer before undergoing terminal deoxynucleotidyl transferase (TdT) reactions for 60 min at 37 °C. Reactions were stopped with 2× saline sodium citrate (SSC), and the slides were washed three times with PBS and mounted. To visualize all nuclei, the slides were incubated with DAPI. Finally, localized green fluorescence of apoptotic cells was detected by fluorescence microscopy.
Cell viability assay
Cell viability was assayed using a Cell Counting Kit (CCK)-8 kit (Beyotime, Haimen, China). Briefly, RPT cells were cultured in a 96-well plate and treated with iopromide (20, 40 and 80 mg/mL) with or without rhMG53. PBS and boiled rhMG53 were used as controls. Cells were then treated with 10 μL CCK-8 solution/well and incubated for another 30 min at 37 °C. The amount of formazan dye generated by cellular dehydrogenase activity was determined by measuring the absorbance at 450 nm with a microplate reader.
Quantification of iopromide-induced lactic dehydrogenase release from RPT cells
RPT cells were cultured in a 24-well plate in the presence of iopromide (20, 40 and 80 mg/mL) for up to 1 h with or without rhMG53. PBS and BSA were used as controls. The amount of lactic dehydrogenase (LDH) released from the cells was measured with an LDH assay kit (Beyotime, Haimen, China) according to the manufacturer’s instructions with modifications. The supernatants were diluted 1:10 in PBS to reduce the concentration of iopromide because a high concentration of iopromide affects the accuracy of LDH detection.
FM1-43 staining and confocal microscopy
RPT cells were seeded in 20 mm glass bottom culture dishes (Nest, Wuxi, China) at a density of 1 × 104 cells/cm2 and incubated for 12 h, after which the cells were incubated with iopromide (80 mg/mL) with or without rhMG53 (10, 50 μg/mL), and PBS was used as the control. After rinsing with Tyrode’s solution, the cells were mixed with FM1-43 dye (Invitrogen Life Technologies, Karlsruhe, Germany). FM1-43 is membrane impermeable and becomes fluorescent when it enters injured cells and binds to cellular lipids. Dye entry into the cells was monitored continuously with a fluorescence confocal microscope (Olympus Co., Tokyo, Japan) immediately after mixing the cells with the dye. Consecutive live cell images were obtained at an interval of 4 s/frame for 120 frames.
For immunofluorescence staining, RPT cells (1 × 104 cells/well) were seeded and grown overnight in 20 mm glass bottom culture dishes (Nest, Wuxi, China) and then treated with iopromide (80 mg/mL) with or without rhMG53 (50 μg/mL); PBS was used as the control. 4 h later, the cells were fixed with 4% paraformaldehyde for 30 min at 4 °C, followed by permeabilization and blocking with immunostaining blocking buffer (Sangon Biotech, Shanghai, China). The cells were then incubated overnight at 4 °C with primary antibodies, including mouse monoclonal Annexin V antibody (1:100, Santa Cruz, CA, USA) and rabbit polyclonal MG53 antibody (1:100). After incubation with the secondary antibody and DAPI, the samples were observed under a confocal microscope (Olympus Co., Tokyo, Japan).
Statistical analysis
The data are presented as the mean ± standard error of the mean (SEM). Statistical significance was determined by ANOVA followed by a post hoc Dunnett’s test for multi-group (>2) comparisons. Student’s t-test was used for two-group comparisons. Differences were considered statistically significant when P < 0.05.
Results
CI-AKI was ameliorated by administration of rhMG53 protein in SD rats
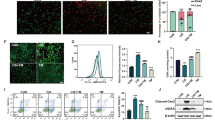
Although MG53 is mainly expressed in skeletal muscle and myocardium, our previous studies also found MG53 expression in the lung [26] and kidney [29]. To investigate whether MG53 plays a role in CI-AKI, we first examined the protein levels of MG53 in the serum and kidney of rats with or without CI-AKI. CI-AKI was verified by increases in SCr and BUN in rats that were administered CM (Fig. 1a, b). Immunoblotting for MG53 showed that MG53 was reduced in the serum of rats with CI-AKI compared to that of controls (Fig. 1c), but was increased in the kidneys (Fig. 1d) of CI-AKI rats relative to those with normal kidney function. IHC staining revealed that endogenous MG53 accumulated in the luminal tubular cell membranes after CI-AKI (Fig. 1e). Immunofluorescent staining of MG53 in frozen sections indicated that exogenous rhodamine-labeled rhMG53 accumulated in the tubules of rats with CI-AKI but not in controls (Fig. 1f). When administered to rats before CI-AKI, rhMG53 attenuated kidney injury, as shown by the reduction in SCr and BUN levels (Fig. 2a, b). In addition, rhMG53 pretreatment reduced histological injury to the renal tubules of CI-AKI rats, as illustrated by H&E staining and semiquantitative scoring of the pathological lesion (Fig. 2c, d). The TUNEL assay showed alleviated tubular cell apoptosis after rhMG53 administration in CI-AKI rats (Fig. 2e, f).

a, b Rats with CI-AKI showed elevated SCr and BUN (*P < 0.05 vs. n = 8). c MG53 protein in the serum decreased significantly in rats subjected to CI-AKI compared with that of control rats (*P < 0.05 vs. n = 8 in control and 7 in CI-AKI). d Western blot showing increased MG53 protein in kidney lysates derived from CI-AKI rats (*P < 0.05 vs. n = 3). e IHC staining showing endogenous MG53 accumulated in the luminal tubular cell membranes in the kidneys of rats subjected to CI-AKI (NC, negative control. Magnification: ×200, top panel; ×400, bottom panel; Scale bar, 50 μm). f Immunofluorescence of rhodamine-rhMG53 in frozen kidney sections showing that exogenous rhodamine-labeled rhMG53 accumulated in the tubules of rats with CI-AKI but not controls (magnification: ×400; scale bar, 50 μm).

a, b Effect of rhMG53 on renal function, as assessed by SCr and BUN. Treatment with rhMG53 (2 mg/kg, intravenous administration) did not impact SCr or BUN in control rats but abrogated the CM-induced increase in SCr and BUN (*P < 0.05 vs. control, #P < 0.05 vs. CI-AKI; n = 8). c Representative images of H&E staining showing that rhMG53 pretreatment alleviated renal pathological injury at 24 h after CI-AKI (magnification of ×400, Scale bar, 50 μm). d Pathological damage scores of H&E-stained sections showing the beneficial effects of rhMG53 in mitigating CI-AKI in rats (*P < 0.05 vs. control, #P < 0.05 vs. CI-AKI; n = 8). e, f Representative images and statistical results of the TUNEL assay showing alleviated tubular cell apoptosis after rhMG53 administration in CI-AKI rats (*P < 0.05 vs. control, #P < 0.05 vs. CI-AKI; n = 4).
MG53 reduced iopromide-induced apoptosis in RPT cells
We used cultured RPT epithelial cells to investigate the protective mechanisms of rhMG53. Cell viability was assayed using CCK-8, and the results suggested that iopromide decreased RPT cell viability in a concentration- and time-dependent manner (Fig. 3a, b). In addition, the TUNEL assay showed that iopromide (80 mg/mL) significantly increased the number of apoptotic RPT cells (Fig. 3c, d). However, in the presence of rhMG53 (50 μg/mL), the reduction in cell viability after iopromide treatment was attenuated (Fig. 3e), and the number of TUNEL-positive cells was dramatically reduced (Fig. 3c, d).

a Iopromide decreased the viability of RPT cells in a dose-dependent manner (*P < 0.05 compared with control, #P < 0.05 compared with iopromide 20 mg/mL, $P < 0.05 compared with iopromide 40 mg/mL; n = 3). b Iopromide decreased the viability of RPT cells in a time-dependent manner (*P < 0.05 compared with control; n = 3). c, d The number of TUNEL-positive apoptotic cells was increased after iopromide treatment, while rhMG53 pretreatment reduced cell apoptosis (magnification: ×200; scale bar, 50 μm; *P < 0.05 vs. control, #P < 0.05 vs. iopromide; n = 3). e Cell viability quantification demonstrating the beneficial effects of rhMG53 treatment (50 µg/mL) (*P < 0.05 vs. control, #P < 0.05 vs. iopromide; n = 3).
MG53 reduced iopromide-induced cell membrane injury in RPT cells
We suspected that injury to the cell membrane was the initial event in iopromide-induced cytotoxicity [21]. To test this hypothesis, LDH release from RPT cells was measured after iopromide treatment. The cell culture supernatants were diluted 1:10 in PBS in the experiments to reduce the concentration of iopromide because a high concentration of iopromide affects the accuracy of LDH detection. Iopromide (80 mg/mL) markedly increased LDH in the cell culture medium in a concentration- and time-dependent manner (Fig. 4a, b). In addition, iopromide-induced cell membrane damage was further confirmed by monitoring entry of the fluorescent dye FM1-43 into RPT cells. Consistent with the LDH release assay results, little FM1-43 entered vehicle-treated cells, but FM1-43 accumulated in iopromide-treated cells in a time-dependent manner (Fig. 4c, d and Supplemental Movies S1–S2). This accumulation was significantly blocked in a dose-dependent fashion by rhMG53 (10 and 50 μg/mL, Fig. 4c, d and Supplemental Movies S1–S4). Additionally, LDH release was blocked by rhMG53 (10 and 50 μg/mL) in a dose-dependent manner (Fig. 4e).

a Iopromide increased LDH release in a dose-dependent manner in RPT cells (*P < 0.05 compared with control, #P < 0.05 compared with iopromide 20 mg/mL, $P < 0.05 compared with iopromide 40 mg/mL; n = 3). b Iopromide caused a time-dependent increase in LDH release in RPT cells (*P < 0.05 compared with control; n = 3). c, d Representative images and statistical results showing iopromide-induced entry of FM1-43 dye into RPT cells. rhMG53 reduced FM1-43 dye entry in a dose-dependent manner (>200 cells were analyzed for each condition and each experiment; magnification: ×800; scale bar, 20 μm). e Iopromide-induced LDH release from RPT cells was attenuated by rhMG53 in a dose-dependent manner (*P < 0.05 compared with the 2nd group, #P < 0.05 compared with the 3rd group; n = 3).
MG53 translocated to the cell membrane and bound to phosphatidylserine to mediate a membrane protective effect
MG53 has been reported to be recruited to sites of membrane damage after cell injury [22, 35]. To investigate the temporal and spatial changes in MG53 expression in RPT cells after iopromide treatment, several experiments were performed. First, qPCR analysis showed that MG53 mRNA was expressed in RPT cells (Fig. 5a) and that iopromide treatment had little effect on mRNA transcription of MG53 (Fig. 5a). In addition, the immunoblotting results suggested that exogenous rhMG53 treatment increased MG53 protein accumulation in iopromide-injured RPT cells (Fig. 5b). The accumulation of MG53 in iopromide-injured RPT cells was further confirmed by laser immunofluorescence microscopy (Fig. 5c). In RPT cells without iopromide treatment, endogenous MG53 was mostly scattered in the cytosol. However, more MG53 was distributed at the cell membrane after iopromide treatment (upper panel) than in vehicle-treated cells. More interestingly, rhodamine-labeled rhMG53 was concentrated at the cell membrane in iopromide-treated RPT cells but not in control cells (lower panel). This result may be specific for rhMG53 because rhodamine-labeled BSA was not concentrated.

a Representative images of agarose gel electrophoresis and RT-PCR showing the presence of MG53 mRNA in RPT cells. No significant changes in MG53 mRNA were detected with iopromide treatment (n = 3). b The total protein level of MG53 was not changed in RPT cells after iopromide treatment, while RPT cells treated with rhMG53 showed abundant uptake of the protein. GAPDH was used as an internal control (*P < 0.05 vs. control, #P < 0.05 vs. iopromide; n = 3). c Laser confocal microscopy showing that MG53 was mostly scattered in the cytosol, while more MG53 was distributed at the cell membrane after iopromide treatment (upper panel). Rhodamine-labeled rhMG53 was concentrated at the cell membrane in iopromide-treated RPT cells but not in control cells (lower panel). This result may be specific for rhMG53 because rhodamine-labeled BSA was not concentrated (magnification: ×400; scale bar, 50 μm).
The membrane repair function of MG53 has been reported to be mediated by interactions with exposed phosphatidylserine (PS) at injury sites [22, 26]. Annexin V is a cellular protein that is commonly used to detect apoptotic cells by binding to PS. To verify whether the protective effect of rhMG53 against cell membrane damage induced by iopromide was mediated by binding to PS, immunofluorescence staining was performed and indicated that rhMG53 bound with Annexin V after iopromide treatment (Fig. 6a). In addition, the membrane protective effect of MG53 on RPT cells was attenuated by exogenous administration of PS but not phosphatidylcholine, as reflected by increased LDH release compared to that of the control groups (Fig. 6b).

a Laser confocal microscopy showing rhodamine-labeled rhMG53 (lower panel) but not rhodamine-labeled BSA (upper panel) bound to Annexin V after iopromide treatment (magnification: ×400; scale bar, 50 μm). b The membrane protective effect of rhMG53 was attenuated by exogenous administration of PS, as indicated by LDH release (*P < 0.05 compared with the 3rd group, #P < 0.05 compared with the 5th group; n = 3).
Discussion
In the present study, we found that MG53 prevents CI-AKI by attenuating proximal tubular cell apoptosis. This conclusion is based on the following findings: (i) endogenous MG53 is present in the serum and kidneys of healthy rats; (ii) endogenous MG53 translocates from the serum to the kidney after exposure to CM and accumulates in the luminal cell membranes of the proximal tubule epithelia; (iii) rhMG53 protein accumulates in the tubulointerstitium of rodents with CI-AKI but not in control rats; (iv) administration of rhMG53 attenuates the decrease in kidney function, histological damage and tubular cell apoptosis during CI-AKI; (v) contrast reduces proximal tubular epithelial cell viability through the induction of apoptosis, which is ameliorated by rhMG53; (vi) rhMG53 alleviates cell membrane damage in contrast-treated RPT cells and prevents increased membrane permeability; and (vii) rhMG53 binds to exposed phosphatidylserine groups on contrast-injured RPT cell membranes.
MG53 is a key component of the membrane resealing machinery [22, 36, 37] that has been regarded as a muscle-specific protein [22, 37,38,39], but it has also been found in the lungs [26] and kidneys [29]. More interestingly, exercise or muscle injury triggers the release of MG53 from skeletal muscle into circulation, and muscle-derived MG53 protects remote organs from injury. Our study found that MG53 is expressed at the transcript and protein levels in an RPT cell line, indicating that MG53 is a ubiquitous cell repair protein. The finding that serum MG53 translocates to the kidney during CI-AKI supports this notion. However, the effect of this endogenous system does not appear to be sufficient to prevent the acute loss of kidney function.
Previous studies have shown that exogenous rhMG53 protects the heart and skeletal muscles from acute ischemia-reperfusion injury [27, 39] and the lung from multiple types of injury [26], and accelerates dermal wound healing [34]. These studies demonstrate that administration of rhMG53 augments the endogenous protective system and opens the possibility that rhMG53 could be used therapeutically. With respect to the therapeutic potential of MG53 in the kidney, rhMG53 has been shown to alleviate ischemia-reperfusion- or cisplatin-induced acute kidney injury [29] and kidney injury after severe burns [40]. Thus, it is reasonable to investigate whether augmenting MG53 prior to CI-AKI could protect the kidney, and our study proved that renal protection was afforded both functionally and histologically. This raises the intriguing possibility that MG53 could be used to pretreat patients at high risk for contrast-induced kidney injury, reducing the length of hospitalization and overall morbidity, because the most important risk factor for CI-AKI in humans is pre-existing chronic kidney disease. However, this remains an open question because our model started with rats with normal kidney function. Our study does not address whether administration of exogenous MG53 can benefit already established CI-AKI and decrease the time to recovery of kidney function, which is an equally relevant clinical question. In addition, SCr and BUN are not very sensitive markers in animal models. These limitations need to be carefully considered and addressed in further studies.
The direct cytotoxic effect of CM on tubular cells is considered to be one of the important mechanisms of CI-AKI [41, 42] because active endocytosis and exocytosis in the brush border of proximal tubular cells are essential for normal kidney function. Our study also found that CM primarily affects tubular cells, as reflected by H&E staining. Furthermore, CM induced tubular cell apoptosis both in vivo and in vitro, which was alleviated by rhMG53 administration. It has been proposed that direct cell toxicity and the proapoptotic effects of CM are initiated by cell membrane damage [21, 43]. When the membrane is damaged, extracellular molecules such as Ca2+ enter the cell freely, which could subsequently trigger the release of intracellular apoptotic signals and eventually lead to cell apoptosis. Thus, MG53 may reduce tubular cell apoptosis through membrane repair. However, another limitation of our study is that we did not investigate the role of MG53 in tubular cell necrosis, which might also contribute to the development of CI-AKI. MG53 is known to bind to phosphatidylserine with some specificity and to interact with caveolin-3 as part of its function in vesicle trafficking [22, 37, 44]. Our data also proved that binding to phosphatidylserine, likely due to increased apoptosis, was a key mechanism by which MG53 mediates the membrane repair process after iopromide treatment.
In conclusion, this study demonstrates that pretreatment with rhMG53 protects against CI-AKI by reducing tubular cell membrane damage and apoptosis. Our study provides new insight into the clinical prevention and treatment of CI-AKI.
References
Calvin AD, Misra S, Pflueger A. Contrast-induced acute kidney injury and diabetic nephropathy. Nat Rev Nephrol. 2010;6:679–88.
Davidson CJ, Hlatky M, Morris KG, Pieper K, Skelton TN, Schwab SJ, et al. Cardiovascular and renal toxicity of a nonionic radiographic contrast agent after cardiac catheterization. A prospective trial. Ann Intern Med. 1989;110:119–24.
McCullough PA. Contrast-induced acute kidney injury. J Am Coll Cardiol. 2008;51:1419–28.
Fähling M, Seeliger E, Patzak A, Persson PB. Understanding and preventing contrast-induced acute kidney injury. Nat Rev Nephrol. 2017;13:169–80.
Murphy SW, Barrett BJ, Parfrey PS. Contrast nephropathy. J Am Soc Nephrol. 2000;11:177–82.
Maeder M, Klein M, Fehr T, Rickli H. Contrast nephropathy: review focusing on prevention. J Am Coll Cardiol. 2004;44:1763–71.
Rancic ZS. Commentary on 'Contrast Induced Nephropathy and Long-term Renal Decline After Percutaneous Transluminal Angioplasty for Symptomatic Peripheral Arterial Disease'. Eur J Vasc Endovasc Surg. 2016;51:394.
Tepel M, Aspelin P, Lameire N. Contrast-induced nephropathy: a clinical and evidence-based approach. Circulation. 2006;113:1799–806.
Seeliger E, Sendeski M, Rihal CS, Persson PB. Contrast-induced kidney injury: mechanisms, risk factors, and prevention. Eur Heart J. 2012;33:2007–15.
Silvain J, Collet JP, Montalescot G. Contrast-induced nephropathy: the sin of primary percutaneous coronary intervention? Eur Heart J. 2014;35:1504–6.
Romano G, Briguori C, Quintavalle C, Zanca C, Rivera NV, Colombo A, et al. Contrast agents and renal cell apoptosis. Eur Heart J. 2008;29:2569–76.
Mamoulakis C, Tsarouhas K, Fragkiadoulaki I, Heretis I, Wilks MF, Spandidos DA, et al. Contrast-induced nephropathy: Basic concepts, pathophysiological implications and prevention strategies. Pharmacol Ther. 2017;180:99–112.
Mathew R, Haque K, Woothipoom W. Acute renal failure induced by contrast medium: steps towards prevention. BMJ. 2006;333:539–40.
ACT Investigators. Acetylcysteine for prevention of renal outcomes in patients undergoing coronary and peripheral vascular angiography: main results from the randomized Acetylcysteine for Contrast-induced nephropathy Trial (ACT). Circulation. 2011;124:1250–9.
Thiele H, Hildebrand L, Schirdewahn C, Eitel I, Adams V, Fuernau G, et al. Impact of high-dose N-acetylcysteine versus placebo on contrast-induced nephropathy and myocardial reperfusion injury in unselected patients with ST-segment elevation myocardial infarction undergoing primary percutaneous coronary intervention. The LIPSIA-N-ACC (Prospective, Single-Blind, Placebo-Controlled, Randomized Leipzig Immediate PercutaneouS Coronary Intervention Acute Myocardial Infarction N-ACC) Trial. J Am Coll Cardiol. 2010;55:2201–9.
Marenzi G, Assanelli E, Marana I, Lauri G, Campodonico J, Grazi M, et al. N-acetylcysteine and contrast-induced nephropathy in primary angioplasty. N Engl J Med. 2006;354:2773–82.
Kelly AM, Dwamena B, Cronin P, Bernstein SJ, Carlos RC. Meta-analysis: effectiveness of drugs for preventing contrast-induced nephropathy. Ann Intern Med. 2008;148:284–94.
Myrvang H. Acute kidney injury: Acetylcysteine does not prevent contrast-induced acute kidney injury. Nat Rev Nephrol. 2011;7:605.
Zhang Y, Chen X, Gueydan C, Han J. Plasma membrane changes during programmed cell deaths. Cell Res. 2018;28:9–21.
Jimenez AJ, Maiuri P, Lafaurie-Janvore J, Divoux S, Piel M, Perez F. ESCRT machinery is required for plasma membrane repair. Science. 2014;343:1247136.
Sendeski MM. Pathophysiology of renal tissue damage by iodinated contrast media. Clin Exp Pharmacol Physiol. 2011;38:292–9.
Cai C, Masumiya H, Weisleder N, Matsuda N, Nishi M, Hwang M, et al. MG53 nucleates assembly of cell membrane repair machinery. Nat Cell Biol. 2009;11:56–64.
Yi JS, Park JS, Ham YM, Nguyen N, Lee NR, Hong J, et al. MG53-induced IRS-1 ubiquitination negatively regulates skeletal myogenesis and insulin signalling. Nat Commun. 2013;4:2354.
Tan T, Ko YG, Ma J. Dual function of MG53 in membrane repair and insulin signaling. BMB Rep. 2016;49:414–23.
Weisleder N, Takizawa N, Lin P, Wang X, Cao C, Zhang Y, et al. Recombinant MG53 protein modulates therapeutic cell membrane repair in treatment of muscular dystrophy. Sci Transl Med. 2012;4:139ra85.
Jia Y, Chen K, Lin P, Lieber G, Nishi M, Yan R, et al. Treatment of acute lung injury by targeting MG53-mediated cell membrane repair. Nat Commun. 2014;5:4387.
Liu J, Zhu H, Zheng Y, Xu Z, Li L, Tan T, et al. Cardioprotection of recombinant human MG53 protein in a porcine model of ischemia and reperfusion injury. J Mol Cell Cardiol. 2015;80:10–9.
Chandler HL, Tan T, Yang C, Gemensky-Metzler AJ, Wehrman RF, Jiang Q, et al. MG53 promotes corneal wound healing and mitigates fibrotic remodeling in rodents. Commun Biol. 2019;2:71.
Pu D, Li H, Lin P, Tan T, Wang Z, Chen K, et al. MG53-mediated cell membrane repair protects against acute kidney injury. Sci Transl Med. 2015;7:279ra36.
Agmon Y, Peleg H, Greenfeld Z, Rosen S, Brezis M. Nitric oxide and prostanoids protect the renal outer medulla from radiocontrast toxicity in the rat. J Clin Invest. 1994;94:1069–75.
Yokomaku Y, Sugimoto T, Kume S, Araki S, Isshiki K, Chin-Kanasaki M, et al. Asialoerythropoietin prevents contrast-induced nephropathy. J Am Soc Nephrol. 2008;19:321–8.
Chen YH, Fu YC, Wu MJ. Does Resveratrol Play a Role in Decreasing the Inflammation Associated with Contrast Induced Nephropathy in Rat Model? J Clin Med. 2019;8:147.
Wang Z, Liu Y, Han Y, Guan W, Kou X, Fu J, et al. Protective effects of aliskiren on ischemia–reperfusion-induced renal injury in rats. Eur J Pharmacol. 2013;718:160–6.
Li H, Duann P, Lin PH, Zhao L, Fan Z, Tan T, et al. Modulation of wound healing and scar formation by MG53 protein-mediated cell membrane repair. J Biol Chem. 2015;290:24592–603.
Yao Y, Zhang B, Zhu H, Li H, Han Y, Chen K, et al. MG53 permeates through blood-brain barrier to protect ischemic brain injury. Oncotarget. 2016;7:22474–85.
Weisleder N, Lin P, Zhao X, Orange M, Zhu H, Ma J. Visualization of MG53-mediated cell membrane repair using in vivo and in vitro systems. J Vis Exp. 2011;52:2717.
Weisleder N, Takeshima H, Ma J. Mitsugumin 53 (MG53) facilitates vesicle trafficking in striated muscle to contribute to cell membrane repair. Commun Integr Biol. 2009;2:225–6.
Cai C, Weisleder N, Ko JK, Komazaki S, Sunada Y, Nishi M, et al. Membrane repair defects in muscular dystrophy are linked to altered interaction between MG53, caveolin-3, and dysferlin. J Biol Chem. 2009;284:15894–902.
Zhang Y, Lv F, Jin L, Peng W, Song R, Ma J, et al. MG53 participates in ischaemic postconditioning through the RISK signalling pathway. Cardiovasc Res. 2011;91:108–15.
Wu Y, Huang J, Liu D, Tan J, Peng Y, Yang J, et al. Mitsugumin 53 protects the kidney from severe burn injury in mice. Burns Trauma. 2013;1:128–33.
Michael A, Faga T, Pisani A, Riccio E, Bramanti P, Sabbatini M, et al. Molecular mechanisms of renal cellular nephrotoxicity due to radiocontrast media. Biomed Res Int. 2014;2014:249810.
Liu ZZ, Schmerbach K, Lu Y, Perlewitz A, Nikitina T, Cantow K, et al. Iodinated contrast media cause direct tubular cell damage, leading to oxidative stress, low nitric oxide, and impairment of tubuloglomerular feedback. Am J Physiol Renal Physiol. 2014;306:F864–72.
Zager RA, Johnson ACM, Hanson SY. Radiographic contrast media-induced tubular injury: Evaluation of oxidant stress and plasma membrane integrity. Kidney Int. 2003;64:128–39.
Zhu H, Lin P, De G, Choi KH, Takeshima H, Weisleder N, et al. Polymerase transcriptase release factor (PTRF) anchors MG53 protein to cell injury site for initiation of membrane repair. J Biol Chem. 2011;286:12820–4.
Acknowledgements
These studies were supported in part by grants to CZ from the National Key R&D Program of China (2018YFC1312700), National Natural Science Foundation of China (31730043, 81600585), Program of Innovative Research Team by National Natural Science Foundation of China (81721001), Chongqing Technology Innovation and Application Demonstration Project (cstc2018jscx-mszdX0024), and Clinical Medical Research Personnel Training Program of the Army Medical University (2018XLC10I2). Research conducted in the laboratories of JJM and BHR was supported by the US National Institutes of Health (R01-DK106394).
Author information
Authors and Affiliations
Contributions
CYZ, YH, and CL designed the experiments. CL, YHH, YBW, YZ, XQZ, DFH, HMR, YKL, PHL, and HCL performed the experiments and analyzed the data. CL, YHH, and HYW prepared the original manuscript. TT, BHR, JJM, and CYZ revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
JJM and TT have an equity interest in TRIM-edicine, Inc., which develops rhMG53 for the treatment of human diseases. Patents on the use of MG53 are held by Rutgers University—Robert Wood Johnson Medical School.
Supplementary information
Rights and permissions
About this article

Cite this article
Liu, C., Hu, Yh., Han, Y. et al. MG53 protects against contrast-induced acute kidney injury by reducing cell membrane damage and apoptosis. Acta Pharmacol Sin 41, 1457–1464 (2020). https://doi.org/10.1038/s41401-020-0420-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-020-0420-8
Keywords
This article is cited by
-
Multipronged regulation of autophagy and apoptosis: emerging role of TRIM proteins
Cellular & Molecular Biology Letters (2024)
-
Structural basis for TRIM72 oligomerization during membrane damage repair
Nature Communications (2023)
-
Increased AT1 receptor expression mediates vasoconstriction leading to hypertension in Snx1−/− mice
Hypertension Research (2021)

{kind=link}
{kind=link}
{kind=link}
{kind=link}