
Abstract
There is an urgent need to develop effective therapies for ischemic stroke, but the complicated pathological processes after ischemia make doing so difficult. In the current study, we identified a novel diaryl acylhydrazone derivative, A11, which has multiple neuroprotective properties in ischemic stroke models. First, A11 was demonstrated to induce neuroprotection against ischemic injury in a dose-dependent manner (from 0.3 to 3 μM) in three in vitro experimental ischemic stroke models: oxygen glucose deprivation (OGD), hydrogen peroxide, and glutamate-stimulated neuronal cell injury models. Moreover, A11 was able to potently alleviate three critical pathological changes, apoptosis, oxidative stress, and mitochondrial dysfunction, following ischemic insult in neuronal cells. Further analysis revealed that A11 upregulated the phosphorylation levels of protein kinase B (AKT) and extracellular signal-regulated kinase (ERK) in OGD-exposed neuronal cells, suggesting joint activation of the phosphoinositide 3-kinase (PI3K)/AKT and mitogen-activated protein kinase (MEK)/ERK pathways. In rats with middle cerebral artery occlusion, single-dose administration of A11 (3 mg/kg per day, i.v.) at the onset of reperfusion significantly reduced the infarct volumes and ameliorated neurological deficits. Our study, for the first time, reports the anti-ischemic effect of diaryl acylhydrazone chemical entities, especially A11, which acts on multiple ischemia-associated pathological processes. Our results may provide new clues for the development of an effective therapeutic agent for ischemic stroke.
Similar content being viewed by others


Introduction
Stroke is one of the leading causes of long-term disability and morbidity worldwide [1]. Currently, recombinant tissue plasminogen activator (rt-PA) is the only drug approved by the US Food and Drug Administration for the treatment of ischemic stroke. Although rt-PA can provide acute recanalization and improve functional outcome, the use of rt-PA is limited due to its short time window and risk of hemorrhage [2]. By contrast, neuroprotective agents are expected to be an increasingly used alternative approach to better combat the progression of the pathological conditions of ischemic stroke [3]. Nevertheless, given the innate complexity of the pathological cascade of events involved in the pathogenesis of ischemic stroke, including oxidative stress, apoptosis, mitochondrial dysfunction, and so on [4,5,6,7], the currently available neuroprotective agents have been demonstrated to have only limited clinical efficacies in the treatment of ischemic stroke. Therefore, it is important to explore active chemical compounds that can simultaneously affect the multiple pathological pathways that are activated following ischemic insult [8]. In fact, an increasing number of multifunctional agents have already been demonstrated to exhibit promising efficacies against ischemic stroke [9,10,11,12].
Phenotypic screening is one of the most successful strategies for discovering drugs that are effective against multifactorial diseases [13], because it takes place within living cells and, therefore, is able to evaluate the breathtaking complexity of in vivo biology [14]. Phenotypic screening has relative high-throughput compared to animal-based drug screening and is also an effective discovery approach for multifunctional agents [15, 16]. Therefore, phenotypic screening allows drug developers to be confident that candidate molecules will have the desired therapeutic efficacy [17, 18].
Based on the background described above, cell-based phenotypic screening was employed to screen our in-house compound library with the aim of discovering novel chemical compounds that can be used to treat ischemia/reperfusion injury. To our delight, a series of diaryl acylhydrazone derivatives was identified with significant neuroprotective effects against oxygen glucose deprivation (OGD) injury (Supplementary Table 1). In particular, diaryl acylhydrazone derivative A11 (Fig. 1) was shown to be one of the best candidates identified through this screening (Supplementary Table 1).

Chemical structure of A11
In the present study, the in vitro anti-ischemic effects of A11 were systematically investigated in neuronal cells stressed with OGD, hydrogen peroxide (H2O2) or glutamate, which are three toxic stimuli that mimic different ischemic insults [19,20,21,22]. Moreover, the multifunctional effects of A11 against apoptosis, oxidative stress, and mitochondrial dysfunction, three critical pathological events following ischemia [4,5,6,7], were assessed in OGD-insulted SH-SY5Y cells. Furthermore, the effects of A11 on phosphorylation of protein kinase B (AKT) and extracellular signal-regulated kinase (ERK) were evaluated in OGD-insulted SH-SY5Y cells to investigate the potential molecular mechanisms of A11. In addition, the in vivo anti-ischemic effect of A11 was studied in a middle cerebral artery occlusion (MCAO) rat model to further evaluate its anti-ischemic effect.
Materials and methods
Cell culture
Human neuroblastoma SH-SY5Y cells were purchased from ATCC (2266) and cultured in a mixture of Eagle’s minimum essential medium and Ham’s F12 medium (MEM/F12, Life Tech, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS, Gibco, Grand Island, NY, USA), 100 U/mL penicillin and 100 µg/mL streptomycin at 37 °C in a humidified incubator with 5% CO2. HT22 mouse hippocampal neuronal cells were obtained from Jennio Biotechnology Co., Ltd. (Guangzhou, China) and cultured in 10% FBS, 100 U/mL penicillin and 100 µg/mL streptomycin supplemented Dulbecco’s modified Eagle’s medium (DMEM, Life Tech, Grand Island, NY, USA) in a humidified 5% CO2 incubator at 37 °C. Cells were grown to 70–80% confluency before experiments.
Drug treatment
All of the diaryl acylhydrazone derivatives, including A11, were dissolved in dimethyl sulfoxide (DMSO) at 10 mM as a stock solution and diluted with cell culture medium. Cells were treated with the indicated concentrations of A11 immediately before OGD exposure or 2 h before H2O2 or glutamate insult. To inhibit the PI3K/AKT or MEK/ERK signaling pathway, SH-SY5Y cells were treated with 10 μM LY294002 (Selleck, Houston, TX, USA) or 5 μM U0126 (Merck, Darmstadt, Germany), respectively, followed by treatment with 1 μM A11. For the in vivo study, A11 was dissolved in saturated Na2CO3 and PEG400 (each at a concentration of 0.075%) and then further diluted to a final concentration of 0.3 mg/mL with saline for intravenous administration. A11 was administered intravenously at the onset of reperfusion immediately after the monofilament was withdrawn, and the neurological deficits and infarct volumes were evaluated at 24 h of reperfusion.
OGD, H2O2, and glutamate insults
SH-SY5Y cells were seeded into 96-well plates at a density of 3 × 105 cells/mL and used on the second day after seeding. To induce ischemia/reperfusion injury, SH-SY5Y cells were washed with glucose-free Earle’s balanced salt solution buffer (117.2 mM NaCl, 5.3 mM KCl, 0.8 mM MgSO4, 26.2 mM NaHCO3, 1.8 mM CaCl2, 1.0 mM NaH2PO4·2H2O) and incubated in glucose and serum free DMEM (Gibco, Grand Island, NY) containing different concentrations of A11 in a hypoxia chamber (H35 hypoxystation, Don Whitley Scientific, Shipley, West Yorkshire, UK) containing a mixture of 85% N2/10% H2/5% CO2 at 37 °C for 2 h. Then, SH-SY5Y cells were returned to normal culture conditions for 24 h of reoxygenation. Control cells without OGD exposure were maintained in complete medium (as described above) under normoxia at 37 °C in 95% air/5% CO2. To mimic oxidative stress, SH-SY5Y cells were transferred to fresh media and pretreated with A11 for 2 h; then, H2O2 freshly prepared from an 8.8 M stock solution was added to the medium to a final concentration at 100 μM.
To mimic glutamate-caused neuronal injury, HT22 cells were seeded into 96-well plates (0.5 × 104 cells per well) and allowed to attach for 12 h. Cells were incubated with the indicated concentrations of A11 for 2 h, followed by 24 h of exposure to l-glutamic acid hydrochloride (Sigma-Aldrich, St. Louis, MO, USA) at a final concentration of 10 mM.
Cell viability assay
Cell viability was evaluated by morphological observation under a microscope (Nikon, TE200, Melville, NY, USA) and with a 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT, BioDee Biotech, Beijing, China) assay. After different treatments, MTT was added to the culture medium at a final concentration of 0.5 mg/mL and incubated at 37 °C for 3 h. Following removal of the MTT solution and addition of 100 μL of DMSO to dissolve the formazan crystals, the absorbance of each well at 490 nm was recorded on a DTX 800 multimode detector (Beckman Coulter, Fullerton, CA, USA). The results are shown as the percentage of the control.
Lactate dehydrogenase assay
Lactate dehydrogenase (LDH) is a cytoplasmic enzyme that is retained by viable cells with intact plasma membranes and released into the culture medium when cell membranes are damaged. Supernatants of media were collected 24 h after OGD insult and centrifuged at 300 × g for 10 min to pellet the debris. Supernatants of media were then evaluated by an LDH assay, which was performed using an LDH kit based on the manufacturer’s instructions (Nanjing Jiancheng, Nanjing, China). The absorbance of each well was measured using a DTX 800 multimode detector (Beckman Coulter, Fullerton, CA, USA) at 450 nm.
Measurement of the cellular ATP levels
Cellular ATP was measured using a bioluminescent ATP detection kit (Promega, Madison, WI, USA). In brief, cells were placed at room temperature for 30 min and then lysed by adding 100 μL of an ATP-releasing reagent. The lysates were incubated with the luciferin substrate and luciferase enzyme in the dark for 10 min, and bioluminescence was measured using a PerkinElmer microplate reader. The bioluminescence intensity was normalized to that of the control group and is presented as the relative ATP level.
Flow cytometry analysis of ROS production
SH-SY5Y cells were seeded in six-well plates and stimulated with OGD for 2 h, followed by 24 h of reoxygenation under normal culture conditions. After cultivation, cells were trypsinized and collected by centrifugation at 300 × g for 5 min. Cells were washed with cold PBS and then incubated in darkness with 10 μM DCFH-DA (Sigma-Aldrich, St. Louis, MO, USA) (to measure intracellular ROS levels) in NaCl-medium (132.0 mM NaCl, 4.0 mM KCl, 1.0 mM CaCl2, 1.4 mM MgCl2, 1.2 mM NaH2PO4·2H2O, 6.0 mM glucose, 10.0 mM HEPES, pH 7.4) for 30 min at 37 °C. Cells were then subjected to fluorescence-activated cell sorting (FACS) analysis (BD Biosciences, San Jose, CA, USA).
Analysis of apoptosis
Fluorescently labeled Annexin V (Annexin V-FITC) and propidium iodide (PI) (Rainbio, Shanghai, China) were used to stain apoptotic cells. The early stage of cell death is identified by the Annexin V, while PI staining is an indicator of necrosis or late-stage apoptosis. Briefly, cells were trypsinized and collected by centrifugation at 300 × g for 5 min. The cells were washed with cold PBS and then resuspended in 100 μL of binding buffer. Each cell suspension was returned to its respective FACS tube along with 5 μL of Annexin V and 5 μL of PI and incubated for 10 min at room temperature in the dark. Another 400 μL of binding buffer was added to the samples, and then, the samples were analyzed by flow cytometry (BD Biosciences, San Jose, CA, USA) within 1 h.
Western blotting analysis
Whole cell extracts were prepared by lysing cells on ice for 20 min with RIPA lysis buffer (50 mM Tris-HCl, 150 mM NaCl, 0.5% sodium deoxycholate, 1% Triton X-100, 0.1% sodium dodecyl sulfate) supplemented with protease inhibitors (1 mM PMSF, 1% P8340) and phosphatase inhibitors (1 mM NaF, 1 mM Na3VO4). The nuclear fraction was extracted with the NE-PER reagents (Thermo Scientific, Rockford, IL, USA) according to the manufacturer’s instructions. The protein concentrations were determined by using a bicinchoninic acid protein assay kit (Thermo Scientific, Rockford, IL, USA). Equal amounts of protein were separated by electrophoresis on SDS-polyacrylamide gels and transferred to nitrocellulose membranes (Whatman, Maidstone, Kent, UK). The membranes were blocked with 5% skimmed milk and incubated with the appropriate primary antibodies overnight at 4 °C. The following antibodies were from Cell Signaling Technology (Beverly, MA, USA): anti-p-ERK1/2 (Thr202/Tyr204, 1:1000), anti-ERK1/2 (1:1000), anti-p-AKT (Ser 473, 1:2000), anti-AKT (1:2000), anti-p53 (1:500), anti-Lamin B (1:1000), anti-Caspase-3 (1:2000), anti-cleaved Caspase-3 (Asp175, 1:500), and anti-β-actin (1:10000) was from Sigma-Aldrich (St.Louis, MO, USA). The membranes were washed with T-TBS, followed by incubation with horseradish peroxidase-conjugated secondary antibodies (1:5000; Kangchen Biotechnology, Shanghai, China); then, the membranes were developed by using an enhanced chemiluminescence reagent (Millipore Corporation, Bedford, MA, USA). Densitometric analysis of each band was performed by using Image J software. Phosphorylated proteins were normalized against the total amount of the respective proteins. Unless otherwise stated, β-actin was used as the loading control.
Middle cerebral artery occlusion
Adult male Sprague–Dawley rats weighing 230–300 g were provided by the Laboratory Animal Center, Chinese Academy of Sciences (Shanghai, China). All experimental procedures followed the National Institutes of Health Guide for the Care and Use of Laboratory Animals as well as the guidelines of the Animal Care and Use Committee of Shanghai Institute of Materia Medica, Chinese Academy of Sciences. Rats were housed on a 12/12-h day and night cycle and had free access to food and water. They were randomly assigned to different groups before surgery. Focal cerebral ischemia was induced by the intraluminal MCAO as described previously with minor modifications [23]. In brief, rats were anesthetized with chloral hydrate (400 mg/kg, i.p.); then, a 4-0 surgical nylon monofilament nylon suture (Beijing Sunbio Biotech Co. Ltd, Beijing, China) was introduced into the left internal carotid artery through the external carotid stump and advanced approximately 20 mm past the carotid bifurcation up the internal carotid. The intraluminal suture blocked the origin of the middle cerebral artery (MCA) and induced transient MCAO injury. The suture was left in place for 2 h and then withdrawn, resulting in reperfusion of the MCA. Sham-operated rats were manipulated with the same procedure without inserting the filament. The rectal temperature of the animals was maintained at 37 ± 0.5 °C with a heating pad throughout the surgical procedure. Rats were then returned to their home cages after surgery (room temperature was constantly maintained at 24–26 °C).
Assessment of neurological function
Neurological deficits were measured at 24 h after the onset of reperfusion according to the modified neurological severity scores (mNSS). The mNSS was used to grade neurological deficits, including motor, visual, and tactile responses; proprioception; performance on a balance beam; reflex responses; and abnormal movements (scale: 0–18; normal score, 0; maximal deficits core, 18); thus, the composite score reflects combined motor, sensory, and reflex functions [24]. An experimenter blinded to the experimental groups performed the neurological scores assessment.
Measurement of cerebral infarct volume
Infarct volumes were measured by triphenyltetrazolium chloride (TTC; Sinophar Chemical Reagent Co. Ltd., Shanghai, China) staining. In brief, rats were decapitated, and their brains were rapidly removed and frozen at −80 °C for 5 min. Each brain was cut into six equal coronal sections (2 mm thick per section), and the sections were stained with 1% TTC for 15 min in a 37 °C chamber in the dark and then fixed in a paraformaldehyde solution (4%) for 24 h before digital images were taken. The infarct volume was determined as the slice thickness times the sum of the infarct area in all of the brain slices. The size of the infarct area was calculated by the formula: (area of normal hemisphere−area of non-infarcted ischemic hemisphere)/area of normal hemisphere × 100%.
Statistical analysis
Data are expressed as the mean ± SEM. Differences between groups were analyzed by Student’s t-test or one-way ANOVA followed by Turkey’s tests for multiple comparisons. Statistical significance was established at P < 0.05.
Results
A11 attenuates neuronal death caused by OGD
To evaluate the effect of A11 on ischemic stimuli, SH-SY5Y cells were treated with A11 immediately before 2 h-exposure to OGD, followed by 24 h of reoxygenation. Exposure of SH-SY5Y cells to OGD alone resulted in cell morphological changes featuring neurite disappearance and body shrinkage. By contrast, the morphological damage to SH-SY5Y cells induced by OGD was remarkably attenuated by treatment with 1 μM A11 (Fig. 2a). Consistent with the results of the morphological observation, OGD-induced cell damage was also shown to lead to significant MTT reduction (51.7%, P < 0.001 vs. the control group) (Fig. 2b) and excessive release of LDH (233.6%, P < 0.001 vs. the control group) (Fig. 2c) compared to the control group. However, treatment with A11 (0.3, 1, and 3 μM) exhibited a dose-dependent attenuation of OGD-induced cell viability reduction, with a maximum protection at 3 μM (restored cell viability to 91.5%, P < 0.001 vs. the OGD group) (Fig. 2b). Similarly, treatment with 1 μM and 3 μM of A11 significantly reduced the LDH levels in the culture media to 186.7% (P < 0.01 vs. the OGD group) and 194.5% (P < 0.05 vs. the OGD group), respectively, in OGD-exposed SH-SY5Y cells (Fig. 2c).

A11 alleviated the SH-SY5Y cell damage induced by OGD exposure. a Phase-contrast micrographs (10×) of cells subjected to different treatments. Scale bar = 100 μm. b SH-SYSY cells were treated with 0.3–3 μM A11 immediately before exposure to OGD. c The LDH levels in the culture media after different treatments were assayed. The results represent the mean ± SEM of three independent experiments. ###P < 0.001 vs. the control group, *P < 0.05, **P < 0.01, ***P < 0.001 vs. the OGD group
A11 attenuates oxidative stress-induced neuronal death
To further evaluate the in vitro anti-ischemia effects of A11, H2O2 and glutamate injured neuronal cells were used, both of which are commonly used as in vitro models of ischemic stroke [21, 22]. We assessed the effects of A11 on oxidative insults by exposing cells to an exogenous source of free radicals, H2O2. Cell viability was greatly reduced when exposed to 100 μM H2O2 (57.3%, P < 0.001 vs. the control group). By contrast, treatment with A11 (0.3, 1, and 3 μM) exhibited a significant attenuation of H2O2-induced cell viability reduction, with a maximum protection at 3 μM (85.7%, P < 0.01 vs. the H2O2 group) (Fig. 3a). Glutamate evokes oxidative death in HT22 cells and is widely used to study non-receptor-mediated oxidative glutamate toxicity [22, 25, 26]. Similarly, A11 at 1 μM and 3 μM (77.8% and 76.3%, respectively, P < 0.001 vs. the glutamate group) remarkably attenuated glutamate-induced neurotoxicity in HT22 cells (Fig. 3b).

A11 increased the viability of neuronal cells induced by H2O2 and glutamate. a SH-SYSY cells were pretreated with A11 for 2 h followed by H2O2 injury. b HT22 cells were pretreated with A11 for 2 h followed by glutamate injury. The results represent the mean ± SEM of three independent experiments. ###P < 0.001 vs. the control group, *P < 0.05, **P < 0.01, ***P < 0.001 vs. the toxic stimuli groups
A11 inhibits the apoptotic changes of SH-SY5Y cells exposed to OGD
The anti-apoptotic effect of A11 was investigated by Annexin-PI staining detected with flow cytometry. Normal and viable cells were distributed in the lower left quadrant, while early and late-stage apoptotic cells were distributed in the lower right quadrant and upper right quadrant, respectively. OGD insult induced an obvious increase in cell numbers in both the upper and lower right quadrant (Fig. 4a) and increased the apoptotic rate to 65.6% (P < 0.001) in the OGD group compared to the control group (27.5%) (Fig. 4b). A11 treatment at concentrations of 1 and 3 μM suppressed the apoptotic ratio to 32.8% and 37.1%, respectively (Fig. 4b, P < 0. 01 for both vs. the OGD group).

A11 inhibited apoptosis induced by OGD. a Apoptotic cells were detected by flow cytometry. b The apoptotic rate is represented as a histogram. Western blot analysis of cleaved Caspase-3 (c) and p53 (d) in SH-SY5Y cells treated with 1 μM A11 followed by exposure to OGD and cultured for 24 h after reoxygenation (n = 4). The results represent the mean ± SEM of three to four independent experiments. ##P < 0.01, ###P < 0.001 vs. the control group, **P < 0.01, ***P < 0.001 vs. the OGD group
To further verify the anti-apoptotic effect of A11, two important apoptosis-related proteins, Caspase-3 and p53, were evaluated in OGD-exposed SH-SY5Y cells. OGD exposure markedly induced Caspase-3 cleavage (P < 0.001 vs. the control group) (Fig. 4c) and upregulated the level of p53 in the nucleus (P < 0.01 vs. the control group) (Fig. 4d), while A11 at 1 μM markedly downregulated OGD-induced Caspase-3 cleavage to 54.4% of that of the OGD group (P < 0.001 vs. the OGD group) (Fig. 4c) and the p53 level to 63.3% of that of the OGD group (P < 0.01 vs. the OGD group) (Fig. 4d).
A11 alleviates OGD-induced mitochondrial dysfunction in SH-SY5Y cells
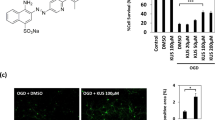
To explore the protective effects of A11 against mitochondrial dysfunction, the intracellular ROS and ATP levels were measured. ROS production in SH-SY5Y cells was assayed by flow cytometry. Production of ROS was increased after OGD stimulation in SH-SY5Y cells (125.6%, P < 0.001 vs. the control group), while A11 at concentrations of 1 and 3 μM significantly reduced the overproduction of ROS almost to basal levels (93.2% and 88.2%, P < 0.05 for both vs. the OGD group) (Fig. 5a).

A11 suppressed ROS production and elevated the ATP level in SH-SY5Y cells. a Changes in ROS production in different treatment groups were measured by assessing the changes in the DCFH-DA fluorescence intensity. b Changes of the intracellular ATP level in different treatment groups. The results represent the mean ± SEM of three independent experiments. ###P < 0.001 vs. the control group, *P < 0.05, ***P < 0.001 vs. the OGD group
ATP is an energy storage and transfer molecule that is obviously reduced following OGD insult [27, 28]. As shown in Fig. 5b, the level of ATP was significantly decreased to 17.5% of that of the control following OGD exposure (P < 0.001 vs. the control group), while treatment with A11 at 0.3, 1, and 3 μM significantly rescued the ATP decrease 48.4%, 71.6% and 66.4% of that of the control (P < 0.05, P < 0.001, P < 0.001 vs. the OGD group, respectively, Fig. 5b).
A11 prevents reduction of ERK phosphorylation and enhances AKT phosphorylation in OGD-exposed SH-SY5Y cells
The potential underlying mechanisms of the protective effects of A11 were further investigated. Previous studies have shown that the MAPK/ERK and PI3K/AKT pathways both play essential roles in regulating cell death after ischemia. Similar to previous reports [29], OGD insult caused a significant reduction of ERK phosphorylation in SH-SY5Y cells (P < 0.05 vs. the control group), while treatment with A11 at 1 and 3 μM (P < 0.05 for both vs. the OGD group) robustly enhanced ERK phosphorylation to 320.8% and 333.2% of that of the OGD group, respectively (Fig. 6a). While ERK phosphorylation was remarkably repressed, the phosphorylation level of AKT was significantly induced after OGD insult in SH-SY5Y cells (P < 0.001 vs. the control group) (Fig. 6b), and incubation with A11 at 0.3, 1, 3 μM further enhanced AKT phosphorylation (P < 0.05 for all vs. the OGD group) to 189.6%, 190.2%, 180.7% of that of the OGD group, respectively (Fig. 6b).

A11 prevented repression of phosphorylation of ERK and enhanced AKT phosphorylation in OGD-exposed SH-SY5Y cells. SH-SY5Y cells were incubated with A11 followed by OGD treatment and cultured for 15 min after reoxygenation. a Expression of ERK phosphorylation in different treatment groups. b Expression of AKT phosphorylation in different treatment groups. The results represent the mean ± SEM of four independent experiments. #P < 0.05, ###P < 0.001 vs. the control group, *P < 0.05 vs. the OGD group
Based on the results shown in Fig. 6, we hypothesized that the neuroprotective effects of A11 might be related to enhanced phosphorylation of ERK and AKT. To test this hypothesis, specific inhibitors of MEK/ERK (U0126) and PI3K/AKT (LY294002) were used. The upregulated phosphorylation levels of ERK and AKT in SH-SY5Y cells were remarkably suppressed by treatment with U0126 and LY294002 (Fig. 7a, b). Consistent with the suppression of ERK and AKT activation, treatment with these inhibitors also significantly inhibited the A11 induced increase in cell viability (Fig. 7c, d).

A11 protected SH-SY5Y cells against OGD insult by activating the MEK/ERK and PI3K/AKT pathways. a Western blot analysis of ERK and p-ERK in hypoxic cells after treatment with MEK inhibitor U0126. b AKT and p-AKT levels in OGD injured cells treated with A11 and PI3K inhibitor LY294002. Cell viability was measured by MTT when treated with A11 and U0126 (c) or LY294002 (d). The results represent the mean ± SEM of four independent experiments. #P < 0.05, ###P < 0.001 vs. the control group, *P < 0.05, **P < 0.01 ***P < 0.001 vs. the OGD group, $$P < 0.01, $$$P < 0.001 vs. the OGD + A11 group
A11 reduces brain infarction volume and improves functional outcomes after MCAO
The potent neuroprotective effects of A11 in vitro prompted us to investigate its protective effects in vivo. Accordingly, we evaluated the protective effects of A11 on rats subjected to MCAO/reperfusion, a reliable and reproducible rodent model of cerebral ischemia and reperfusion-induced brain injury [30]. Infarction was determined by the appearance of a white region after TTC staining (Fig. 8a). Intravenous administration of A11 (3 mg/kg) at the onset of reperfusion significantly reduced the infarct volume by 13.1% (P < 0.01 vs. the vehicle-treated MCAO group) (Fig. 8b). Consistent with protection against brain infarction, MCAO-induced neurological deficits were significantly ameliorated by treatment with 3 mg/kg A11 (P < 0.05 vs. the vehicle-treated MCAO group) (Fig. 8c).

Intravenous administration of A11 (3 mg/kg) at the onset of reperfusion reduced the cerebral infarct volumes and ameliorated neurological deficits in MCAO rats. a Representative photographs of TTC-stained brain slices of rats. b Quantitative analysis of the cerebral infarct volume. c Quantitative analysis of neurological scores. The results represent the mean ± SEM. ###P < 0.001 vs. the sham group, *P < 0.05, **P < 0.01 vs. the vehicle-treated MCAO group, n = 16–19
Discussion
In the current study, application of cell-based phenotypic screening led to the discovery of the promising anti-ischemic potency of a class of diaryl acylhydrazone derivatives, especially A11, which has neuroprotective effects against both OGD/reperfusion and MCAO/reperfusion-induced injuries. To the best of our knowledge, diaryl acylhydrazone derivatives have not been previously reported to have neuroprotection against ischemia/reperfusion injury. The representative diaryl acylhydrazone derivative A11 was demonstrated to possess multifunctional neuroprotection effects against ischemia-reperfusion related pathological changes in neuronal cells, including oxidative stress, apoptotic changes, and mitochondrial dysfunction. Mechanistic studies revealed that the neuroprotective effects of A11 might be associated with triggering of the MEK/ERK and PI3K/AKT pathways in ischemic neuronal cells. Importantly, intravenous administration of A11 markedly reduced infarct volumes and ameliorated neurological deficits of experimental rats suffering from MCAO/reperfusion injury.
An OGD-stimulated SH-SY5Y neuronal model was employed to mimic the lack of energy supply to neuronal cells after ischemia insult, which is a classic experimental model that is used to mimic ischemic stroke in vitro [19, 20]. OGD stimulation caused a significant reduction in cell survival, as observed by morphological changes and MTT and LDH assays, which was alleviated by treatment with A11. MTT is converted to colored formazan by live cells, the amount of which indicates the number of metabolically active cells [31]. LDH is an indicator of the cell membrane integrity and is released from damaged cells into the medium [32]. Measurements of the amount of released LDH and MTT are widely used to evaluate cell viability and reflect the degree of injury in cells [33]. The results of the MTT and LDH measurements indicated the neuroprotective effects of A11 against ischemic-induced neuronal death. Treatment with 1 μM or 3 μM of A11 alone exerted no obvious effects on SH-SY5Y cell viability (data not shown), indicating that the protective effects of A11 are more likely mediated by resisting the OGD injury and not simply enhancing cell viability.
The potential efficacy of A11 against the multiple pathological changes triggered by ischemia was further evaluated in neuronal cells. It is well known that three key pathological changes, oxidative stress, apoptosis, and mitochondrial dysfunction, are powerful mediators of ischemic injury, resulting in profound neuronal damage. First, due to its limited antioxidation ability, the brain is particularly vulnerable to ROS attack, which acts directly as an executor of cell death [34]. Moreover, apoptosis, characterized by shrinkage of the cell and its nucleus, with maintenance of the plasma membrane integrity, is a typical type of cell death that occurs after ischemic injury, which persists until late in the injury [35,36,37]. Caspase-3 is a member of the cysteine-aspartic acid protease family, which is activated by cleavage and plays a central role in cell apoptosis [38]. p53 is one of the key upstream factors that activate apoptosis [39]. We found that A11 significantly attenuated neuronal death induced by oxidative stress and markedly inhibited the apoptotic changes in neuronal cells exposed to OGD, as shown by inhibition of Caspase-3 cleavage and the decreased level of p53 in the nucleus. Finally, mitochondrial function is severely impaired during brain ischemia, as shown by intracellular overproduction of ROS, mitochondrial membrane depolarization, and inhibition of ATP synthesis [4, 5, 37], while increased production of ROS is recognized as a major contributor to the whole process of ischemic injury, including ischemia, reperfusion and the later post-reperfusion phase of ischemia/reperfusion injury [40]. Similarly, A11 decreased intracellular ROS generation and ameliorated mitochondrial dysfunction of ischemic neuronal cells. Taken together, the above-mentioned beneficial effects of A11 on neuronal cells suggest that A11 has the ability to combat the above-mentioned three key pathological events triggered by ischemic injury.
It is, therefore, interesting to determine the potential regulatory mechanisms that underlie the neuroprotective effect of A11. Numerous studies have shown that PI3K/AKT and MEK/ERK are two important cascades that are involved in ischemia/reperfusion-induced apoptosis and oxidative stress [41,42,43,44]. Moreover, activation of ERK and AKT signaling confers neuroprotection to reduce ischemic damage [45,46,47,48]. Considering that a cell model is a relatively simple environment and that the current in vitro anti-ischemic results of A11 were obtained in neuronal cells, we employed neuronal cells to study the potential mechanisms. Interestingly, we observed that A11 upregulated the phosphorylation levels of ERK and AKT in OGD-exposed SH-SY5Y cells. Moreover, the neuroprotective effects of A11 against OGD-induced cytotoxicity were markedly blocked after treatment with PI3K and MEK inhibitors accompanied by a decreased level of phosphorylated ERK and AKT, suggesting that triggering the ERK and AKT pathways may contribute to the protection of neuronal cells conferred by A11 in hypoxic environments.
More importantly, A11 effectively alleviated the brain damage induced by focal brain ischemia in rats when intravenously administered at the onset of reperfusion. A single-dose of 3 mg/kg of A11 significantly reduced the infarct volume. Furthermore, in agreement with the reduced lesion size, A11 also provided beneficial effects on the functional outcomes of MCAO rats, as assessed by the neurological score. Given that MCAO is a widely employed animal model to mimic acute ischemic injury in the brain, the above in vivo anti-ischemic results suggest A11 is a potential candidate for acute intervention in ischemic stroke.
In contrast to the relatively simple cellular system, it is more difficult to clarify the mechanism of A11 in the ischemic animal model because the ischemic brain is complicated system that consists of various cell types, including neurons and glial cells, and each type of cell exhibits a wide variety of alterations during pathologic processes, which are interrelated and interact with each other and, consequently, lead to neuronal damage. Currently, we can only conclude that the ERK and AKT signaling pathways may contribute to the multiple protective effects of A11 on neuronal cells under ischemic insult because our mechanistic studies were only performed in neuronal cell lines. Moreover, although we observed the neuroprotective effects of A11 both in vitro and in vivo, we still could not conclude whether they are closely associated with each other.
In summary, this work reveals the multi-aspect neuroprotective effects of a novel diaryl acylhydrazone derivative, A11, against ischemic injury and describes its interesting regulation of both the ERK and AKT signaling pathways in neuronal cells, which could provide important clues for the development of drug leads/candidates against ischemic stroke. However, further systematic study of A11, including determination of its pharmacokinetic profiles, is needed to better understand the correlation between its in vitro and in vivo efficacies and to understand the precise molecular mechanisms of A11 against ischemic injury.
References
Krishnamurthi RV, Feigin VL, Forouzanfar MH, Mensah GA, Connor M, Bennett DA, et al. Global and regional burden of first-ever ischaemic and haemorrhagic stroke during 1990–2010: findings from the Global Burden of Disease Study 2010. Lancet Glob Health. 2013;1:e259–e81.
Wardlaw JM, Murray V, Berge E, del Zoppo G, Sandercock P, Lindley RL, et al. Recombinant tissue plasminogen activator for acute ischaemic stroke: an updated systematic review and meta-analysis. Lancet. 2012;379:2364–72.
Moretti A, Ferrari F, Villa RF. Neuroprotection for ischaemic stroke: current status and challenges. Pharmacol Ther. 2015;146:23–34.
Buendia I, Tenti G, Michalska P, Mendez-Lopez I, Luengo E, Satriani M, et al. ITH14001, a CGP37157-nimodipine hybrid designed to regulate calcium homeostasis and oxidative stress, exerts neuroprotection in cerebral ischemia. ACS Chem Neurosci. 2017;8:67–81.
Ham PB 3rd, Raju R. Mitochondrial function in hypoxic ischemic injury and influence of aging. Prog Neurobiol. 2017;157:92–116.
Moskowitz MA, Lo EH, Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron. 2010;67:181–98.
Iadecola C, Anrather J. Stroke research at a crossroad: asking the brain for directions. Nat Neurosci. 2011;14:1363–8.
Lapchak PA. Emerging therapies: pleiotropic multi-target drugs to treat stroke victims. Transl Stroke Res. 2011;2:129–35.
Lapchak PA. A critical assessment of edaravone acute ischemic stroke efficacy trials: is edaravone an effective neuroprotective therapy? Expert Opin Pharmacother. 2010;11:1753–63.
Leger PL, De Paulis D, Branco S, Bonnin P, Couture-Lepetit E, Baud O, et al. Evaluation of cyclosporine A in a stroke model in the immature rat brain. Exp Neurol. 2011;230:58–66.
Lapchak PA. Neuroprotective and neurotrophic curcuminoids to treat stroke: a translational perspective. Expert Opin Investig Drugs. 2011;20:13–22.
Nader-Kawachi J, Gongora-Rivera F, Santos-Zambrano J, Calzada P, Rios C. Neuroprotective effect of dapsone in patients with acute ischemic stroke: a pilot study. Neurol Res. 2007;29:331–4.
Swinney DC, Anthony J. How were new medicines discovered? Nat Rev Drug Discov. 2011;10:507–19.
Khurana V, Tardiff DF, Chung CY, Lindquist S. Toward stem cell-based phenotypic screens for neurodegenerative diseases. Nat Rev Neurol. 2015;11:339–50.
Schubert D, Maher P. An alternative approach to drug discovery for Alzheimer’s disease dementia. Future Med Chem. 2012;4:1681–8.
Talevi A. Tailored multi-target agents. Applications and design considerations. Curr Pharm Des. 2016;22:3164–70.
Lee JA, Uhlik MT, Moxham CM, Tomandl D, Sall DJ. Modern phenotypic drug discovery is a viable, neoclassic pharma strategy. J Med Chem. 2012;55:4527–38.
Lee JA, Berg EL. Neoclassic drug discovery: the case for lead generation using phenotypic and functional approaches. J Biomol Screen. 2013;18:1143–55.
Cho EY, Lee SJ, Nam KW, Shin J, Oh KB, Kim KH, et al. Amelioration of oxygen and glucose deprivation-induced neuronal death by chloroform fraction of bay leaves (Laurus nobilis). Biosci Biotechnol Biochem. 2010;74:2029–35.
Hossmann KA. Cerebral ischemia: models, methods and outcomes. Neuropharmacology. 2008;55:257–70.
Liu P, Zhao H, Wang R, Wang P, Tao Z, Gao L, et al. MicroRNA-424 protects against focal cerebral ischemia and reperfusion injury in mice by suppressing oxidative stress. Stroke. 2015;46:513–9.
Rai G, Joshi N, Jung JE, Liu Y, Schultz L, Yasgar A, et al. Potent and selective inhibitors of human reticulocyte 12/15-lipoxygenase as anti-stroke therapies. J Med Chem. 2014;57:4035–48.
Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91.
Chen J, Sanberg PR, Li Y, Wang L, Lu M, Willing AE, et al. Intravenous administration of human umbilical cord blood reduces behavioral deficits after stroke in rats. Stroke. 2001;32:2682–8.
Fukui M, Choi HJ, Zhu BT. Mechanism for the protective effect of resveratrol against oxidative stress-induced neuronal death. Free Radic Biol Med. 2010;49:800–13.
Fukui M, Song JH, Choi J, Choi HJ, Zhu BT. Mechanism of glutamate-induced neurotoxicity in HT22 mouse hippocampal cells. Eur J Pharmacol. 2009;617:1–11.
Iijima T, Mishima T, Tohyama M, Akagawa K, Iwao Y. Mitochondrial membrane potential and intracellular ATP content after transient experimental ischemia in the cultured hippocampal neuron. Neurochem Int. 2003;43:263–9.
Almeida A, Delgado-Esteban M, Bolanos JP, Medina JM. Oxygen and glucose deprivation induces mitochondrial dysfunction and oxidative stress in neurones but not in astrocytes in primary culture. J Neurochem. 2002;81:207–17.
Liu Y, Lu Z, Cui M, Yang Q, Tang Y, Dong Q. Tissue kallikrein protects SH-SY5Y neuronal cells against oxygen and glucose deprivation-induced injury through bradykinin B2 receptor-dependent regulation of autophagy induction. J Neurochem. 2016;139:208–20.
Fluri F, Schuhmann MK, Kleinschnitz C. Animal models of ischemic stroke and their application in clinical research. Drug Des Devel Ther. 2015;9:3445–54.
Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63.
Burd JF, Usategui-Gomez M. A colorimetric assay for serum lactate dehydrogenase. Clin Chim Acta. 1973;46:223–7.
Abe K, Matsuki N. Measurement of cellular 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction activity and lactate dehydrogenase release using MTT. Neurosci Res. 2000;38:325–9.
Adibhatla RM, Hatcher JF. Lipid oxidation and peroxidation in CNS health and disease: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal. 2010;12:125–69.
Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med. 2009;361:1570–83.
Yuan J. Neuroprotective strategies targeting apoptotic and necrotic cell death for stroke. Apoptosis. 2009;14:469–77.
Khoshnam SE, Winlow W, Farzaneh M, Farbood Y, Moghaddam HF. Pathogenic mechanisms following ischemic stroke. Neurol Sci. 2017;38:1167–86.
Lavrik IN, Golks A, Krammer PH. Caspases: pharmacological manipulation of cell death. J Clin Invest. 2005;115:2665–72.
Fridman JS, Lowe SW. Control of apoptosis by p53. Oncogene. 2003;22:9030–40.
Kalogeris T, Bao Y, Korthuis RJ. Mitochondrial reactive oxygen species: a double edged sword in ischemia/reperfusion vs preconditioning. Redox Biol. 2014;2:702–14.
Mullonkal CJ, Toledo-Pereyra LH. Akt in ischemia and reperfusion. J Invest Surg. 2007;20:195–203.
Song Q, Gou WL, Zhang R. FAM3A protects HT22 cells against hydrogen peroxide-induced oxidative stress through activation of PI3K/Akt but not MEK/ERK pathway. Cell Physiol Biochem. 2015;37:1431–41.
Mehta SL, Manhas N, Raghubir R. Molecular targets in cerebral ischemia for developing novel therapeutics. Brain Res Rev. 2007;54:34–66.
Maiese K, Chong ZZ, Wang S, Shang YC. Oxidant stress and signal transduction in the nervous system with the PI 3-K, Akt, and mTOR cascade. Int J Mol Sci. 2012;13:13830–66.
Zhao H, Wang R, Tao Z, Yan F, Gao L, Liu X, et al. Activation of T-LAK-cell-originated protein kinase-mediated antioxidation protects against focal cerebral ischemia-reperfusion injury. FEBS J. 2014;281:4411–20.
Abd El-Aal SA, Abd El-Fattah MA, El-Abhar HS. CoQ10 Augments rosuvastatin neuroprotective effect in a model of global ischemia via inhibition of NF-kappaB/JNK3/Bax and activation of Akt/FOXO3A/Bim cues. Front Pharmacol. 2017;8:735.
Feng Y, Lu S, Wang J, Kumar P, Zhang L, Bhatt AJ. Dexamethasone-induced neuroprotection in hypoxic-ischemic brain injury in newborn rats is partly mediated via Akt activation. Brain Res. 2014;1589:68–77.
Li X, Zhang J, Chai S, Wang X. Progesterone alleviates hypoxic-ischemic brain injury via the Akt/GSK-3beta signaling pathway. Exp Ther Med. 2014;8:1241–6.
Acknowledgements
This work was funded by grants from the National Natural Science Foundation of China (No 81522045, 31400932, 81602975). We thank Ms. Sha-sha Ji and Ms. Feng-xia Bao for their contribution in assessing the in vitro anti-ischemic effects of A11.
Author contributions
H-xF and C-pL are co-first authors. H-yZ and HL are co-corresponding authors. H-yZ, H-xF, and HL designed the study; H-xF performed the pharmacological assays; C-pL and S-jS synthesized the diaryl acylhydrazone derivatives, including A11; HL instructed the chemical synthesis; H-xF and H-yZ analyzed pharmacological data; and H-xF and H-yZ wrote the manuscript with input from all of the authors
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Feng, Hx., Li, Cp., Shu, Sj. et al. A11, a novel diaryl acylhydrazone derivative, exerts neuroprotection against ischemic injury in vitro and in vivo. Acta Pharmacol Sin 40, 160–169 (2019). https://doi.org/10.1038/s41401-018-0028-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-018-0028-4
Keywords
This article is cited by
-
Endothelial cell-derived RSPO3 activates Gαi1/3-Erk signaling and protects neurons from ischemia/reperfusion injury
Cell Death & Disease (2023)
-
An optically active isochroman-2H-chromene conjugate potently suppresses neuronal oxidative injuries associated with the PI3K/Akt and MAPK signaling pathways
Acta Pharmacologica Sinica (2021)
-
Deregulated Protein Kinases: Friend and Foe in Ischemic Stroke
Molecular Neurobiology (2021)
-
Neuroprotective Effect of Natural Alkaloid Fangchinoline Against Oxidative Glutamate Toxicity: Involvement of Keap1-Nrf2 Axis Regulation
Cellular and Molecular Neurobiology (2019)

{kind=link}