
Abstract
The α7 nicotinic acetylcholine receptor (α7 nAChR) is a ligand-gated Ca2+-permeable homopentameric ion channel implicated in cognition and neuropsychiatric disorders. Pharmacological enhancement of α7 nAChR function has been suggested for improvement of cognitive deficits. In the present study, we characterized a thiazolyl heterocyclic derivative, 6-(2-chloro-6-methylphenyl)-2-((3-fluoro-4-methylphenyl)amino)thiazolo[4,5-d]pyrimidin-7(6H)-one (JWX-A0108), as a novel type I α7 nAChR positive allosteric modulator (PAM), and evaluated its ability to reverse auditory gating and spatial working memory deficits in mice. In Xenopus oocytes expressing human nAChR channels, application of JWX-A0108 selectively enhanced α7 nAChR-mediated inward current in the presence of the agonist ACh (EC50 value = 4.35 ± 0.12 µM). In hippocampal slices, co-application of ACh and JWX-A0108 (10 µM for each) markedly increased both the frequency and amplitude of spontaneous inhibitory postsynaptic currents (sIPSCs) recorded in pyramidal neurons, but JWX-A0108 did not affect GABA-induced current in oocytes expressing human GABAA receptor α1β3γ2 and α5β3γ2 subtypes. In mice with MK-801-induced deficits in auditory gating, administration of JWX-A0108 (1, 3, and 10 mg/kg, i.p.) dose-dependently attenuates MK-801-induced auditory gating deficits in five prepulse intensities (72, 76, 80, 84, and 88 dB). Furthermore, administration of JWX-A0108 (0.03, 0.1, or 0.3 mg/kg, i.p.) significantly reversed MK-801-induced impaired spatial working memory in mice. Our results demonstrate that JWX-A0108 is a novel type I PAM of α7 nAChR, which may be beneficial for improvement of cognitive deficits commonly found in neuropsychiatric disorders such as schizophrenia and Alzheimer’s disease.
Similar content being viewed by others


Introduction
Schizophrenia is a chronic and debilitating mental illness characterized by abnormal social behavior and cognitive deficits, which presents severe health and social problems worldwide [1]. The symptoms of schizophrenia are divided into three categories: positive symptoms (hallucinations and delusions), negative symptoms (anhedonia and social withdrawal), and cognitive impairments (executive function and working memory) [2]. Among them, cognitive deficits are the core feature of schizophrenia [3,4,5]. In addition, auditory gating deficits are ubiquitous in people with schizophrenia, and these deficits are associated with attentional impairment and can lead to cognitive dysfunction [6, 7]. However, clinical therapy for the treatment of cognitive impairment in schizophrenia or Alzheimer’s disease remains extremely unsatisfactory and manifests an urgent unmet medical need.
Nicotinic acetylcholine receptors (nAChRs) are ligand-gated ion channels that are activated by the neurotransmitter acetylcholine for signaling, and they also respond to drugs including the nicotinic receptor agonist nicotine. The homopentameric α7 nAChR is a major neuronal subtype of the family of nAChRs in the central nervous system (CNS) with limited peripheral expression, thus leading to reductions in potential side effects. In the brain, α7 nAChRs are abundantly and predominantly expressed in the regions such as the hippocampus and frontal cortex critical for cognition and memory [8,9,10]. In neurons, α7 nAChRs are also physiologically more important due to their presynaptic localizations, although they are both pre- and postsynaptically localized [11, 12]. Therefore, the normal function of α7 nAChR is critical for cognition, sensory processing, and memory, indicating that α7 nAChR is a potential therapeutic target for cognitive impairment [13].
The genetic defect of schizophrenia is linked to the chromosome 15q13–14 site where the CHRNA7 gene encoding α7 nAChR is located, supporting the hypothesis that the α7 gene may be responsible for inheritance of some of the pathophysiological aspects of the illness [14]. α7 nAChR has been shown to be involved in regulating auditory stimuli and memory function, and disruption of α7 nAChR can cause memory impairment [15, 16]. In an α7-deficient model, mice exhibit impaired attention, which is central to the cognitive deficit [17]. In accordance with these findings, the level of α7 nAChR is markedly reduced in postmortem brains of schizophrenic patients [18]. In contrast, enhancement of α7 nAChR activity normalizes the cognitive deficits and sensory gating deficit [19]. All of these investigations support the hypothesis that selective activation of α7 nAChR may lead to therapeutic benefits for the improvement of the cognitive deficits that are a common feature of neuropsychiatric disorders such as schizophrenia and Alzheimer’s disease.
To date, a number of distinct α7 agonists have been identified in vitro and in vivo [8, 13]. DMXB-A (also named GTS-21), a partial agonist of α7 nAChR, was discovered as a derivative of marine anabaseine, displaying memory-enhancing effects [20] and improvements in auditory gating deficits [21,22,23]. In phase I clinical trials, DMXB-A significantly improved P50 auditory inhibition in schizophrenic patients [24]. However, in phase II clinical studies, DMXB-A failed to achieve the end point for improvement of cognition in schizophrenia [25], suggesting that a strategy for direct activation of α7 nAChR by agonists may need further validation.
Positive allosteric modulators (PAMs) of α7 nAChRs have been proposed to be an attractive therapeutic strategy to treat cognitive disorders. Unlike agonists, PAMs are active only in the presence of endogenous ligands or exogenous agonists. Based on their macroscopic effects on channel gating kinetics, α7 nAChR PAMs are divided into type I and type II [5, 13, 26]. PNU-120596, the first identified selective type II PAM, not only greatly enhances ACh-evoked currents but also improves the auditory gating deficit caused by amphetamine [27]. Although PNU-120596 augments the effects of acetylcholinesterase inhibitors on learning and memory in rodents and nonhuman primates, it has not been beneficial in clinical trials due to its cytotoxicity [28, 29].
As one of the typical representative type I PAMs, AVL-3288 (also named XY4083 or CCMI) has been shown to reverse schizophrenia-like behavior induced by ketamine in rats [30, 31]. AVL-3288 has also been tested to be safe for administration in humans for potential positive neurocognitive effects in CNS disorders [26]. These investigations suggest that α7 type I PAM has potential positive neurocognitive effects in CNS disorders.
In the present study, we evaluated a novel, selective α7 type I PAM, 6-(2-chloro-6-methylphenyl)-2-((3-fluoro-4-methylphenyl)amino)thiazolo[4,5-d]pyrimidin-7(6H)-one (JWX-A0108) for reversal of mouse auditory gating and spatial working memory deficits caused by the N-methyl-d-aspartic acid (NMDA) receptor antagonist MK-801. JWX-A0108 selectively enhances activation of α7 nAChR in both heteroexpression systems and native neurons and facilitates synaptic transmission recorded from hippocampal brain slices. Our findings demonstrate that JWX-A0108 serves not only as a pharmacological tool for the study of α7 nAChR functionality but also may lead to the development of therapy for cognitive deficits commonly shared by neuropsychiatric disorders such as schizophrenia and Alzheimer’s disease.
Materials and methods
Materials
Compound JWX-A0108 was synthesized in the State Key Laboratory of Natural and Biomimetic Drugs Medicinal Chemistry Laboratory, Peking University, China. All other chemicals were purchased from Sigma (St. Louis, MO, USA).
Animals
Adult C57BL/6J male mice and male Sprague–Dawley rats (240–260 g) from Beijing Vital River Laboratory Animal Technology Co. Ltd. (Beijing, China) were used in the study. Animals were housed in groups (four to five per cage) under a controlled temperature (23 ± 2 °C) and humidity (50 ± 5%) environment with ad libitum access to food and water. Animals were maintained on a reverse 12-h/12-h light/dark cycle (lights on at 7:00 am and off at 19:00 pm). The animal experimental protocols were approved by the Animal Use and Care Committee of Qingdao University and were consistent with the Ethical Guidelines of the International Association.
Two-electrode voltage-clamp (TEVC) recordings in Xenopus oocytes
Oocytes were harvested from Xenopus laevis female clawed frogs after being digested with collagenase (2 mg/mL) in a 25-mL tube containing Ca2+-free OR2 solution (82.5 mM NaCl, 2.5 mM KCl, 1 mM MgCl2, and 5 mM HEPES, pH 7.4) for 20 min at 20–25 °C under gentle rotation [32]. Oocytes from stage V to VI were selected and injected with 46 nL of cRNA solution containing ~20 ng of human α7 nAChR cRNAs or 1.5 ng of human GABAR cRNAs (accession number: GABRA1: NM-000806; GABRA5: NM-000810) using a microinjector (Drummond Scientific, Broomall, PA, USA). All cRNAs were transcribed in vitro from linearized plasmids in pBluescript KSM vectors by using the T3 mMESSAGEmMACHINE Kit (Ambion). After injection of cRNAs, oocytes were transferred into ND96 solution (96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, and 5 mM HEPES, pH 7.4 adjusted with NaOH), and incubated at 17 °C in an incubator for 2–5 days. Electrical currents were recorded in Ringer’s solution (115 mM NaCl, 2.5 mM KCl, 10 mM HEPES, 1.8 mM CaCl2, 1 mM MgCl2, and 0.5 µM atropine) at room temperature (22 ± 1 °C) using a GeneClamp 500B amplifier (Molecular Devices, Sunnyvale, CA, USA) [33, 34].
Culture of hippocampal neurons and whole-cell patch-clamp recordings
Hippocampi were isolated from the brains of 18-day-old Sprague–Dawley rat embryos and digested with 0.25% trypsin for 15–30 min at 37 °C. Hippocampal neurons were dissociated gently by passing them through a Pasteur pipette (~25 times) in a medium (DMEM with 10% FBS). The dispersed neurons were plated onto poly-d-lysine-coated coverslips in 35-mm dishes at a density of 1.5 × 106 cells per dish. After 4 h, the medium was replaced with neurobasal medium supplemented with 2% B27, 0.5 mM GlutaMAX-I and 1‰ primocin [35].
Whole-cell patch-clamp recordings were performed using a HEKA EPC10 amplifier with PatchMaster software. Neurons were held at −80 mV and perfused with the external solution containing the following (in mM): 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, and 10 glucose, pH 7.4, atropine (5 µM), CNQX (5 µM), bicuculline (10 µM), and tetrodotoxin (0.5 µM). The patch pipette was filled with internal pipette solution containing the following (in mM): 126 CsCH3SO3, 10 CsCl, 4 NaCl, 1 MgCl2, 0.5 CaCl2, 5 EGTA, 10 HEPES, 3 ATP-Mg, 0.3 GTP-Na, and 4 phosphocreatine, pH 7.2 [36].
Hippocampal brain slice recordings
Brains of male C57BL/6 mice (~13 days old) were quickly removed and submerged in ice-cold artificial cerebrospinal fluid (ACSF) containing the following (in mM): 119 NaCl, 2.5 KCl, 2.5 CaCl2, 1.3 MgSO4, 1 NaH2PO4, 26.2 NaHCO3, and 11 glucose, and bubbled with 95% O2/5% CO2. Transverse hippocampal slices (300 µm) were obtained using a Vibratome Leica VT 1200S (Leica, Wetzlar, Germany). The slices were then incubated for 1 h at room temperature with ACSF. Whole-cell patch-clamp recordings were performed from pyramidal neurons of the CA1 region of the hippocampus. The hippocampus slices were transferred to a chamber and continuously perfused with oxygenated (95% O2 and 5% CO2) ACSF at a rate of 2–3 mL/min. To record the IPSCs, patch pipettes were pulled with resistances of 3–5 MΩ when filled with intracellular solution of the following composition (in mM): 136 CsCl, 4 NaCl, 1 MgCl2, 0.5 CaCl2, 5 EGTA, 10 HEPES, 3 Mg-ATP, 0.3 GTP-Na, and 4-phosphocreatine. To block the fast excitatory synaptic current (EPSC), 5 µM CNQX and 10 µM APV were added to the ACSF. The neurons were voltage-clamped at −70 mV using a MultiClamp 700B Amplifier (Molecular Devices, Sunnyvale, CA, USA). The sampling frequency was set at 10 kHz, and the filter frequency was 1 kHz [36, 37].
Liquid chromatography–tandem mass spectrometry (LC–MS/MS) analyses
The analysis was performed using a Waters ACQUITY UPLC system coupled to the TQ detector. Chromatographic separation was achieved on a Luna C18(2) column (150 mm × 4.6 mm, 5 μm) at 40 °C with a mobile phase consisting of acetonitrile and 0.1% formic acid in H2O (80:20, v:v) at a flow rate of 0.5 mL/min. The autosampler was maintained at 4 °C.
The mass spectrometer was operated in positive ion mode using an electrospray ionization (ESI) source. The multiple reaction monitoring modes were used to detect a specific transition of the precursor ion to the product ion at m/z 419.0 to m/z 169.0 for LD486 and m/z 401.0 to 115.0 for JWX-A0108. The optimal ESI–MS/MS parameters were as follows: positive ESI with capillary voltage of 3.0 kV, cone voltage of 44 V, desolvation temperature of 400 °C, source temperature of 150 °C, desolvation gas flow of 600 L/h, and cone gas flow of 50 L/h.
Pharmacokinetic parameters
Food was stopped 12 h before the experiment, but free access to water was maintained. Compound JWX-A0108 was dissolved in 40% PEG 400 saline. Rats were divided randomly into three groups (six rats for each group) for oral administration (i.g.) at a single dose of 10 mg/kg or intravenous injection (i.v.) at a single dose of 1 mg/kg. Blood samples were collected from the jugular vein into 1.5-mL heparinized Eppendorf tubes at 0.083, 0.25, 0.5, 1, 1.5, 2, 4, 8, and 12 h after i.v. dosing and 0.083, 0.333, 0.667, 1, 1.5, 2, 3, 4, 5, 6, 8, 12, and 24 h after i.g. administration before being centrifuged immediately at 4000 r/min for 5 min. The upper plasma was separated and stored at −20 °C until analysis.
A noncompartmental model was employed to calculate the parameters. The maximum plasma concentration (Cmax) and the time to reach it (Tmax) were directly obtained from the experimental data [38]. The terminal elimination rate constant (Ke) was determined by linear regression of the terminal portion of the plasma concentration–time data, and the elimination half-life (T1/2) was calculated as 0.693/Ke. The area under the plasma concentration curve (AUC) versus time (AUC0−t) from time zero to the time of the last measured concentration (Clast) was calculated by the linear trapezoidal rule. The AUC from time zero to infinity (AUC0−∞) was obtained by the addition of AUC0−t and the extrapolated area determined by Clast/Ke. The total body clearance (CL) was calculated as the quotient of the dose and AUC0−∞. The apparent volume of distribution (Vz) was calculated by the equation: Vz = CL/Ke, and the mean residence time (MRT) was calculated by the equation: MRT = AUMC/AUC. Oral bioavailability was calculated according to the following equation [38]:
Prepulse inhibition (PPI) test
The PPI test was measured in four standard startle chambers for amplitude reduction of a subsequent strong starting stimulus (pulse) after a weaker acoustic prestimulus (prepulse) in mice that received 0.1 mg/kg MK-801 intraperitoneally (i.p.) 30 min before the test. The mouse holder was rested on a platform in each sound-attenuated chamber. The motor response of the mouse was detected by a vibration sensor on the platform and recorded by a computer using Pan Lab software (Pan Lab Harvard Apparatus). Mice were placed in the laboratory for at least 2 days for adaptation to the environment. One day before the test, the mice were habituated to the environment in the Plexiglas cylinder for 15 min with 68-dB background noise. On the following day, after a 5-min acclimatization period, mice were exposed to 70-test trials composed of 10 pulse-only trials, 50 prepulse–pulse trials, and 10 null trials. A 68-dB background noise was present during testing. Pulse-only trials consisted of a single 40-ms 120-dB burst of white noise. Prepulse–pulse trials consisted of a 20-ms prepulse of white noise that was 72, 76, 80, 84, or 88 dB, followed by an 80-ms 120-dB pulse. The white noise background was the only stimulus present during the null-only trials. All trials were presented in pseudorandom order, with an average of 15-s (range from 8 to 22 s) interval between the trials. The percent PPI for each prepulse intensity was calculated as the ratio of the average response during the prepulse–pulse trials to the average pulse-only responses subtracted from 1, multiplied by 100, and expressed as a percentage. Null activity and pulse-only responses are reported in arbitrary units, as measured by the Pan Lab Startle Response system.
Y-maze test
The Y maze for assessing spatial working memory was made up with three gray arms at an angle of 120° from each other. The test consisted of a forced-choice trial followed by a free-choice trial. During the test, no reward or punishment was given. For the forced-choice trial, the start arm and one test arm were open, while another test arm was blocked. The mice were placed at the end of the start arm and then allowed to explore the open arms for 8 min, after which they were removed from the maze. For the free-choice trial, mice were allowed to explore freely for 4 min [39].
Locomotor activity
Mice were placed in clear open-field chambers for 30 min to acclimate to the environment. Twenty-four hours later, the mice were brought back to the chamber, and locomotor activity was measured to evaluate the animal’s basal activity using infrared photosensors. The mice received MK-801 (0.1 mg/kg, i.p.) 30 min before the test. JWX-A0108 (0.03–0.3 mg/kg) or clozapine (1 mg/kg) was administered intraperitoneally 60 min prior to the test. Locomotor activity was recorded for 60 min [40].
Statistical analysis
All data were expressed as the means ± SEM except pharmacokinetics and statistically analyzed with one-way ANOVA or two-way ANOVA using Prism version 5.0 software. In TEVC and whole-cell patch-clamp recordings, responses were quantified by measuring peak current amplitude and data were collected and analyzed using PatchMaster and Origin 8.0 software. For hippocampal brain slice electrophysiology, the data were collected and analyzed using Clampfit and Origin 9.0 software. The pharmacokinetic parameters were evaluated in each individual rat by a noncompartmental approach with the software PK solver 2.0.
Results
Selective enhancement of α7 currents expressed in Xenopus oocytes by compound JWX-A0108
The representative compound JWX-A0108 (Fig. 1a) was synthesized and selected based on screening of a series of thiazolyl heterocyclic derivatives using TEVC recordings of Xenopus oocytes injected with α7 nAChR cRNA. Bath application of different concentrations of JWX-A0108 resulted in a fast dose-dependent activation and rapid inactivation/desensitization of α7 currents in the presence of ACh (100 µM) with maximum current enhancement (Emax) of ~30 fold compared with that in response to 100 µM ACh alone (Fig. 1b) or 10 µM JWX-A0108 alone, which gave rise to no current. The EC50 value of JWX-A0108 was 4.35 ± 0.12 µM with the maximum enhancement of current amplitude ~30 fold and a Hill coefficient of 2.0 (Fig. 1c). We also evaluated the selectivity of JWX-A0108 on other nAChRs and 5-HT3A receptors individually expressed in oocytes. As shown in Fig. 1d, JWX-A0108 had no potentiation effect on α3β4 nAChR, α4β2 nAChR, or 5-HT3AR. These results indicate that JWX-A0108, as a type I α7 PAM, selectively enhances the α7 current functionally expressed in oocytes.

Chemical structure of compound JWX-A0108 and its allosteric and dose-dependent activation of human α7 nAChR channels expressed in Xenopus oocytes. a Chemical structure of compound 6-(2-chloro-6-methylphenyl)-2-((3-fluoro-4-methylphenyl)amino)thiazolo[4,5-d]pyrimidin-7(6H)-one (JWX-A0108, molecular weight of 400.06). b Representative α7 currents recorded from an oocyte expressing human α7 nAChR in response to 100 µM ACh in the absence or presence of different concentrations of JWX-A0108 (0.1–30 µM). c Concentration–response relationship for JWX-A0108. Peak current amplitudes were normalized to the amplitude of current elicited by 100 µM ACh alone, and EC50 = 4.35 ± 0.12 µM, fold increase: 23.7 ± 2.36, and nHill = 2.08 ± 0.03, n = 5 for all data. d Fold-increase in α7 nAChR current, α3β4 nAChR current, and α4β2 nAChR current in the presence of 100 μM or 5-HT3A receptor current evoked by 10 μM 5-HT after incubation with 10 μM JWX-A0108, n = 5 for all experiments. Data are expressed as the means ± SEM
JWX-A0108 potentiates the endogenous α7 current of cultured rat hippocampal neurons
To assess if JWX-A0108 was capable of activating the native α7 current, we examined the effects of JWX-A0108 on hippocampus neurons. As shown in Fig. 2a, application of 10 µM JWX-A0108 alone caused no activation of current, unlike 10 µM ACh, which generated a small fast-activation current with rapid desensitization. In contrast, co-application of JWX-A0108 (3 or 10 µM) and 10 µM ACh gave rise to a dose-dependent increase in α7 current characteristic of fast activation and rapid desensitization (Fig. 2b). Together with data shown in Fig. 1b, these results demonstrate that JWX-A0108 acts as a type I α7 nAChR PAM.

JWX-A0108 potentiates endogenous α7 nAChRs of cultured rat hippocampal neurons. a Representative current traces evoked by PAM JWX-A0108 (10 µM) and agonist ACh (10 µM) in hippocampal neurons (n = 3). The neurons were held at –80 mV. b Representative current traces evoked by a concentration of ACh (10 µM) and increasing concentrations of JWX-A0108 (1–10 µM)
JWX-A0108 enhances GABAergic synaptic transmission in hippocampal brain slices
The α7 nAChR is mainly expressed in interneurons in the CA1 region of the hippocampus and is capable of regulating hippocampal circuits by exciting interneurons and subsequently inhibiting or disinhibiting the pyramidal neurons that express GABA receptors [41]. To test if compound JWX-A0108 can increase the synaptic transmission of GABA neurotransmitters in pyramidal neurons via α7 nAChRs acting on interneurons, we recorded the spontaneous inhibitory postsynaptic currents (sIPSCs) in pyramidal neurons from the hippocampal CA1 region that receives input from interneurons. In response to challenges with ACh (10 µM), JWX-A0108 (10 µM), or a combination of ACh and JWX-A0108 (Fig. 3a, b), JWX-A0108 caused no detectable change in the frequency or amplitude of sIPSCs or spontaneously occurring synaptic events when applied alone (Fig. 3c, d). In the same configuration, continuous bath application of ACh (10 µM) produced a transient increase in synaptic activity (Fig. 3c, d). In contrast, JWX-A0108 markedly enhanced the actions of ACh on synaptic transmission, resulting in a large increase in GABAergic synaptic activity (Fig. 3).

JWX-A0108 enhances GABAergic synaptic transmission in brain slices. a Representative raw traces showing that co-application of JWX-A0108 and ACh significantly increased both the frequency and amplitude of sIPSCs compared with application of JWX-A0108 or ACh alone. The neurons were held at –70 mV. b Peak amplitude distributions of all IPSC events detected in a. c Normalized average IPSC peak amplitudes from all recordings. Co-application of JWX-A0108 and ACh increased IPSC amplitude in each individual neuron (n = 5). d Statistical analysis of average IPSC frequency from all neurons recorded (n = 6). Co-application of JWX-A0108 and ACh significantly increased the average frequency of the IPSCs. Data are expressed as the means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. A0108 JWX-A0108
To further examine if the enhancement of IPSCs resulted from an increase in GABAergic synaptic transmission, we tested the effect of JWX-A0108 on subtypes of GABAA receptors. The α1β3γ2 is a major subtype in the brain, and the α5β3γ2 is predominantly found on hippocampal cells [42]. As shown in Fig. 4, co-application of 1 μM GABA with 10 μM JWX-A0108 had no observable effects on these two subtypes compared with application of 1 μM GABA alone.

Selectivity assessments of JWX-A0108 on subtypes of GABAA receptors. Representative current traces recorded in oocytes expressing human α1β3γ2 (a) and α5β3γ2 (b) subtypes, showing the effects of JWX-A0108 on GABAA activity in the presence of 1 µM GABA (left), the co-application of 1 µM GABA and 10 μM JWX-A0108 (middle), and statistical analysis of fold changes between control and co-application of GABA and JWX-A0108 (right), showing that 10 μM JWX-A0108 had no significant effect on GABAA currents mediated by α1β3γ2 (98.94 ± 13.12%, P > 0.05, n = 5) or α5β3γ2 (114.17 ± 16.98%, P > 0.05, n = 3) subtypes compared with the control. Data are expressed as the means ± SEM
Pharmacokinetic analysis of JWX-A0108
To gain insight into the pharmacokinetics of compound JWX-A0108 for in vivo evaluation, we determined the elimination half-life in rat plasma following intravenous administration of JWX-A0108 (1 mg/kg). As shown in Fig. 5a and Table 1, intravenous administration of JWX-A0108 (1 mg/kg) gave rise to an elimination half-life (T1/2) of 2.7 h. When orally administered at 10 mg/kg, JWX-A0108 reached its maximum concentration at 1.3 h and elimination half-life (T1/2) at 1.9 h (Fig. 5b and Table 1). The Cmax of JWX-A0108 was 231.5 ± 24.2 mg/L and 145.1 ± 36.3 mg/L, while the value of the area under the curve (AUC0–∞) was 293.8 ± 68.6 mg h/L and 395.2 ± 127.3 mg h/L at a dosage of 1 mg/kg (i.v.) and 10 mg/kg (i.g.), respectively (Table 1).

Plasma concentration–time profile for compound JWX-A0108. a The compound was administered intravenously at 1 mg/kg in SD rats (n = 3). b JWX-A0108 was given through oral administration at 10 mg/kg in SD rats (n = 6). Data are expressed as the means ± SD
JWX-A0108 reverses MK-801-induced deficits in auditory gating
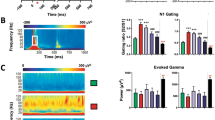
To further evaluate the effect of JWX-A0108 on behavior, we utilized the schizophrenia-like behavioral mouse model of auditory gating deficit induced by the NMDA antagonist MK-801 and measured the PPI in acoustic startle chambers. During the prepulse–pulse test, five different intensities of prepulse (72, 76, 80, 84, and 88 dB) were presented for 20 ms with an 80-ms delay before the application of the startle stimulus (pulse alone) at 120 dB for 40 ms. As shown in Fig. 6a, intraperitoneal injection of MK-801 (0.1 mg/kg) resulted in a significant reduction in PPI from 36% to 19%. In contrast, administration of different concentrations (1–10 mg/kg) of JWX-A0108 reversed the reduction in PPI to 25, 32, and 37% in a dose-dependent manner, as compared with the positive control clozapine (1 mg/kg, i.p.), which reversed the reduced PPI to 31% (Fig. 6a). The average percent PPI across five different prepulse levels is shown in Fig. 6b. MK-801 (0.1 mg/kg, i.p.) significantly diminished PPI, which was reversed by the positive control clozapine (1 mg/kg, i.p.). Administration of JWX-A0108 (1–10 mg/kg, i.p.) attenuated the MK-801-induced deficit in PPI in mice in a dose-dependent manner (Fig. 6b). These results indicate that the type I α7 PAM JWX-A0108 can rescue the auditory gating deficit of schizophrenia-like behavior in mice.

JWX-A0108 reverses MK-801-induced deficits in auditory gating. a Administration of JWX-A0108 (1, 3, and 10 mg/kg, i.p.) significantly attenuates auditory gating deficits induced by MK-801 in five prepulse intensities (72, 76, 80, 84, and 88 dB). Clozapine (CLO) was administered as the positive control. b Average percent of PPI across the prepulse level. JWX-A0108 dose-dependently reversed MK-801-induced deficits in PPI (n = 12–16). Data are expressed as the means ± SEM. **P < 0.01, ***P < 0.001 vs. control group. #P < 0.05, ##P < 0.01, ###P < 0.001 vs. MK-801 group. VEH vehicle
Improvement of spatial working memory by JWX-A0108 in the Y-maze
Spatial working memory was examined by recording the time spent in the novel arm in the Y-maze. Intraperitoneal injection of MK-801 (0.1 mg/kg) resulted in a significant reduction in time in the novel arm from 62 to 45% (Fig. 7a), indicating a behavioral deficit in spatial working memory. In contrast, the administration of different concentrations (0.03, 0.1, or 0.3 mg/kg) of JWX-A0108 reversed the reduction in time in the novel arm by approximately 49%, 53%, and 61%, respectively (Fig. 7a), demonstrating an improvement in spatial working memory.

Effects of JWX-A0108 on spatial working memory and locomotor activity. a Administration of JWX-A0108 (0.03, 0.1, and 0.3 mg/kg, i.p.) significantly improved the time spent in the novel arm in the Y-maze (n = 12–15). b Administration of JWX-A0108 (0.03 mg/kg, i.p.) reduced the total distance traveled during locomotor activity (n = 10–12). Data are expressed as the means ± SEM. *P < 0.05, ***P < 0.001 vs. control group. #P < 0.05, ###P < 0.001 vs. MK-801 group
Effect of JWX-A0108 on locomotor activity
JWX-A0108 was also assessed for its effects on locomotor activity. Intraperitoneal injection of MK-801 (0.1 mg/kg) resulted in a significant potentiation of total travel distance from 24,928 ± 617.5 cm to 34,825 ± 1469 cm (Fig. 7b). In contrast, intraperitoneal injections of 0.03 mg/kg JWX-A0108 attenuated the increase in total travel distance caused by MK-801, compared with the positive control clozapine (1 mg/kg, i.p.), which also reversed the total distance to 28,374 ± 852.0 cm (Fig. 7b). However, administration of higher doses of JWX-A0108 (0.1 and 0.3 mg/kg) had no significant effect on the increase in total travel distance induced by MK-801, suggesting that a higher dose of JWX-A0108 may have a trend of increasing locomotion.
Discussion
The goal of this study was to characterize both the in vitro and in vivo activities of a novel α7 nAChR PAM. Using electrophysiology as a primary screen, we found that compound JWX-A0108 as a type I PAM enhances the activity of hippocampal neurons with a native α7 nAChR current and synaptic transmission in vitro and reverses auditory gating deficits and spatial working memory in vivo.
Within rat hippocampus, α7 nAChRs are located on inhibitory interneurons, and activation of those receptors can increase hippocampal GABAergic neurotransmission [43,44,45]. GABA is an inhibitory synaptic transmitter, and decreased release of GABA can lead to disinhibition of sensory gating [36]. To evaluate the ability of JWX-A0108 to modulate GABAergic synaptic transmission, we recorded the spontaneous IPSCs in pyramidal neurons from acutely isolated rat hippocampal slices. The spontaneous GABAergic synaptic events were recorded for 3–6 min under baseline conditions. In agreement with the previously reported role of α7 nAChR in the hippocampus, there was no detectable change in synaptic activity after the application of JWX-A0108 alone, whereas the bath application of 10 µM ACh increased the frequency and amplitude of sIPSCs, and in particular, the frequency increased significantly.
Sensory gating defects are a common clinical manifestation in schizophrenia patients [46]. Insufficient function of inhibitory neurons is thought to be one of the causes underlying the pathogenesis of schizophrenia. Deficits in auditory sensory gating can lead to an inability to judge and filter out irrelevant content from meaningful sensory inputs, resulting in an overloaded sensory state, which contributes to the attentional and cognitive deficits in CNS diseases, most convincingly in schizophrenia [47].
Type I α7 PAMs have received more attention in recent years. Previous work has shown that agonists of α7 nAChR can improve gating deficits in rats regardless of whether the impairment is induced pharmacologically [48, 49], genetically [50], or behaviorally [21]. However, most clinical trials conducted for AD or schizophrenia with α7 nAChR agonists such as EVP-6124 and MEM3454 were either suspended in phase II and III or showed a paucity of effects. With limited data and reports, we can only assume the lack of sufficient selectivity against 5-HT3 receptors since almost all of the α7 nAChR agonists show cross-activity with 5-HT3 receptors, and improper designation of clinical trials might be the cause of the discontinued compounds and clinical failure [13]. When compared with agonists, α7 nAChR PAMs are more promising because of their maintenance of endogenous activation characteristics, better selectivity profile, higher structural diversity, and better neuroprotective effect [51]. Currently, a few type I α7 nAChR PAMs are either in the preclinical late stage or a phase I trial [13], whereas a potent type II α7 PAMs PNU-120596 has exhibited a cytotoxic effect, suggesting that a too-potent activity of type II α7 PAM may not be favorable in drug discovery [28, 29]. In the present study, our α7 nAChR type I PAM JWX-A0108 reversed the MK-801-induced auditory gating deficit and improved spatial working memory, suggesting a developmental potential for improvement of cognitive impairment.
In summary, the data presented in this study demonstrate that a novel type I α7 nAChR PAM JWX-A0108 enhances α7 current and GABAergic synaptic transmission in hippocampal brain slices in the presence of the neurotransmitter acetylcholine. JWX-A0108 also reverses MK-801-induced deficits in auditory gating. JWX-A0108 can serve as a suitable tool for further characterizing the pharmacological role of α7 nAChR in brain function and may also have the potential for development into a new therapy for neuropsychiatric disorders characterized by cognitive deficits and α7 nAChR dysfunction.
References
Wallace TL, Bertrand D. Alpha7 neuronal nicotinic receptors as a drug target in schizophrenia. Expert Opin Ther Targets. 2013;17:139–55.
Tamminga CA, Holcomb HH. Phenotype of schizophrenia: a review and formulation. Mol Psychiatry. 2005;10:27–39.
Elvevag B, Goldberg TE. Cognitive impairment in schizophrenia is the core of the disorder. Crit Rev Neurobiol. 2000;14:1–21.
Ochoa EL, Lasalde-Dominicci J. Cognitive deficits in schizophrenia: focus on neuronal nicotinic acetylcholine receptors and smoking. Cell Mol Neurobiol. 2007;27:609–39.
Potasiewicz A, Holuj M, Kos T, Popik P, Arias HR, Nikiforuk A. 3-Furan-2-yl-N-p-tolyl-acrylamide, a positive allosteric modulator of the alpha7 nicotinic receptor, reverses schizophrenia-like cognitive and social deficits in rats. Neuropharmacology. 2017;113:188–97.
Martin LF, Kem WR, Freedman R. Alpha-7 nicotinic receptor agonists: potential new candidates for the treatment of schizophrenia. Psychopharmacol (Berl). 2004;174:54–64.
Martin LF, Freedman R. Schizophrenia and the alpha7 nicotinic acetylcholine receptor. Int Rev Neurobiol. 2007;78:225–46.
Freedman R. alpha7-nicotinic acetylcholine receptor agonists for cognitive enhancement in schizophrenia. Annu Rev Med. 2014;65:245–61.
Taly A, Corringer PJ, Guedin D, Lestage P, Changeux JP. Nicotinic receptors: allosteric transitions and therapeutic targets in the nervous system. Nat Rev Drug Discov. 2009;8:733–50.
Gotti C, Zoli M, Clementi F. Brain nicotinic acetylcholine receptors: native subtypes and their relevance. Trends Pharmacol Sci. 2006;27:482–91.
Dineley KT, Pandya AA, Yakel JL. Nicotinic ACh receptors as therapeutic targets in CNS disorders. Trends Pharmacol Sci. 2015;36:96–108.
Wonnacott S. Presynaptic nicotinic ACh receptors. Trends Neurosci. 1997;20:92–8.
Yang T, Xiao T, Sun Q, Wang K. The current agonists and positive allosteric modulators of alpha7 nAChR for CNS indications in clinical trials. Acta Pharm Sin B. 2017;7:611–22.
Freedman R, Coon H, Myles-Worsley M, Orr-Urtreger A, Olincy A, Davis A, et al. Linkage of a neurophysiological deficit in schizophrenia to a chromosome 15 locus. Proc Natl Acad Sci USA. 1997;94:587–92.
Stevens KE, Freedman R, Collins AC, Hall M, Leonard S, Marks MJ, et al. Genetic correlation of inhibitory gating of hippocampal auditory evoked response and alpha-bungarotoxin-binding nicotinic cholinergic receptors in inbred mouse strains. Neuropsychopharmacology. 1996;15:152–62.
Felix R, Levin ED. Nicotinic antagonist administration into the ventral hippocampus and spatial working memory in rats. Neuroscience. 1997;81:1009–17.
Young JW, Crawford N, Kelly JS, Kerr LE, Marston HM, Spratt C, et al. Impaired attention is central to the cognitive deficits observed in alpha 7 deficient mice. Eur Neuropsychopharmacol. 2007;17:145–55.
Freedman R, Hall M, Adler LE, Leonard S. Evidence in postmortem brain tissue for decreased numbers of hippocampal nicotinic receptors in schizophrenia. Biol Psychiatry. 1995;38:22–33.
Cui R, Suemaru K, Li B, Kohnomi S, Araki H. Tropisetron attenuates naloxone-induced place aversion in single-dose morphine-treated rats: role of alpha7 nicotinic receptors. Eur J Pharmacol. 2009;609:74–7.
de Fiebre CM, Meyer EM, Henry JC, Muraskin SI, Kem WR, Papke RL. Characterization of a series of anabaseine-derived compounds reveals that the 3-(4)-dimethylaminocinnamylidine derivative is a selective agonist at neuronal nicotinic alpha 7/125I-alpha-bungarotoxin receptor subtypes. Mol Pharmacol. 1995;47:164–71.
O’Neill HC, Rieger K, Kem WR, Stevens KE. DMXB, an alpha7 nicotinic agonist, normalizes auditory gating in isolation-reared rats. Psychopharmacol (Berl). 2003;169:332–9.
Callahan PM, Terry AV Jr., Tehim A. Effects of the nicotinic alpha7 receptor partial agonist GTS-21 on NMDA-glutamatergic receptor related deficits in sensorimotor gating and recognition memory in rats. Psychopharmacol (Berl). 2014;231:3695–706.
Simosky JK, Stevens KE, Kem WR, Freedman R. Intragastric DMXB-A, an alpha7 nicotinic agonist, improves deficient sensory inhibition in DBA/2 mice. Biol Psychiatry. 2001;50:493–500.
Olincy A, Harris JG, Johnson LL, Pender V, Kongs S, Allensworth D, et al. Proof-of-concept trial of an alpha7 nicotinic agonist in schizophrenia. Arch Gen Psychiatry. 2006;63:630–8.
Freedman R, Olincy A, Buchanan RW, Harris JG, Gold JM, Johnson L, et al. Initial phase 2 trial of a nicotinic agonist in schizophrenia. Am J Psychiatry. 2008;165:1040–7.
Gee KW, Olincy A, Kanner R, Johnson L, Hogenkamp D, Harris J, et al. First in human trial of a type I positive allosteric modulator of alpha7-nicotinic acetylcholine receptors: pharmacokinetics, safety, and evidence for neurocognitive effect of AVL-3288. J Psychopharmacol. 2017;31:434–41.
Hurst RS, Hajos M, Raggenbass M, Wall TM, Higdon NR, Lawson JA, et al. A novel positive allosteric modulator of the alpha7 neuronal nicotinic acetylcholine receptor: in vitro and in vivo characterization. J Neurosci. 2005;25:4396–405.
Callahan PM, Hutchings EJ, Kille NJ, Chapman JM, Terry AV Jr.. Positive allosteric modulator of alpha7 nicotinic-acetylcholine receptors, PNU-120596 augments the effects of donepezil on learning and memory in aged rodents and non-human primates. Neuropharmacology. 2013;67:201–12.
Ng HJ, Whittemore ER, Tran MB, Hogenkamp DJ, Broide RS, Johnstone TB, et al. Nootropic alpha7 nicotinic receptor allosteric modulator derived from GABAA receptor modulators. Proc Natl Acad Sci USA. 2007;104:8059–64.
Nikiforuk A, Kos T, Potasiewicz A, Popik P. Positive allosteric modulation of alpha 7 nicotinic acetylcholine receptors enhances recognition memory and cognitive flexibility in rats. Eur Neuropsychopharmacol. 2015;25:1300–13.
Nikiforuk A, Kos T, Holuj M, Potasiewicz A, Popik P. Positive allosteric modulators of alpha 7 nicotinic acetylcholine receptors reverse ketamine-induced schizophrenia-like deficits in rats. Neuropharmacology. 2016;101:389–400.
Liang P, Wang H, Chen H, Cui Y, Gu L, Chai J, et al. Structural Insights into KChIP4a Modulation of Kv4.3 Inactivation. J Biol Chem. 2009;284:4960–7.
Dascal N. Voltage clamp recordings from Xenopus oocytes. Curr Protoc Neurosci. 2001; Chapter 6:Unit 6.12:5–7
Guan B, Chen X, Zhang H. Two-electrode voltage clamp. Methods Mol Biol. 2013;998:79–89.
Kaech S, Banker G. Culturing hippocampal neurons. Nat Protoc. 2006;1:2406–15.
Tang JS, Xie BX, Bian XL, Xue Y, Wei NN, Zhou JH, et al. Identification and in vitro pharmacological characterization of a novel and selective alpha7 nicotinic acetylcholine receptor agonist, Br-IQ17B. Acta Pharmacol Sin. 2015;36:800–12.
Townsend M, Whyment A, Walczak JS, Jeggo R, van den Top M, Flood DG, et al. alpha7-nAChR agonist enhances neural plasticity in the hippocampus via a GABAergic circuit. J Neurophysiol. 2016;116:2663–75.
Huang X, Jiao W, Sun Q. Pharmacokinetic characterization of a novel α7 nicotinic acetylcholine receptor (nAChR) positive allosteric modulator LD486 in rat plasma using a validated LC-MS/MS assay. J Chin Pharm Sci. 2016;25:517–25. KW
Bristow LJ, Easton AE, Li YW, Sivarao DV, Lidge R, Jones KM, et al. The novel, nicotinic Alpha7 receptor partial agonist, BMS-933043, improves cognition and sensory processing in preclinical models of schizophrenia. PLoS ONE. 2016;11:e0159996.
Wallace TL, Callahan PM, Tehim A, Bertrand D, Tombaugh G, Wang S, et al. RG3487, a novel nicotinic alpha7 receptor partial agonist, improves cognition and sensorimotor gating in rodents. J Pharmacol Exp Ther. 2011;336:242–53.
Ji D, Dani JA. Inhibition and disinhibition of pyramidal neurons by activation of nicotinic receptors on hippocampal interneurons. J Neurophysiol. 2000;83:2682–90.
McKernan RM, Whiting PJ. Which GABAA-receptor subtypes really occur in the brain? Trends Neurosci. 1996;19:139–43.
Alkondon M, Pereira EF, Barbosa CT, Albuquerque EX. Neuronal nicotinic acetylcholine receptor activation modulates gamma-aminobutyric acid release from CA1 neurons of rat hippocampal slices. J Pharmacol Exp Ther. 1997;283:1396–411.
Radcliffe KA, Fisher JL, Gray R, Dani JA. Nicotinic modulation of glutamate and GABA synaptic transmission of hippocampal neurons. Ann N Y Acad Sci. 1999;868:591–610.
Buhler AV, Dunwiddie TV. Alpha7 nicotinic acetylcholine receptors on GABAergic interneurons evoke dendritic and somatic inhibition of hippocampal neurons. J Neurophysiol. 2002;87:548–57.
Wallace TL, Porter RH. Targeting the nicotinic alpha7 acetylcholine receptor to enhance cognition in disease. Biochem Pharmacol. 2011;82:891–903.
Leiser SC, Bowlby MR, Comery TA, Dunlop J. A cog in cognition: how the alpha 7 nicotinic acetylcholine receptor is geared towards improving cognitive deficits. Pharmacol Ther. 2009;122:302–11.
Stevens KE, Kem WR, Mahnir VM, Freedman R. Selective alpha7-nicotinic agonists normalize inhibition of auditory response in DBA mice. Psychopharmacol (Berl). 1998;136:320–7.
Hajos M, Hurst RS, Hoffmann WE, Krause M, Wall TM, Higdon NR, et al. The selective alpha7 nicotinic acetylcholine receptor agonist PNU-282987 [N-[(3R)-1-Azabicyclo[2.2.2]oct-3-yl]-4-chlorobenzamide hydrochloride] enhances GABAergic synaptic activity in brain slices and restores auditory gating deficits in anesthetized rats. J Pharmacol Exp Ther. 2005;312:1213–22.
Stevens KE, Wear KD. Normalizing effects of nicotine and a novel nicotinic agonist on hippocampal auditory gating in two animal models. Pharmacol Biochem Behav. 1997;57:869–74.
Corradi J, Bouzat C. Understanding the bases of function and modulation of alpha7 nicotinic receptors: implications for drug discovery. Mol Pharmacol. 2016;90:288–99.
Acknowledgements
This project is supported by research grants to KWW and QS from the National Natural Science Foundation of China (NSFC 81573410, 31370741, and 21572011), and from the Ministry of Science and Technology of China (2013CB531302). We thank F Hu and W Lu for technical assistance. KWW wishes to thank JM Wang for her consistent support during this research.
Author contributions
L-lS performed behavioral experiments; T-yY and N-nW performed electrophysiological experiments; WL made the recordings of brain slices; W-xJ synthesized compound JWX-A0108; Q-qZ, Y-zM, and QG performed some behavioral experiments; X-tW analyzed the data of pharmacokinetics; L-lS analyzed the data and drafted the manuscript; QS and KWW supervised the project and finalized the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Sun, Ll., Yang, Ty., Wei, Nn. et al. Pharmacological characterization of JWX-A0108 as a novel type I positive allosteric modulator of α7 nAChR that can reverse acoustic gating deficits in a mouse prepulse inhibition model. Acta Pharmacol Sin 40, 737–745 (2019). https://doi.org/10.1038/s41401-018-0163-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-018-0163-y
Keywords
This article is cited by
-
α7 Nicotinic acetylcholine receptor: a key receptor in the cholinergic anti-inflammatory pathway exerting an antidepressant effect
Journal of Neuroinflammation (2023)
