
Abstract
Circular RNAs (circRNAs) are emerging species of mRNA splicing products with largely unknown functions. Although several computational pipelines for circRNA identification have been developed, these methods strictly rely on uniquely mapped reads overlapping back-splice junctions (BSJs) and lack approaches to model the statistical significance of the identified circRNAs. Here, we reported a systematic computational approach to identify circRNAs by simultaneously utilizing BSJ overlapping reads and discordant BSJ spanning reads to identify circRNAs. Moreover, we developed a novel procedure to estimate the P-values of the identified circRNAs. A computational cross-validation and experimental validations demonstrated that our method performed favorably compared to existing circRNA detection tools. We created a standalone tool, CircRNAFisher, to implement the method, which might be valuable to computational and experimental scientists studying circRNAs.
Similar content being viewed by others

Introduction
Circular RNAs (circRNAs) are unorthodox RNA species produced by exon circularization during alternative splicing. Although earlier studies report circRNAs in viroids [1,2,3] and in animals [4,5,6,7], circRNAs have only gained mainstream acceptance recently, largely due to the accumulation of high-throughput sequencing data and computational approaches that uncovered the ubiquitous existence of circRNAs [8,9,10,11,12]. Although the biological functions of the vast majority of circRNAs remain unknown, circRNAs demonstrate a strong correlation with the risk of human diseases, [13] suggesting that circRNAs may not be mere transcriptional noise but may play essential roles in biological processes and may represent novel therapeutic opportunities.
A premise to understand the function of circRNAs is their thorough and robust identification. Memczak et al. [10] identified circRNAs by splitting single reads into two fragments and demanding that the mapped fragments be aligned in a reverse orientation. Gao et al developed a comprehensive computational tool, CIRI, for circRNA identification [14, 15]. CIRI employed a paired chiastic clipping (PCC) signal detection followed by systematic filtering to remove false positives. Instead of mapping reads to the entire human genome, several methods attempted to identify circRNAs by mapping the reads to a list of scrambled exon–exon candidate junctions [9, 16, 17]. Finally, computational and experimental approaches, such as RNase R digestion, have been combined to identify circRNAs [11, 18,19,20]. Current approaches for circRNA identification are summarized in recent reviews [21, 22]. A limitation of the existing methods is that only reads uniquely mapped to back-splice junctions (BSJs) are used in circRNA detection. Moreover, the current methods do not provide statistical significance for the identified circRNAs. These key issues remain to be resolved to enable the thorough and robust detection of circRNAs.
Here, we present a systematic computational pipeline, CircRNAFisher, for de novo genome-wide circRNA identification and annotation. CircRNAFisher combines BSJ searching with a series of statistical filters to detect candidate circRNAs. CircRNAFisher also provides a formal method to estimate the P-values of the identified circRNAs by combining BSJ overlapping reads with discordant BSJ spanning reads. We analyzed the performance of CircRNAFisher by computation analyses and experimental validations. The results confirmed that CircRNAFisher performed favorably compared to existing methods and identified novel circRNAs with high confidence. CircRNAFisher was developed into an open source tool, which is valuable for the study of circRNAs.
Materials and methods
RNA-Seq data
RNA-Seq data for two cancer cell lines (A549 and MCF7) and one normal skin fibroblast cell line (BJ) were downloaded from the ENCODE project [23]. The reference genome (human hg19) and reference transcripts (UCSC known genes) were downloaded from the UCSC genome browser website [24].
Cell culture
A549 cells and HeLa cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Corning, MA, USA) supplemented with 10% fetal bovine serum (Gibco, Australia).
RNA isolation and reverse transcription PCR
Total RNA was isolated from the cells using the TRIzol reagent (TaKaRa, Kusatsu, Japan). Reverse transcription was performed using the PrimeScriptTM RT Reagent Kit (TaKaRa, Kusatsu, Japan) according to the manufacturer’s instructions.
Considering the unique circular structure of circRNAs, we designed corresponding divergent primers and negative control primers as described previously [10]. Briefly, the divergent primers for the circRNAs were designed to amplify a target sequence, with a length of ~150–200 bp, by joining the 100 nt sequence from the 3′ end to the 100 nt sequence at the 5′ end. The primers for the negative controls were designed from the 5′ end to the 3′ end, did not span the junction and did not amply the target 200 bp circRNA sequences. The primer sequences are shown in Supplementary Tables S1 and S2. These primers were synthesized by the GENEray Biotechnology Company (Shanghai, China).
Circular back splicing junction determination
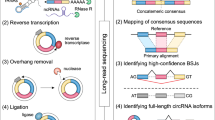
To determine the exact positions of BSJs supported by the reads spanning them, pairs of anchors were generated by extracting the k-mers (default k = 20) from both ends of the unmapped reads. These k-mers were then aligned separately by Bowtie2 [25] to find anchor pairs that align in a circular way. The alignment process allows no more than three mismatches and no gaps by default. After aligning the anchor pairs, an extension process followed, in which the aligned anchor pairs were extended against the reference sequences until the complete read was aligned. This strategy was similar to Memczak’s [10] method but with important improvements (Fig. 1). First, in addition to the BSJ overlapping reads (Fig. 2a), we also included discordant BSJ spanning reads, and these reads enclosed but did not overlap the BSJs (Fig. 2b). The BSJ overlapping reads were used to determine the exact BSJ positions, and the BSJ spanning reads helped to estimate the significance of the putative circRNA. Second, we considered the linear junctions caused by the introns during linear splicing, and the reads mapped to the linear splicing junctions were removed. Finally, we designed a scoring strategy to determine the precise positions of the BSJs considering both the non-uniquely mapped reads and the paired-end reads (Supplementary Material).

Overview of the CircRNAFisher pipeline

Types of read supporting circRNAs. a BSJ overlapping reads. b Discordant BSJ spanning reads. The paired reads enclose the back-splice junction, but their alignments are in reverse orientation
Filtering by mapping quality
To address the non-unique mapping problem and further reduce the false discovery rate (FDR) of the circRNA identification, CircRNAFisher assigned a mapping quality score to each read-circRNA alignment, and circRNAs with inferior mapping quality scores were discarded. For a read pair (read1, read2) that supports a circular BSJ, all the reasonable combinations of their alignments (circular alignments and linear alignments) were considered. For a particular alignment i, SUM_BASE_Q(i) was defined as the sum of the base quality scores at the mismatched bases from each read’s alignment of that read pair. Then, the mapping quality score for the read-circRNA alignment k was defined as:
For a particular circRNA, we calculated its mean mapping quality score by averaging all the corresponding mapping quality scores. We use 10 as the averaged mapping quality score threshold by default, and circRNAs below this threshold were discarded.
Estimating expression significance
To obtain P-values quantifying the expression significance of the circRNAs, we combined the BSJ overlapping reads with the discordant BSJ spanning reads and evaluated the enrichment of such a combination within a small window compared to the background distribution. The putative BSJ overlapping reads and the discordant BSJ spanning reads formed paired peaks spanning the putative BSJs (Supplementary Figure S2), the height of which was directly proportional to the expression level of corresponding circRNAs. Inspired by John et al’s work [26], we gauged the enrichment of the BSJ overlapping reads and the discordant BSJ spanning reads in a 500 bp window (250 bp at each end of BSJ) over a binomial background distribution. We supposed that n observed reads were mapped to the small window and N total reads were mapped to the corresponding chromosome, which was considered the background window (N ≥ n). Each read in the background window was considered as an ‘experiment’, and reads that fell within the small window were considered a “success”. Assuming that the reads were randomly distributed along the background window, the probability of a “success” was therefore p = 500/(length of background window). According to the binomial distribution, the expected number of reads falling within the smaller window is μ = Np, and the standard deviation of this expected value is \(\sigma = \sqrt {Np(1 - p)}\). We then used this model to calculate the P-value for each pair of peaks and corrected for multiple testing using the Bonferroni correction procedure.
RNA lariat filtering
An RNA lariat is another circle product created during RNA splicing. It contains intronic sequences and a 2′–5′ phosphodiester linkage at the branch point [27]. To lower the risk of identifying lariats as exonic circRNAs, we discarded candidate circRNAs for which >80% of the supporting reads have mismatches around branch point or located in the introns. This procedure is optional in the pipeline. It can be left out when users want to detect both intronic and exonic circRNAs.
Results
Overview of the CircRNAFisher pipeline
CircRNAFisher consists of two main phases: (1) identification of candidate BSJ reads and (2) filtering the candidate circRNAs through a series of statistical filters (Fig. 1). CircRNAFisher starts by discarding the reads that are entirely and contiguously aligned to the reference genome or transcriptomes (e.g., hg19 or UCSC known genes) and retains the candidate circRNA reads (referred to as unmapped reads) for downstream analyses. The BSJs supported by the unmapped reads are then determined by a custom realignment and filtering process (Materials and methods). After the candidate BSJs were found during the first phase, two statistical filters were employed to reduce the FDR of the identified circRNAs. Table 1 displays some essential elements of the circRNAs determined by CircRNAFisher, including the positions of the circRNAs on the genome, the number of BSJ-overlapping reads and discordant BSJ-spanning reads, and the inferred internal structure of the predicted circRNAs according to the UCSC annotated transcripts.
CircRNAFisher identifies circRNAs at a prescribed expression significant level
A unique feature of CircRNAFisher is to assign P-values to the identified circRNAs, which allows users to control the number of identified circRNAs at a prescribed expression significance level. The number of identified circRNAs under different P-value thresholds using A549 cell line data is shown in Fig. 3a, which shows that the P-value has a significant impact on the number of identified circRNAs. To gain a better understanding of the impact of the sequencing depth on the identification of circRNAs, we simulated a series of RNA-seq libraries by downsampling the A549 cell line data. We then identified circRNAs from these simulated libraries using CircRNAFisher (Fig. 3b). As expected, CircRNAFisher identified more circRNAs as the library size increased. Importantly, the number of identified circRNAs reached a plateau at ~100 million reads for all the tested P-value cutoffs, suggesting that the sequencing depth has a limited impact on circRNA identification beyond 100 million reads.

Identification of circRNAs at a prescribed expression significant level. a The number of identified circRNAs at different P-value levels. The x-axis represents the multi-testing corrected P-value, and the y-axis represents the number of identified circRNAs. The vertical dashed line represents the default P-value cut-off (0.01). b CircRNA coverage vs. different sequencing depth at different P-value conditions. The x-axis in millions represents the sequencing depth simulated by selecting the reads randomly. The y-axis is the number of the identified circRNAs under different sequencing depths and different P-value conditions. The horizontal dashed lines show the coverage of the detected circRNAs
CircRNAFisher effectively detects circRNAs in A549, MCF7, and BJ cell lines
We next utilized CircRNAFisher to identify circRNAs using three published RNA-seq datasets (A549, MCF7, and BJ cell lines). Previous studies identified circRNAs using these data sets, which allowed us to systematically compare the performance of CircRNAFisher with existing methods. CircRNAFisher identified 3676 candidate circRNAs in the A549, 670 in MCF7 cells and 3026 in the BJ cell lines (Fig. 4a). Although each pair of cell line demonstrated significant overlap (415 between A547 and MCF7, 1488 between A549 and BJ, and 376 between MCF7 and BJ), the three cell lines only shared 330 circRNAs, which represented 6.10% of all the identified circRNAs (5423) in the three cell lines. This result confirmed previous reports that circRNAs are regulated in a cell-type specific manner [28]. To gain further insight into the robustness of the identified circRNAs, we compared the CircRNAFisher results with previously reported circRNAs in the same three cell lines. Among the 5423 distinct circRNAs identified by CircRNAFisher in the three cell lines, approximately 61% were also be reported by other studies (Fig. 4b), including Jeck et al. [11], Salzman et al. [9], and Memczak et al. [10] Moreover, among the 5423 distinct circRNAs identified by CircRNAFisher, 4182 had consistent exon boundaries according to the UCSC annotation, and only 1241 had unannotated exon boundaries. Interestingly, CircRNAFisher did not identify 604 high confidence circRNAs, which were previously reported by two or more different methods. To explore the possible mechanisms, we realigned the initial unmapped reads to the BSJs of the 604 circRNAs and found that there were ~18% BSJs not supported by any reads, suggesting that these are unlikely to be true circRNAs. Taken together, these results suggest that CircRNAFisher is a robust method for circRNA identification.

CircRNAs identified by CircRNAFisher. a The number of circRNAs identified by CircRNAFisher in the A549, MCF7, and BJ cell lines. b Comparison of the circRNAs identified by CircRNAFisher with previously reported circRNAs
Discordant BSJ spanning reads improve circRNA detection
A key feature of CircRNAFisher is to utilize the discordant BSJ spanning reads for circRNA identification (Fig. 2). To gain a better understanding of the relationship between the BSJ overlapping reads and the BSJ spanning reads, we compared the number of reads in each type for the 3676 circRNAs identified in A549 cell line. For each detected circRNA, we plotted the number of BSJ overlapping reads vs. the number of discordant BSJ spanning reads (Fig. 5a). The two types of reads demonstrated a highly significant Pearson correlation coefficient (0.6055, P-value < 2.2e−16), indicating that the number of BSJ overlapping reads and discordant BSJ spanning reads were highly correlated. These data suggest that including discordant BSJ spanning reads substantially increases the number of reads contributing to the detection of circRNAs; this increase may lead to the improved detection of circRNAs. To test this idea, we compared CircRNAFisher’s performance with or without the discordant BSJ spanning reads using the A549 RNAseq data (Fig. 5b). CircRNAFisher reported 2911 circRNAs when only considering the BSJ overlapping reads, which was significantly lower than the number of circRNAs identified using both the BSJ overlapping reads and the BSJ spanning reads (3676). This result clearly demonstrated the advantage of utilizing the discordant BSJ spanning reads and suggested that CircRNAFisher might command improved sensitivity and specificity compared to methods only using the BSJ overlapping reads for circRNA detection.

The relationship between the BSJ overlapping reads and the discordant BSJ spanning reads. a Log–log plot showing the correlation of the number of BSJ overlapping reads with the number of discordant BSJ spanning reads for circRNAs identified in the A549 cells. b Bar chart showing the number of circRNAs detected with or without the discordant BSJ spanning reads
Performance comparison with the existing methods
To further evaluate CircRNAFisher, we compared its performance with two publically available tools, CIRI and find_circ. We first generated simulation datasets with different coverages of the circular transcripts using CIRI-simulator, which is part of the CIRI program. We then applied each of the tested programs to predict the circRNAs using the simulated datasets. The FDR and sensitivity for the three methods are shown in Table 2. Compared with CIRI, CircRNAFisher demonstrated improved FDR and comparable sensitivity. Furthermore, CircRNAFisher demonstrated improved sensitivity and comparable FDR when compared with find_circ. Interestingly, CircRNAFisher demonstrated a significant improvement in sensitivity using data with low coverage, suggesting that CircRNAFisher excelled in circRNA detection when only limited sequencing data was available.
Next, we compared CIRI and CircRNAFisher using a HeLa dataset treated with RNase R (SRR1636985), which was used to evaluate CIRI. CircRNAFisher and CIRI detected 2727 and 3551 candidate circRNAs, respectively, with a large fraction (2375) in common (Fig. 6a). Interestingly, CircRNAFisher detected fewer circRNAs than CIRI in both the RNase R positive and RNase R negative samples, suggesting that CircRNAFisher was more conservative in calling candidate circRNAs (Fig. 6b). We further compared the three methods using an A549 RNA-seq dataset from the ENCODE database. CircRNAFisher detected the vast majority of the circRNAs reported by CIRI and find_circ (Fig. 6c). Moreover, CircRNAFisher reported a large number of candidate circRNAs that were found by neither CIRI nor find_circ, suggesting that CircRNAFisher was much more sensitive. CircRNAFisher identified more circRNAs using the ENCODE dataset compared to the HeLa dataset, which was expected considering that the ENCODE dataset has more reads than the HeLa dataset (89 848 834 reads vs. 35 685 310), and consequently more BSJ overlapping reads (147 499 vs. 19 139).

Performance comparison of CircRNAFisher, CIRI and find_circ. a Venn diagram showing the circRNAs identified by CircRNAFisher and CIRI using the RNase R+ HeLa data (SRR1636985). b Venn diagram showing the circRNAs identified by CircRNAFisher and CIRI using the RNase R- HeLa data (SRR1637089). c Venn diagram showing the circRNAs identified by CircRNAFisher, CIRI and find_circ using an ENCODE dataset
To further verify the candidate circRNAs predicted by the three algorithms, we performed PCR assays, which is the gold standard for circRNAs verification, in the HeLa cells and A549 cells. To gain a systematic evaluation, we first ranked candidate circRNAs by their prediction scores. For CircRNAFisher and CIRI, both the A549 and HeLa datasets were used. The candidate circRNAs were then divided into 5 levels according to their ranks, and one representative circRNA was randomly selected from each level for experimental validation, resulting in 10 circRNA for both algorithms. For the candidates from the find_circ algorithm, we divided the candidates in the A549 dataset into 10 classes and randomly selected 1 candidate in each class. In summary, 10 random candidate circRNAs were selected for each algorithm for the PCR validations (Table 3). The validation results (Fig. 7, Supplementary Figure S3) demonstrated that the vast majority of the randomly selected targets were verified (8 out of 10 for CIRI and find_circ and 9 out of 10 for CircRNAFisher). Importantly, CircRNAFisher demonstrated comparable validation rates with CIRI and find_circ, suggesting that the more sensitive CircRNAFisher identified many more potential circRNAs.

PCR validation of 30 circRNA candidates identified by CircRNAFisher, CIRI and find_circ. a Validation results of CIRI. b Validation results of Find_circ. c Validation results of CircRNAFisher
Genomic features of the identified CircRNAs
We next investigated the genomic features of the detected circRNAs. To enable a robust analysis, we focused on the 1625 selected circRNAs that were highly expressed and contained consistent BSJs according to UCSC gene annotation. The vast majority of the circular spliced exons were located in the middle of the UCSC genes, while only a fraction of the circularly spliced exons was of the first or the last exons of the corresponding genes (Fig. 8a). Interestingly, the number of exons in the circRNAs appeared to follow a negative binomial distribution, with a P-value of 9.4e−14 by the Kolmogorov–Smirnov test [29], suggesting that multiple exons were the dominant form of circRNAs (Fig. 8b). Moreover, the lengths of these exons in multi-exon circRNAs were much shorter than the exons from the single-exon circRNAs (Fig. 8c). Similar to previously reported results [16], there were, on average, 4 Alu elements in the flanking convergent (Fig. 8d) or divergent introns (Fig. 8e). Finally, we confirmed that the flanking introns of circRNAs were much longer than the randomly selected introns (Fig. 8f), which was consistent with previously reported results [11, 16]

Genomic features of the circRNAs. a A majority of the circular spliced exons were located in the middle of genes. b The distribution of the number of circular spliced exons in the circRNAs. c The distribution of the exon length (y-axis) from the circRNAs with different numbers of exons (x-axis). d The number of Alu elements in the flanking convergent introns. e The number of Alu elements in the flanking divergent introns. f The length distribution of the flanking introns
.
Discussion
The increasing interest in circRNAs has inspired the development of numerous circRNA identification algorithms, and a thorough comparison of 11 circRNA identification algorithms was recently published [22]. The authors analyzed the respective algorithms and proposed that additional care should be taken in the validation of long circRNAs because long circRNAs are more prone to unspecific RNAseR decay. The general recommendation provided in their paper is to combine two or more prediction algorithms to compensate the respective weaknesses and minimize the overall false positives.
There are several novel improvements of CircRNAFisher over the existing methods. First, CircRNAFisher calculates the P-values for the identified circRNAs, which provides a statistically principled metric to select circRNAs for downstream validation and biological function analysis. Second, CircRNAFisher is the first circRNA detection method that combines the BSJ overlapping reads and the discordant BSJ spanning reads. We would like to point out that the discordant BSJ spanning reads alone did not determine the exact BSJ sites, therefore they did not improve the variety of the circRNAs. Instead, by assisting the BSJ overlapping reads, this approach led to an improved sensitivity for circRNA identification over the existing methods, which typically only consider the BSJ overlapping reads. This capability is especially important for the detection of circRNAs when only limited sequencing data is available. Finally, the CircRNAFisher pipeline consisted of a series of tunable statistical filters, which allowed for the flexible detection of circRNAs.
We performed a thorough evaluation of CircRNAFisher by comparing it with two established methods using simulated data, RNA-Seq data from three different cell lines, and RNase R treated/untreated data. Furthermore, we experimentally validated selected circRNAs, including those with low prediction scores, to enable a relatively unbiased evaluation. This setup allowed for a systematic evaluation of CircRNAFisher. The evaluation results demonstrated that CircRNAFisher robustly identified circRNAs (Fig. 3b) and identified a significantly higher number of expressed circRNAs at a relatively low sequencing depth (Fig. 4b). In summary, CircRNAFisher represents an improved circRNA identification pipeline that may be of great value to the circRNA research community.
Software availability
CircRNAFisher is freely available at https://github.com/duolinwang/CircRNAFisher.
References
Hsu MT, Kung HJ, Davidson N. An electron microscope study of Sindbis virus RNA. Cold Spring Harb Symp Quant Biol. 1974;38:943–50.
Hewlett MJ, Pettersson RF, Baltimore D. Circular forms of Uukuniemi virion RNA: an electron microscopic study. J Virol. 1977;21:1085–93.
Hsu MT, Coca-Prados M. Electron microscopic evidence for the circular form of RNA in the cytoplasm of eukaryotic cells. Nature. 1979;280:339–40.
Cocquerelle C, Daubersies P, Majerus MA, Kerckaert JP, Bailleul B. Splicing with inverted order of exons occurs proximal to large introns. EMBO J. 1992;11:1095–8.
Saad FA, Vitiello L, Merlini L, Mostacciuolo ML, Oliviero S, Danieli GA. A 3′ consensus splice mutation in the human dystrophin gene detected by a screening for intra-exonic deletions. Hum Mol Genet. 1992;1:345–6.
Bailleul B. During in vivo maturation of eukaryotic nuclear mRNA, splicing yields excised exon circles. Nucleic Acids Res. 1996;24:1015–9.
Zaphiropoulos PG. Circular RNAs from transcripts of the rat cytochrome P450 2C24 gene: correlation with exon skipping. Proc Natl Acad Sci USA. 1996;93:6536–41.
Danan M, Schwartz S, Edelheit S, Sorek R. Transcriptome-wide discovery of circular RNAs in Archaea. Nucleic Acids Res. 2011;40:3131–42. gkr1009
Salzman J, Gawad C, Wang PL, Lacayo N, Brown PO. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS ONE. 2012;7:e30733.
Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333–8.
Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J, et al. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA. 2013;19:141–57.
Glazar P, Papavasileiou P, Rajewsky N. circBase: a database for circular RNAs. RNA. 2014;20:1666–70.
Burd CE, Jeck WR, Liu Y, Sanoff HK, Wang Z, Sharpless NE. Expression of linear and novel circular forms of an INK4/ARF-associated non-coding RNA correlates with atherosclerosis risk. PLoS Genet. 2010;6:e1001233.
Gao Y, Wang J, Zhao F. CIRI: an efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol. 2015;16:4.
Gao Y, Zhang J, Zhao F. Circular RNA identification based on multiple seed matching. Brief Bioinf. (2017) https://doi.org/10.1093/bib/bbx014.
Zhang XO, Wang HB, Zhang Y, Lu X, Chen LL, Yang L. Complementary sequence-mediated exon circularization. Cell. 2014;159:134–47.
Kim D, Salzberg SL. TopHat-Fusion: an algorithm for discovery of novel fusion transcripts. Genome Biol. 2011;12:R72.
Danan M, Schwartz S, Edelheit S, Sorek R. Transcriptome-wide discovery of circular RNAs in Archaea. Nucleic Acids Res. 2012;40:3131–42.
Jeck WR, Sharpless NE. Detecting and characterizing circular RNAs. Nat Biotechnol. 2014;32:453–61.
Wang K, Singh D, Zeng Z, Coleman SJ, Huang Y, Savich GL, et al. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 2010;38:e178.
Gao Y, Zhao F. Computational strategies for exploring circular RNAs. Trends Genet. 2018;34:389–400.
Hansen TB. Improved circRNA identification by combining prediction algorithms. Front Cell Dev Biol. 2018;6:20.
Consortium EP. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 2004;306:636–40.
Karolchik D, Baertsch R, Diekhans M, Furey TS, Hinrichs A, Lu YT, et al. The UCSC Genome Browser Database. Nucleic Acids Res. 2003;31:51–4.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9.
John S, Sabo PJ, Thurman RE, Sung MH, Biddie SC, Johnson TA, et al. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat Genet. 2011;43:264–8.
Keller W. The RNA lariat: a new ring to the splicing of mRNA precursors. Cell. 1984;39:423–5.
Salzman J, Chen RE, Olsen MN, Wang PL, Brown PO. Cell-type specific features of circular RNA expression. PLoS Genet. 2013;9:e1003777.
Chakravarti IM, Laha RG. Handbook of Methods of Applied Statistics. New York: Wiley; 1967.
Acknowledgements
This work was supported in part by the National Natural Science Foundation of China (NSFC) grant 31671380.
Author contributions
P.W. and Y.-C.L. conceived and designed the experiments; G.-Y.J., D.-l.W., and M.-Z.X. performed the experiments, analyzed the data and wrote the paper; and Y.-W.L., Y.-C.P., Y.-Q.Y., and J.-M.X. discussed and edited the manuscript. All the authors have read and approved the final manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Jia, Gy., Wang, Dl., Xue, Mz. et al. CircRNAFisher: a systematic computational approach for de novo circular RNA identification. Acta Pharmacol Sin 40, 55–63 (2019). https://doi.org/10.1038/s41401-018-0063-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-018-0063-1
Keywords
This article is cited by
-
New insight into circRNAs: characterization, strategies, and biomedical applications
Experimental Hematology & Oncology (2023)
-
A novel CircRNA Circ_0001722 regulates proliferation and invasion of osteosarcoma cells through targeting miR-204-5p/RUNX2 axis
Journal of Cancer Research and Clinical Oncology (2023)
-
Circular RNA circ_001422 promotes the progression and metastasis of osteosarcoma via the miR-195-5p/FGF2/PI3K/Akt axis
Journal of Experimental & Clinical Cancer Research (2021)
-
Circular RNAs and their role in renal cell carcinoma: a current perspective
Cancer Cell International (2021)
