
Abstract
Keloid is a benign skin tumor characterized by its cell hyperproliferative activity, invasion into normal skin, uncontrolled growth, overproduction and deposition of extracellular matrices and high recurrence rate after various therapies. Nintedanib is a receptor tyrosine kinase inhibitor targeting VEGF, PDGF, FGF, and TGF-β receptors with proved efficacy in anti-angiogenesis and in treating various types of cancers. In this study, we investigated the effects of nintedanib on keloid fibroblasts in both in vitro and ex vivo models. Keloid fibroblasts were prepared from 54 keloid scar samples in active stages collected from 49 patients. We found that nintedanib (1−4 μM) dose-dependently suppressed cell proliferation, induced G0/G1 cell cycle arrest, and inhibited migration and invasion of keloid fibroblasts. The drug also significantly inhibited the gene and protein expression of collagen I (COL-1) and III (COL-3), fibronectin (FN), and connective growth factor (CTGF), as well as the gene expression of other pathological factors, such as alpha smooth muscle actin (α-SMA), plasminogen activator inhibitor-1 (PAI-1), FK506-binding protein 10 (FKBP10), and heat shock protein 47 (HSP47) in keloid fibroblasts. Furthermore, nintedanib treatment significantly suppressed the phosphorylation of p38, JNK, ERK, STAT3, and Smad, enhanced endocytosis of various growth factor receptors. Using an ex vivo tissue explant model, we showed that nintedanib significantly suppressed cell proliferation, migration, and collagen production. The drug also significantly disrupted microvessel structure ex vivo. In summary, our results demonstrate that nintedanib is likely to become a potential targeted drug for keloid systemic therapy.
Similar content being viewed by others

Introduction
Keloids are characterized by uncontrolled growth beyond the original lesion, hyperproliferative fibroblasts and overproduction and deposition of extracellular matrices (ECM). Thus, keloids are considered as benign skin tumors [1, 2], and are suitable for anticancer strategies [3, 4]. In addition, chemotherapy agents targeting signaling pathways are good drug candidates for cancer therapy.
Previous studies showed the involvement of abnormal signaling pathways, including the transforming growth factor-β (TGF-β)/Smad, phosphatidylinositol 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) and mitogen-activated protein kinase (MAPK)-signaling pathways, in keloid development [5, 6]. Additionally, enhanced cell proliferative and migratory abilities, abnormal angiogenesis, and vascular hyperpermeability have also been found to be enhanced by vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), and fibroblast growth factor (FGF) in keloid development [4, 7]. Recent progress in this field also demonstrated that inhibitors of multiple signaling pathways are the preferred targeted drugs due to their synergistic mechanisms of antagonizing different signaling pathways [8]. Nintedanib (BIBF-1120) is a receptor tyrosine kinase inhibitor (TKI) targeting VEGF receptors (VEGFRs), FGF receptors (FGFRs), and PDGF receptors (PDGFRs) [9] with proven efficacy in antiangiogenesis and in treating various types of cancers, including hepatocellular carcinoma (HCC), renal cell carcinoma (RCC), colorectal cancer (CRC), prostate cancer, and gynecologic malignancies [10]. For example, nintedanib reduced vessel density, vessel permeability, and tumor growth by inhibiting the PI3K-AKT and MAPK-signaling pathways [10]. Nintedanib was also reported to reduce fibrotic progression by inhibiting early events in TGF-β signaling, involving phosphorylation of the type II TGF-β receptor, activation of Smad3, and p38 MAPK phosphorylation [11]. In particular, nintedanib is the first targeted chemotherapy drug for idiopathic pulmonary fibrosis (IPF) therapy in patients, indicating the therapeutic potential of TKIs in the treatment of nonmalignant diseases.
Previous studies have demonstrated that PDGFR, VEGFR, and FGFR are usually localized at low-density, detergent-resistant membrane domains called lipid rafts/caveolae [12, 13]. Lipid rafts/caveolae are small cholesterol-enriched domains present on the cell membrane. When cholesterol is depleted, receptors are retained in the residual elements of the caveolae, and remain able to bind the ligand, but the downstream cascades are blocked due to steric inhibition and the inability to form a signaling complex [12].
It has been shown that the keloid mechanism involves multiple signaling pathways, such as VEGF, PDGF, FGF, and TGF-β, but there has been no report on a drug that could target more than three growth factors among the studies on drugs targeting keloid cell behavior [5, 14, 15]. This study aimed to explore the targeted drug effect of nintedanib on antagonizing multiple signaling pathways related to VEGF-R, FGF-R, PDGF-R, and TGF-β-R and examine its therapeutic effect on keloid fibroblast behaviors, including cell proliferation, cell cycle progression, overproduction of extracellular matrix, cell migration, and cell invasion, using in vitro and ex vivo models. In addition, we also investigated whether disruption of the lipid raft/caveolae structure is involved in the drug effects.
Materials and methods
Patients and keloid samples
Fifty-four keloid scar samples in active stages were collected from 49 patients without any previous treatment before surgery (see details listed in Table 1). Keloid specimens derived from surgical excision were donated by the patients for research purposes only with written informed consent. The procedures for processing human tissues and cells were approved by the Ethics Committee of the Shanghai Jiao Tong University School of Medicine.
Chemical reagents
Nintedanib (Selleck Chemicals, Houston, TX, USA) was dissolved in 100% dimethyl sulfoxide (DMSO) to a stock concentration of 5 mM and stored at −20 °C under light-protected conditions. The reagent was diluted directly to the desired dose upon use, with an identical final concentration of DMSO in both the experimental and control groups. The final concentration of DMSO was maintained below 0.1% (v/v) in all of the following experiments to ensure no cytotoxic effect on the cultured KFs.
Isolation and culture of keloid fibroblasts
Isolation and culture of keloid fibroblasts were performed according to a previous protocol [5]. Keloid tissues were treated with collagenase NB4 (SERVA Electrophoresis, Heidelberg, Germany) dissolved in Dulbecco’s modified Eagle’s medium (DMEM, HyClone, Logan, UT, USA, 0.3% v/v) for 4 h at 37 °C with rotation. After digestion, cells were collected and resuspended in DMEM supplemented with 10% fetal bovine serum (FBS, Gibco-Invitrogen Corp., Grand Island, NY, USA) and penicillin/streptomycin (Gibco) and seeded onto 10-cm culture dishes. The primary, first-passage cells were utilized in the subsequent experiments. To better represent the keloid cases, KFs derived from three patient tissue samples were mixed as a pooled cell sample.
Cell counting kit-8 (CCK-8) assay
The drug effect on KF proliferation was analyzed by CCK-8 (Dojindo Laboratories, Mashiki, Japan). Briefly, 1000 primary cultured KFs were seeded in 96-well plates per well and starved for 24 h. Then, the medium was replaced with culture medium with or without nintedanib, and the cells were tested with CCK-8 reagent at days 1, 3, 5, 7, and 9. The medium optical density value of each well was measured at 450 nm using a microplate reader (Thermo Fisher Scientific, Waltham, MA, USA). The assay was carried out in quadruplicate and repeated with three independent pooled cell samples.
Cell cycle analysis
As previously described [5], KFs treated with nintedanib at different concentrations (0, 0.5, 1, 2, 4 μM) for 5 days were collected. The cells were then fixed in 70% ethanol at 4 °C overnight followed by staining with a cell cycle analysis kit (7sea Biotech, Shanghai, China). Afterwards, a flow cytometer (Beckman Coulter, Fullerton, CA, USA) equipped with ModiFit LT v2.0 software was used for flow cytometric analyses, and the experiment was repeated in four independent pooled cell samples.
EdU incorporation assay
After KFs were treated with different concentrations of nintedanib for 72 h, 5-ethynyl-20-deoxyuridine (EdU) was added to the culture medium and incubated for 3 h. Detection of EdU incorporation was performed with the Click-iT™ EdU Alexa Fluor Imaging kit (Invitrogen/Molecular Probes, Eugene, OR, USA) following the manufacturer’s instructions. The EdU-positive cells (red) and Hoechst-positive cells (blue) were imaged under a fluorescence microscope (Olympus, Tokyo, Japan) and counted using Image-Pro Plus (IPP) 6.0 software (Media Cybernetics, Silver Spring, MD, USA). The incorporation ratio of EdU-positive cells is shown as the percentage of EdU-positive cells of the total Hoechst-positive cells. The results were derived from the average cell counts in five randomly selected fields in a well and repeated with three independent pooled cell samples.
Cell migration assay
For the scratch assay, as previously described [5], the cell monolayer was scratched using a 200-μL pipette tip when the cells reached more than 90% confluence, and the cells were then cultured in serum-free medium with or without nintedanib for 48 h. Images were acquired at 0, 24, and 48 h after scratching. The results were derived from measured areas in six randomly selected fields in a well and repeated with six independent pooled cell samples.
A transwell system (8-μm pore size) was also utilized for the migration assay as reported [5]. In brief, 3 × 104 serum-starved KFs were seeded in the upper compartment of the Boyden chamber in a 24-well plate in serum-free medium containing nintedanib or vehicle control. After incubation for 24 h, the migrated cells were fixed with 4% paraformaldehyde and stained with 4′,6-diamidino-2-phenylindole (DAPI, Sigma, Chemical Co., St. Louis, MO, USA). Five high-power fields were randomly selected, and the migrated cells were counted by Image-Pro Plus. This assay was performed in three independent pooled cell samples.
Furthermore, an OrisTM cell migration assay kit (Platypus Technologies, Madison, WI, USA) was used as reported [14]. Briefly, 2.5 × 104 KFs were seeded in each well, and after 24 h, the medium was replaced with 100 μL serum-free medium containing different concentrations of nintedanib after removing the stoppers. After another 24-h incubation, cells were stained with calcein-AM (Maiyuer Bio, Shanghai, China). The cell number in the migration zone was counted by Image-Pro Plus. This assay was performed with three independent pooled cell samples.
Cell invasion assay
For the transwell assay, 6 × 104 serum-starved KFs were placed in the upper well of a Boyden chamber coated with Matrigel (Corning). After a 48-h incubation, invaded cells were fixed and stained with DAPI. Cell numbers in five randomly selected high-power fields were counted by Image-Pro Plus. The assay was repeated with three independent pooled cell samples.
Furthermore, an OrisTM 3D embedded invasion assay (Platypus Technologies) was performed to confirm the inhibitory effect of the drug on KF invasive ability as previously described [16]. Briefly, 2.5 × 104 KFs per well were seeded, and after 24 h, the medium was replaced by 40 μL rat-tail collagen I (COL-1) solution. After incubation for 30 min, serum-free medium containing nintedanib or vehicle control was added to each well. Images of KFs stained with calcein-AM were obtained at 48 h post treatment. Cells in the detection zone were counted by Image-Pro Plus. This assay was performed with three independent pooled cell samples.
Ex vivo explant culture of human keloid tissues
Keloid tissue after epidermal removal was minced into 3 mm × 2 mm × 2 mm fragments. The dermal fragments were equally treated, seeded onto three 10-cm culture dishes and incubated in DMEM containing 10% FBS until attachment. Then, the medium was replaced with fresh medium containing nintedanib or vehicle control. Representative images were acquired at the same location on days 0, 3, 5, and 7 after the KFs migrated from the edge of the tissue. On day 7, the tissue fragments were collected for RT-qPCR and Western blotting assays, and the KFs growing in the dishes were collected and counted using a hematocytometer.
RNA isolation and real-time quantitative PCR
After treatment with nintedanib or vehicle control for 72 h in DMEM plus 5% FBS, the cultured KFs were harvested for total RNA extraction using TRIzol reagent (Invitrogen Life Technologies Inc., Grand Island, NY, USA) as previously reported [17]. For the ex vivo explants, the tissues were minced with scissors and ground by a Pro-200 tissue homogenizer (Pro Scientific, Monroe, CT, USA) to a homogeneous lysate and prepared for RNA isolation using TRIzol reagent.
Complementary DNA (cDNA) was synthesized from 1 µg total RNA per sample using AMV reverse transcriptase (Promega, Madison, WI, USA). cDNA was amplified using RT-qPCR in a real-time thermal cycler (Stratagene, La Jolla, CA, USA) with Power SYBR Green PCR master mix (2×) (Applied Biosystems, Foster City, CA, USA) as previously described [18]. The housekeeping gene β-actin was used as an internal control. Each assay was performed in triplicate and repeated with three independent pooled cell samples. The human primers for real-time qPCR analysis are displayed in Table 2.
Immunofluorescence assay
Briefly, 1 × 105 KFs per well (for alpha smooth muscle actin, α-SMA detection) or 2 × 105 KFs (for COL-1 detection) were seeded in six-well plates and preincubated for 24 h in regular culture medium. Afterwards, the medium was replaced with DMEM containing 5% FBS with or without nintedanib for 72 h (for α-SMA detection) or 7 days (for COL-1 detection) followed by fixation in 4% paraformaldehyde overnight.
The primary and secondary antibodies (Table 3) were diluted according to the instructions. DAPI was used for nuclear counterstaining. Images of α-SMA-positive or COL-1-positive (green) and DAPI nuclear-stained (blue) cells were obtained under a fluorescence microscope (Olympus). This assay was performed with three independent pooled cell samples.
Immunohistochemical analyses
Keloid specimens with a size of 3 mm × 2 mm × 2 mm were fixed in 4% paraformaldehyde at 4 °C overnight after being cultured in DMEM plus 10% FBS with nintedanib or vehicle control for 7 days. Afterwards, the specimens were embedded in paraffin and sectioned to 5 μm thickness. Following incubation of the sections with antibodies against COL-1, fibronectin (FN), CD31, and CD34 (see details in Table 3), 3,3′-diaminobenzidine (DAB, G1211, Servicebio, Wuhan, China) was used as a chromogen to visualize the bound antibodies, and the slides were counterstained with hematoxylin. CD31+ and CD34+ vessels were counted in five randomly selected fields under a microscope for semiquantification. This assay was performed with four independent samples.
Western blotting analysis
For ex vivo explants, 50 mg/each of treated or nontreated tissues were washed and cut into small pieces in PBS. The tissues were then homogenized with a Pro-200 tissue homogenizer (Pro Scientific) in lysis buffer supplemented with a protease inhibitor cocktail (Thermo Fisher Scientific, Rockford, IL, USA). The lysate was centrifuged at 12,000 × g for 10 min at 4 °C. For protein production in cells, KFs were treated with or without nintedanib (1, 2, 4 μM) for 6 or 72 h to examine cellular signaling molecules or other antigens. Protein extraction and Western blotting analysis were performed as previously described [19]. Detailed information on the antibodies is listed in Table 3. This assay was performed with three independent samples.
Cholesterol assay
To determine whether lipid rafts/caveolae were related to the inhibitory effect of nintedanib on keloid fibroblasts, disruption of lipid rafts/caveolae was carried out by incubating cells in the presence of 1 mM MβCD (Sigma) dissolved in double-distilled water (ddH2O). CCK-8 and scratch assays were used to measure cell proliferation and migration, respectively. Western blotting was also performed to examine the expression of receptors. Cells were treated with vehicle control, MβCD (1 mM) or nintedanib (4 μM) alone or in combination in growth medium. For the combination treatment, the cells were pretreated with 1 mM MβCD for 30 min at 37 °C before the experiment. For the CCK-8 assay, cells were tested on days 1, 3, and 5, and for the scratch assay, images were obtained at 0 and 48 h after scratching. For Western blotting assays, cells were collected 3 days after treatment. Detailed methods are described above.
Statistical analysis
All the data are presented as the mean ± standard deviation (SD) and were statistically analyzed with one-way ANOVA followed by Student–Newman–Keuls (S–N–K) post hoc test after confirming the normal distribution of the data. For ratio data of the EdU assay, nonparametric Kruskal–Wallis test plus Dunn’s post hoc test with Bonferroni correction was employed. All statistical analyses were performed with the statistical software SPSS (version 22.0; SPSS, Inc., Chicago, IL, USA). A P-value < 0.05 was considered statistically significant.
Results
Nintedanib suppressed KF proliferation and induced G0/G1 cell cycle arrest
As shown in Fig. 1a, the CCK-8 assay demonstrated a significant inhibitory effect of nintedanib on KF proliferation in a dose-dependent manner during the 9-day time period compared to the vehicle control group. ANOVA plus the S–N–K test demonstrated significant differences among the groups (Supplementary Fig. S1, P < 0.05).

a The CCK-8 assay was used to analyze the proliferation of KFs treated with different concentrations of nintedanib at days 1, 3, 5, 7, and 9. b Flow cytometry analysis of the cell cycle of treated keloid fibroblasts. c Representative cell cycle data were plotted for each group. d After treatment with nintedanib (1, 2, 4 μM) or vehicle control for 72 h, KFs were incubated with EdU for an additional 3 h. The samples were imaged under a fluorescence microscope at ×200 (bar = 100 μm). e The ratio of EdU-positive cells to DAPI-labeled cells in each group was determined. *P < 0.05 vs control, #P < 0.05 vs 0.5 μM, $P < 0.05 vs 1 μM. Abbreviations: G0/G1, period between the end of M phase and the start of S phase; S, DNA duplication phase; G2, period between the end of S phase and the start of M phase; M, mitosis.
Next, cell cycle analysis revealed a dose-dependent increase in the cell percentage in G0/G1 phase, whereas a dose-dependent decrease in the cell percentage was found in S phase with significant differences among the groups treated with 1, 2 and 4 μM nintedanib (P < 0.05, Fig. 1b, c), indicating that nintedanib induced cell cycle arrest at G0/G1 phase. Additionally, an EdU-incorporation assay also showed significant inhibition of DNA synthesis in KFs by nintedanib treatment because much less EdU labeling was observed in the treated cells than in the control cells (Fig. 1d). Semiquantitative analysis showed a reduction in EdU labeling among the groups (P < 0.05, Fig. 1e).
Nintedanib attenuated the migratory capacity of cultured KFs
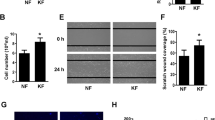
After 24 and 48 h of nintedanib treatment, the scratch assay demonstrated a significant inhibitory effect on KF migration compared to that in nontreated cells when observed under a microscope (Fig. 2a). At both time points, quantitative analysis further confirmed a significant dose-dependent reduction in cell migration in the treated groups (P < 0.05, Fig. 2b).

a Confluent monolayers of cells were wounded with a scratch and incubated in the presence or absence of nintedanib (0.5, 1, 2, 4 μM) as indicated for 24 and 48 h. Images indicate a relative wound closure as monitored by visual examination using a Nikon phase-contrast microscope at ×40 (bar = 200 μm). b Semiquantitative analysis of the scratch assay results with the indicated significant differences. c Cells that penetrated through the micropore membrane to the lower surface of the filter were stained with DAPI and observed under a light microscope at ×200 (bar = 100 μm). d Semiquantitative analysis of cells in the lower chambers. e A COL-1-coated Oris migration assay was used to show the effect of nintedanib on keloid fibroblast migration (×40, bar = 200 μm). f Semiquantitative analysis of the Oris migration assay results with the indicated significant differences. *P < 0.05 vs control, #P < 0.05 vs 0.5 μM, $P < 0.05 vs 1 μM, &P < 0.05 vs 2 μM.
Similarly, the transwell assay also revealed the inhibitory effect of nintedanib on cell migration by reducing the number of cells that migrated to the bottom surface of the upper chamber in the treated group at 24 h post treatment (Fig. 2c). Semiquantitative analysis showed a dose-dependent reduction in the migrated cell numbers with significant differences among the groups, as revealed by ANOVA plus the S–N–K test (P < 0.05, Fig. 2d). Furthermore, the Oris migration assay also confirmed the inhibitory effect of nintedanib on cell migration by both microscopic observation (Fig. 2e) and quantitative analysis (Fig. 2f), which revealed a dose-dependent reduction in migrated cell numbers with significant differences among the groups after 24 h of nintedanib treatment (P < 0.05).
Nintedanib inhibited the invasive capacity of KFs
Both the transwell assay and Oris invasion assay combined with matrix coating were also performed to investigate the drug effect. As shown in Fig. 3a, b, much fewer cells were able to successfully traverse the Matrigel-coated chamber after nintedanib treatment compared to the control group at 24 h post treatment, and the effect was dose-dependent.

a Cells that penetrated through the Matrigel to the lower surface of the filter were stained with DAPI and observed under a light microscope at ×200 (bar = 100 μm). b Semiquantification of cells in the lower chambers. c Keloid fibroblasts crawled into the 2-mm invasion zone in an in vitro three-dimensional basement membrane extract model (×40, bar = 200 μm). d Semiquantitative analysis of the cell numbers inside the invasion zone. e Representative images of tissue explants cultured with or without different concentrations of nintedanib at days 0, 3, 5, and 7 (×40, bar = 200 μm). f Semiquantification of the cell numbers that migrated out of the tissue explants at day 7. g and h qPCR analysis revealed decreased gene expression of MMP1 and MMP3 with nintedanib treatment. *P < 0.05 vs control, $P < 0.05 vs 1 μM, &P < 0.05 vs 2 μM.
A similar trend was also confirmed in the Oris invasion assay, which used COL-1 coating as the matrix material. As shown in Fig. 3c (microscopic view) and Fig. 3d (quantitative analysis), a dose-dependent decrease in cell numbers in the detection zone was found with increasing drug concentrations (P < 0.05).
Nintedanib suppressed the migration and proliferation of cultured keloid explants
To further verify the inhibitory effect of nintedanib on cell migration and proliferation, an ex vivo keloid explant culture model was also established. As shown in Fig. 3e, f, spindle-shaped KFs of the vehicle group migrated out of the edges of the cultured keloid explants and gradually spread over the Petri dish within a week with a total cell number of 1.84 × 105 after 7 days of culture. However, treatment with increasing concentrations of nintedanib for 7 days significantly inhibited KFs from migrating out of the tissue explants in a dose-dependent manner with significant differences from that of the control group (P < 0.05).
Interestingly, the inhibitory effect of the drug was also supported by decreased gene expression of matrix metalloproteinases (MMPs). As shown in Fig. 3g, h, MMP1 and MMP3 gene levels were significantly reduced in the drug-treated groups, as demonstrated by RT-qPCR analysis (P < 0.05).
Nintedanib suppressed the expression of fibrotic factors related to keloid pathogenesis
Using an in vitro cell culture model, the drug effects on the expression of fibrotic factors at both the gene and protein levels were investigated. As presented in Fig. 4a, at the transcriptional level, treatment with the drug at concentrations of 1, 2, and 4 μM for 72 h significantly suppressed the gene expression of COL-1, FN, connective tissue growth factor (CTGF), plasminogen activator inhibitor-1 (PAI-1), and FK506-binding protein 10 (FKBP10) in a dose-dependent manner, with significant differences among the groups (P < 0.05). The suppression of α-SMA and heat shock protein 47 (HSP47) occurred at higher concentrations, and no significant effect was found on COL-3.

a qPCR analysis of COL-1, COL-3, FN, CTGF, α-SMA, PAI-1, FKBP10, and HSP47 expression in keloid fibroblasts in the presence or absence of nintedanib (1, 2, 4 μM) after 72 h of treatment. b Western blotting analysis of COL-1, FN, and CTGF production at 72 h post treatment. c Semiquantification of the Western blotting results. d Immunofluorescence analysis of α‑SMA expression at 72 h post treatment (×200, bar = 100 μm). e Fluorescence microscopy image of intracellular COL-1 expression at 72 h post treatment (×40, bar = 200 μm). *P < 0.05 vs control, $P < 0.05 vs 1 μM, &P < 0.05 vs 2 μM.
Consistent with the gene expression results, Western blotting also showed significant suppression of the protein production of COL-1, FN, and CTGF, as shown in Fig. 4b, and semiquantitative analysis revealed significant differences among the different groups (Fig. 4c). The suppressed protein production of α-SMA and COL-1 was also confirmed by immunofluorescence staining of KFs treated with different concentrations of nintedanib (Fig. 4d, e), which showed decreasing protein expression with increasing drug concentrations. In addition, the drug treatment also disrupted the intracellular collagen pattern (Fig. 4e).
Nintedanib inhibited collagen production and disrupted angiogenesis in keloid tissue explants
To better mimic the clinical scenario, an ex vivo model of tissue explants was generated for the investigation. Immunohistochemical staining of the keloid tissue explants exposed to nintedanib showed a marked decrease in the intensity of COL-1 staining and a reduced FN staining area, which indicated significantly decreased production of COL-1 and FN (Fig. 5a). Consistent with these findings, qPCR and Western blotting analyses of tissue explants also showed a reduction in COL-1 and FN expression at both the transcriptional and translational levels (Fig. 5b–d). Furthermore, a reduced number of CD31-positive and CD34-positive microvessels as well as disrupted capillary structure were also observed with nintedanib treatment (Fig. 5a, e, f).

a Immunohistochemical analysis of COL-1, FN, CD31, and CD34 expression in cultured keloid tissue explants (×200, bar = 100 μm). b and c qPCR analysis of COL-1 and FN expression in keloid tissue explants in the presence or absence of nintedanib (2, 4 μM) treatment at day 7. d Western blot analysis of COL-1 and FN expression in keloid tissue explants in the presence or absence of nintedanib (2, 4 μM) treatment at day 7. e and f Semiquantitative analysis of CD31+ and CD34+ microvessel numbers in immunohistochemically stained tissue sections. *P < 0.05 vs control.
Nintedanib attenuated various intracellular signaling pathways in treated KFs
To further define the potential mechanism of the above findings, the effect of nintedanib on various signaling pathways was investigated using an in vitro cell culture model. As shown in Fig. 6, treatment of KFs with nintedanib significantly inhibited the phosphorylation of Smad2 and Smad3 at higher concentrations. It also significantly inhibited the phosphorylation of MAPK pathway components including p38, JNK, and ERK, as well as signal transducer and activator of transcription 3 (STAT3), in a dose-dependent manner (Fig. 6a).

a Western blotting showed decreased phosphorylation of various signaling molecules at 6 h post-treatment in groups which were treated with different drug concentrations (1, 2, 4 μM). b and c CCK-8 and scratch assays, respectively, showed partial rescue by MβCD of the inhibitory effect of nintedanib on cell proliferation and migration. d Semiquantitative data analysis of the scratch assay results. e Western blotting showed partial rescue by MβCD of the inhibitory effect nintedanib on internalization of multiple growth factor receptors after 72 h of drug treatment. *P < 0.05 vs control, #P < 0.05 vs M, $P < 0.05 vs N. Abbreviations: M, MβCD (1 mM) group; N, nintedanib (4 μM) group; M + N, cotreatment with MβCD (1 mM) and nintedanib (4 μM) group.
Disruption of lipid rafts decreased the inhibitory effect of nintedanib on KFs
Cholesterol is the major structural component of lipid rafts/caveolae; thus, cholesterol depletion was performed to determine whether lipid rafts/caveolae mediate the cellular response to nintedanib. Among various approaches, MβCD is the most common method for removing cellular cholesterol [12].
CCK-8 and scratch assays were used to measure proliferation and migration. The results showed that MβCD treatment alone could have a minor inhibitory effect on cell proliferation and migration, which was significantly different from the control treatment. Nevertheless, MβCD was able to rescue to a certain degree the inhibitory effect of nintedanib on KF proliferation and migration when compared to treatment with nintedanib alone (Fig. 6b–d). Interestingly, Western blotting assays demonstrated increased protein expression of growth factor receptors in the MβCD and nintedanib combined treatment group relative to the expression levels of nintedanib only treatment group (Fig. 6e), providing supporting evidence that nintedanib may affect the functions of growth factor receptors by enhancing their endocytosis, which can be disrupted by removing cellular cholesterol in the lipid raft domain.
Discussion
Keloids are benign skin tumors in which keloid fibroblasts display tumor cell-like biological behaviors [1, 4]. Keloids inevitably recur after various treatments, and thus, new therapeutic approaches are needed. Targeted therapy is considered an effective strategy for cancer treatment and includes various targets for drug intervention, such as rhodopsin-like G protein-coupled receptors (GPCRs), ion channels, protein kinases, and nuclear hormone receptors, with kinase inhibitors being an important part of targeted drugs for cancer therapy [20, 21].
Keloid formation is driven by multiple growth factors, including TGF-β, PDGF, and VEGF. TGF-β promotes keloid fibroblast proliferation, collagen production, inhibition of MMPs [22, 23], and the Smad-signaling pathway [24]. PDGF and VEGF also contribute to keloid pathogenesis by stimulating collagen production and angiogenesis [4]. Additionally, PDGF contributes to the chemotaxis of fibroblasts, cell proliferation, migration, and collagen synthesis during wound healing [25].
Studies also demonstrated elevated PDGF receptor expression in keloid fibroblasts, which responded to PDGF more potently than normal dermal fibroblasts [7].
Elevated VEGF in keloid tissue and circulation of keloid patients [26] could lead to imbalanced angiogenesis and contribute to keloid formation [26, 27]. Moreover, bFGF is also indicated to be involved in keloid angiogenesis [4]. Preclinical data also demonstrated that the inhibition of FGFR signaling could suppress the proliferation of cells derived from many tissue types [28,29,30]. This collected evidence provides an excellent strategy for targeted drug therapy of keloids.
There have been several studies on the targeted therapy of keloids. Palomid 529 (P529), which inhibits the PI3K/Akt/mTOR pathway by targeting both mTOR complex 1 (mTORC-1) and mTORC-2 signaling, caused keloid tissue shrinkage, growth arrest, and apoptosis [6]. Sorafenib, a potent TKI targeting VEGFR2 and PDGFRβ, has been shown to effectively inhibit keloid cell proliferation, migration, invasion, and collagen production [5]. Antisense therapy specific to TGF-β mRNA transcripts demonstrated downregulation of MMP-9 in keloid fibroblasts [15]. However, these studies focused on only one or two targets. We thus hypothesized that the use of a chemical compound that targets multiple signaling pathways would be a more efficient approach to keloid-targeted therapy.
Nintedanib was reported as an angiogenesis inhibitor of PDGFR-α and β, VEGFR-1, 2, and 3 and FGFR-1, 2, and 3 and was proven effective in the therapy of many types of cancers [10]. Moreover, the antifibrotic effects of nintedanib have also been utilized in treating several fibrosis-related disorders, such as IPF and systemic sclerosis [9, 31]. Recent studies also showed that nintedanib likely targets the TGF-β-signaling pathway [11].
As TGF-β, VEGF, PDGF, and bFGF are involved in keloid mechanisms [4, 26], and nintedanib is a good candidate for targeted drug therapy for keloids, it would be interesting to explore the roles of nintedanib in inhibiting various pathological activities of keloid fibroblasts as preclinical experimental evidence for future clinical studies.
This study demonstrated that nintedanib could inhibit various pathological phenotypes, including inhibition of cell proliferation, cell cycle arrest at G0/G1 phase in vitro (Fig. 1) and inhibition of KF migration and invasion in vitro and ex vivo (Figs. 2 and 3). In addition, it also significantly inhibited the gene expression of COL-1, FN, α-SMA, and their protein production in vitro (Fig. 4). The reduced protein production was also confirmed in an ex vivo model (Fig. 5). In addition, matrix degradation is likely to be enhanced by drug treatment, as the gene expression of PAI-1, a serine protease inhibitor that is often highly expressed in keloid fibroblasts [32], was significantly inhibited (Fig. 4). In addition, nintedanib also significantly inhibited the gene expression of FKBP10 and HSP47, both of which are highly expressed in fibrotic tissues [33, 34]. Moreover, this study showed that MMP-1 and MMP-3 gene expression was inhibited by the drug, which may also partially account for the inhibited cell migration in cultured tissue explants (Fig. 3), in which matrix degradation was also required for cell migration out of the tissue explants.
As these pathological aspects of keloids are mediated by growth factors, including TGF-β, PDGF, VEGF, and bFGF, drug treatments are likely to inhibit the functions of these factors. For example, CTGF, a growth factor downstream of TGF-β [35, 36], was found to be downregulated at both the gene and protein levels, which may account for the inhibition of cell proliferation and matrix production.
As further evidence of the inhibition of multiple signaling pathways, the results of Fig. 6 demonstrated that nintedanib noticeably inhibited the phosphorylation of Smad2 and Smad3 at higher concentrations and the MAPK pathway dose-dependently, including the pathway components p38, ERK, and JNK. A previous study showed that the MAPK pathway is involved in TGF-β transcription, in which p38 participates in the phosphorylation of Smad2/3 in KFs, while ERK and JNK promote Smad2/3/4 complex translocation and then regulate the expression of related genes, such as PAI-1 [37, 38].
Consistent with the elevated Smad2 and Smad3 phosphorylation levels in keloid fibroblasts which were related to cell proliferation, matrix synthesis, and MMP regulation in keloids [5, 39], downregulation of Smad2 and Smad3 by RNA interference resulted in a significant decrease in procollagen expression in keloids [40, 41]. Thus, the observed inhibition of cell migration and invasion as well as reduced matrix production is likely mediated by drug inhibition of Smad-related pathways. Moreover, increase in STAT3 activation was also found in KFs and may contribute to keloid formation by promoting cell proliferation, migration, and collagen production [42], while STAT3 phosphorylation was inhibited by the drug (Fig. 6a).
In addition, VEGF, PDGF, and bFGF are also involved in keloid pathogenesis by enhancing angiogenesis, cell proliferation, and migration through the p38, PI3K/Akt, and MAPK pathways. The ex vivo model study demonstrated that nintedanib significantly inhibited the expression of CD31 and CD34 and disrupted microvessel formation (Fig. 5), as previously reported [43]. Overall, this study demonstrated that nintedanib could exert its therapeutic effects on various aspects of keloid pathogenesis likely by attenuating various signaling pathways and thus could serve as a blocker of multiple signaling pathways.
According to the literature, most protein kinase antagonists are competitive inhibitors that interact with the ATP-binding pocket to interfere with receptor dimerization and block the phosphorylation of kinases and downstream signaling cascades [10, 21]. However, it has also been reported recently that certain drugs indirectly block signaling pathways by promoting receptor internalization and degradation [44]. Most of the protein kinases are localized in regions of cholesterol-rich lipid rafts/caveolae on cell membranes [12]. Studies have shown that MβCD can block receptor internalization by removing cholesterol [12]. Our results also demonstrated that MβCD was able to rescue nintedanib-mediated receptor internalization to a certain degree, suggesting a dual mechanism for nintedanib-mediated inhibition of signaling pathways, i.e., it blocked the kinase ATP-binding sites as the main mechanism, while it also promoted kinase internalization.
The drawback of this drug is that it also inhibited the proliferation of normal dermal fibroblasts (data not shown) and likely affected other physiological activities. A previous study on pulmonary fibrosis also showed that nintedanib had similar inhibitory effects on both normal pulmonary fibroblasts and fibroblasts in IPF [45] in terms of cell proliferation, α-SMA expression, and myofibroblast appearance. This evidence suggests that nintedanib is less likely to be keloid cell specific and that certain toxic effects on normal cells are possible. Therefore, localized use of this drug for keloid treatment would be an ideal method.
In summary, this study demonstrates that inhibition of the TGF-β/Smad and MAPK-signaling pathways by nintedanib can effectively inhibit the proliferation, migration, invasion, and ECM deposition of KFs by blocking kinase ATP binding and by enhancing kinase internalization. Although further exploration of the detailed mechanism is still needed, the results revealed by the current study support the conclusion that nintedanib is likely to become a promising drug candidate for keloid-targeted therapy, which deserves clinical study in the future.
References
Vincent AS, Phan TT, Mukhopadhyay A, Lim HY, Halliwell B, Wong KP. Human skin keloid fibroblasts display bioenergetics of cancer cells. J Invest Dermatol. 2008;128:702–9.
Yoshimoto H, Ishihara H, Ohtsuru A, Akino K, Murakami R, Kuroda H, et al. Overexpression of insulin-like growth factor-1 (IGF-I) receptor and the invasiveness of cultured keloid fibroblasts. Am J Pathol. 1999;154:883–9.
Slemp AE, Kirschner RE. Keloids and scars: a review of keloids and scars, their pathogenesis, risk factors, and management. Curr Opin Pediatr. 2006;18:396–402.
Andrews JP, Marttala J, Macarak E, Rosenbloom J, Uitto J. Keloids: the paradigm of skin fibrosis-pathomechanisms and treatment. Matrix Biol 2016;51:37–46.
Wang W, Qu M, Xu L, Wu X, Gao Z, Gu T, et al. Sorafenib exerts an anti-keloid activity by antagonizing TGF-β/Smad and MAPK/ERK signaling pathways. J Mol Med. 2016;94:1181–94.
Syed F, Sherris D, Paus R, Varmeh S, Pandolfi PP, Bayat A. Keloid disease can be inhibited by antagonizing excessive mTOR signaling with a novel dual TORC1/2 inhibitor. Am J Pathol. 2012;181:1642–58.
Haisa M, Okochi H, Grotendorst GR. Elevated levels of PDGF alpha receptors in keloid fibroblasts contribute to an enhanced response to PDGF. J Invest Dermatol. 1994;103:560–3.
Csermely P, Agoston V, Pongor S. The efficiency of multi-target drugs: the network approach might help drug design. Trends Pharmacol Sci. 2005;26:178–82.
Roth GJ, Binder R, Colbatzky F, Dallinger C, Schlenker-Herceg R, Hilberg F, et al. Nintedanib: from discovery to the clinic. J Med Chem. 2015;58:1053–63.
Awasthi N, Schwarz R. Profile of nintedanib in the treatment of solid tumors: the evidence to date. Onco Targets Ther. 2015;8:3691.
Rangarajan S, Kurundkar A, Kurundkar D, Bernard K, Sanders YY, Ding Q, et al. Novel mechanisms for the antifibrotic action of nintedanib. Am J Respir Cell Mol Biol. 2016;54:51–9.
Pike LJ. Growth factor receptors, lipid rafts and caveolae: an evolving story. Biochim Biophys Acta. 2005;1746:260–73.
Mukherjee S, Tessema M, Wandinger-Ness A. Vesicular trafficking of tyrosine kinase receptors and associated proteins in the regulation of signaling and vascular function. Circ Res. 2006;98:743–56.
Syed F, Sanganee HJ, Bahl A, Bayat A. Potent dual inhibitors of TORC1 and TORC2 complexes (KU-0063794 and KU-0068650) demonstrate in vitro and ex vivo anti-keloid scar activity. J Invest Dermatol. 2013;133:1340–50.
Sadick H, Herberger A, Riedel K, Bran G, Goessler U, Hoermann K, et al. TGF-beta1 antisense therapy modulates expression of matrix metalloproteinases in keloid-derived fibroblasts. Int J Mol Med. 2008;22:55–60.
He S, Yang Y, Liu X, Huang W, Yang S, Zhang X. Compound astragalus and salvia miltiorrhiza extract inhibits cell proliferation, invasion and collagen synthesis in keloid fibroblasts by mediating transforming growth factor-β / Smad pathway. Br J Dermatol. 2012;166:564–74.
Chen B, Wang B, Zhang WJ, Zhou G, Cao Y, Liu W. In vivo tendon engineering with skeletal muscle derived cells in a mouse model. Biomaterials. 2012;33:6086–97.
Jiang Y, Shi Y, He J, Zhang Z, Zhou G, Zhang W, et al. Enhanced tenogenic differentiation and tendon-like tissue formation by tenomodulin overexpression in murine mesenchymal stem cells. J Tissue Eng Regen Med. 2016;11:2525–36.
Chen YL, Zhang X, Bai J, Gai L, Ye XL, Zhang L, et al. Sorafenib ameliorates bleomycin-induced pulmonary fibrosis: potential roles in the inhibition of epithelial-mesenchymal transition and fibroblast activation. Cell Death Dis. 2013;4:e665–11.
Santos R, Ursu O, Gaulton A, Bento AP, Donadi RS, Bologa CG, et al. A comprehensive map of molecular drug targets. Nat Rev Drug Discov. 2017;16:19.
Roskoski R. Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol Res. 2016;103:26–48.
Bettinger DA, Yager DR, Diegelmann RF, Cohen IK. The effect of TGF-beta on keloid fibroblast proliferation and collagen synthesis. Plast Reconstr Surg. 1996;98:827–33.
Pakyari M, Farrokhi A, Maharlooei MK, Ghahary A. Critical role of transforming growth factor beta in different phases of wound healing. Adv Wound Care. 2013;2:215–24.
Cutroneo KR. TGF-beta-induced fibrosis and SMAD signaling: oligo decoys as natural therapeutics for inhibition of tissue fibrosis and scarring. Wound Repair Regen. 2007;15:S54–60.
Niessen FB, Spauwen PH, Schalkwijk J, Kon M. On the nature of hypertrophic scars and keloids: a review. Plast Reconstr Surg. 1999;104:1435–58.
Mogili NS, Krishnaswamy VR, Jayaraman M, Rajaram R, Venkatraman A, Korrapati PS. Altered angiogenic balance in keloids: a key to therapeutic intervention. Transl Res. 2012;159:182–9.
Fujiwara M, Muragaki Y, Ooshima A. Upregulation of transforming growth factor-β1 and vascular endothelial growth factor in cultured keloid fibroblasts: relevance to angiogenic activity. Arch Dermatol Res. 2005;297:161–9.
Brooks AN, Kilgour E, Smith PD. Molecular pathways: fibroblast growth factor signaling: a new therapeutic opportunity in cancer. Clin Cancer Res. 2012;18:1855–62.
Presta M, Chiodelli P, Giacomini A, Rusnati M, Ronca R. Fibroblast growth factors (FGFs) in cancer: FGF traps as a new therapeutic approach. Pharmacol Ther. 2017;179:171–87.
Chaudhary NI, Roth GJ, Hilberg F, Muller-Quernheim J, Prasse A, Zissel G, et al. Inhibition of PDGF, VEGF and FGF signalling attenuates fibrosis. Eur Respir J. 2007;29:976–85.
Huang J, Beyer C, Palumbo-Zerr K, Zhang Y, Ramming A, Distler A, et al. Nintedanib inhibits fibroblast activation and ameliorates fibrosis in preclinical models of systemic sclerosis. Ann Rheum Dis. 2016;75:883–90.
Ghosh AK, Vaughan DE. PAI-1 in tissue fibrosis. J Cell Physiol. 2011;227:493–507.
Staab-Weijnitz CA, Fernandez IE, Knüppel L, Maul J, Heinzelmann K, Juan-Guardela BM, et al. FK506-binding protein 10, a potential novel drug target for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2015;192:455–67.
Chen JJ, Zhao S, Cen Y, Liu XX, Yu R, Wu DM. Effect of heat shock protein 47 on collagen accumulation in keloid fibroblast cells. Br J Dermatol. 2007;156:1188–95.
Ihn H. Pathogenesis of fibrosis: role of TGF-beta and CTGF. Curr Opin Rheumatol. 2002;14:681–5.
Luo L, Li J, Liu H, Jian X, Zou Q, Zhao Q, et al. Adiponectin is involved in connective tissue growth factor-induced proliferation, migration and overproduction of the extracellular matrix in keloid fibroblasts. Int J Mol Sci. 2017;18:1044–21.
Xia W, Longaker MT, Yang GP. p38 MAP kinase mediates transforming growth factor-β2 transcription in human keloid fibroblasts. Am J Physiol Regul Integr Comp Physiol. 2006;290:R501–8.
He S, Liu X, Yang Y, Huang W, Xu S, Yang S, et al. Mechanisms of transforming growth factor β1/Smad signaling mediated by mitogen-activated protein kinase pathways in keloid fibroblasts. Br J Dermatol. 2010;162:538–46.
Song R, Li G, Li S. Aspidin PB, a novel natural anti-fibrotic compound, inhibited fibrogenesis in TGF-β1-stimulated keloid fibroblasts via PI-3K/Akt and Smad signaling pathways. Chem Biol Interact. 2015;238:66–73.
Wang Z, Gao Z, Shi Y, Sun Y, Lin Z, Jiang H, et al. Inhibition of Smad3 expression decreases collagen synthesis in keloid disease fibroblasts. J Plast Reconstr Aesthet Surg. 2007;60:1193–9.
Gao Z, Wang Z, Shi Y, Lin Z, Jiang H, Hou T, et al. Modulation of collagen synthesis in keloid fibroblasts by silencing Smad2 with siRNA. Plast Reconstr Surg. 2006;118:1328–37.
Lim CP, Phan TT, Lim IJ, Cao X. Stat3 contributes to keloid pathogenesis via promoting collagen production, cell proliferation and migration. Oncogene 2006;25:5416–25.
Wollin L, Pautsch A, Schnapp G, Hostettler K, Stowasser S, Kolb M. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur Respir J. 2015;45:1434–45.
Chung CL, Wang SW, Sun WC, Shu CW, Kao YC, Shiao MS, et al. Sorafenib suppresses TGF-β responses by inducing caveolae/lipid raft-mediated internalization/degradation of cell-surface type II TGF-β receptors_ Implications in development of effective adjunctive therapy for hepatocellular carcinoma. Biochem Pharmacol. 2018;154:39–53.
Lehtonen ST, Veijola A, Karvonen H, Lappi-Blanco E, Sormunen R, Korpela S, et al. Pirfenidone and nintedanib modulate properties of fibroblasts and myofibroblasts in idiopathic pulmonary fibrosis. Respir Res. 2016;17:14.
Acknowledgements
This study was financially supported by the National Natural Science Foundation of China (No. 81671921). The authors also thank Prof. Jung Huang from the Biochemistry Department of St. Louis University School of Medicine (USA) for his valuable suggestion on part of the experimental design and valuable discussion on related scientific issues.
Author information
Authors and Affiliations
Contributions
BYZ and WBW performed research, analyzed the data and drafted the manuscript; XLW, WJZ and GDZ provided valuable insight into the experimental design, data review and critical advice on manuscript preparation; WL and ZG designed research, reviewed the experimental data; WL edited and finalized the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Zhou, By., Wang, Wb., Wu, Xl. et al. Nintedanib inhibits keloid fibroblast functions by blocking the phosphorylation of multiple kinases and enhancing receptor internalization. Acta Pharmacol Sin 41, 1234–1245 (2020). https://doi.org/10.1038/s41401-020-0381-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-020-0381-y
Keywords
This article is cited by
-
Regulatory T cells in skin regeneration and wound healing
Military Medical Research (2023)

{kind=link}