
Abstract
Genetic analyses of psychiatric illnesses, such as bipolar disorder (BPD), have revealed essential information regarding the underlying pathological mechanisms. While such studies in populations of European ancestry have achieved prominent success, understanding the genetic risk factors of these illnesses (especially BPD) in Chinese population remains an urgent task. Given the lack of genome-wide association study (GWAS) of BPD in Chinese population from Mainland China, replicating the previously reported GWAS hits in distinct populations will provide valuable information for future GWAS analysis in Han Chinese. In the present study, we have recruited 1146 BPD cases and 1956 controls from Mainland China for genetic analyses, as well as 65 Han Chinese brain amygdala tissues for mRNA expression analyses. Using this clinical sample, one of the largest Han Chinese BPD samples till now, we have conducted replication analyses of 21 single nucleotide polymorphisms (SNPs) extracted from previous GWAS of distinct populations. Among the 21 tested SNPs, 16 showed the same direction of allelic effects in our samples compared with previous studies; 6 SNPs achieved nominal significance (p < 0.05) at one-tailed test, and 2 additional SNPs showed marginal significance (p < 0.10). Aside from replicating previously reported BPD risk SNPs, we herein also report several intriguing findings: (1) the SNP rs174576 was associated with BPD in our Chinese sample and in the overall global meta-analysis, and was significantly correlated with FADS1 mRNA in diverse public RNA-seq datasets as well as our in house collected Chinese amygdala samples; (2) two (partially) independent SNPs in MAD1L1 were both significantly associated with BPD in our Chinese sample, which was also supported by haplotype analysis; (3) a rare SNP rs78089757 in 10q26.13 region was a genome-wide significant variant for BPD in East Asians, and this SNP was near monomorphic in Europeans. In sum, these results confirmed several significant BPD risk genes. We hope this Chinese BPD case–control sample and the current brain amygdala tissues (with continuous increasing sample size in the near future) will provide helpful resources in elucidating the genetic and molecular basis of BPD in this major world population.
Similar content being viewed by others

Introduction
Bipolar disorder (BPD) is a complex mental illness with considerable genetic heritability1,2,3,4. The neurobiology of BPD is not yet fully understood, but accumulating studies have suggested the essential roles of aberrant synaptic function in specific brain regions relevant to emotion and cognition in BPD pathogenesis5,6,7. In lieu with the putative roles of genetic risk factors in the pathogenesis of BPD, genome-wide association studies (GWAS), the approach widely recognized as effective in identifying genetic risk loci for psychiatric disorders8,9,10,11,12, have been performed in European and Japanese populations and have reported a number of BPD susceptibility loci spanning genes including ANK3, CACNA1C, NCAN, and others13,14,15,16,17,18,19,20,21,22. The largest BPD GWAS so far is performed by the Psychiatric Genomics Consortium Bipolar Disorder (PGC-BP) group. They conducted a meta-analysis of multiple GWAS datasets including 20,352 BPD patients and 31,358 controls in European populations, followed by replication analyses of the significant loci (p < 1 ⨯ 10−4) in an additional 9412 cases and 137,760 controls14. This combined meta-analysis of the GWAS discovery and replication samples (named BPD PGC2 GWAS) yielded 30 leading single nucleotide polymorphisms (SNPs) showing genome-wide significance14. Before BPD PGC2 GWAS, another two BPD GWAS of smaller samples (these samples were partially overlapped with that of BPD PGC2 GWAS) recruited from European populations have also found several SNPs showing genome-wide level of statistical significance15,18. The progress of BPD genetic studies in Asians have also recently emerged, as a GWAS carried out in Japanese have lately identified additional novel risk variants for this illness, suggesting the potential genetic overlap and heterogeneity of BPD between distinct continental populations17. These discoveries motivated further characterization of BPD genetic risk factors in other populations, as well as validation of the identified risk associations to understand the generalizability of current GWAS findings.
For these purposes, we thus sought to explore the unclear genetic basis of BPD in Chinese populations. While Chinese populations make up around one fifth of the total world, the progress of genetic analyses for BPD in Han Chinese has been much slower compared to that in other cohorts. Till now, no genome-wide significant risk variants for BPD in Han Chinese population have been reported23, and replication studies of European GWAS loci in Chinese BPD sample have been barely published. We recruited 1146 BPD patients and 1956 control subjects from Mainland China to replicate the associations of the 21 risk variants highlighted in previous BPD GWAS (Table 1). Given the hypothesis that risk variants usually contribute to the illnesses by modulating the expression of nearby genes24,25, we have also collected 65 brain amygdala tissues from nonpsychiatric individuals of Han Chinese ancestry, and tested whether the BPD risk SNPs affected expression levels of particular genes.
Methods
Chinese BPD case–control sample
With the aim to build a DNA resource pool for investigating the genetic bases of BPD in Han Chinese population, we have initially recruited a total of 1146 BPD cases and 1956 controls of Han Chinese origin from Mainland China. The protocol was approved by the institutional review board of the Kunming Institute of Zoology, Chinese Academy of Sciences and the ethics committees of all participating hospitals and universities. All participants provided written informed consents before any study-related procedures were performed.
The BPD patients were collected from multiple provinces of Mainland China, including mental health centers and psychiatric departments of general hospitals. Each patient was diagnosed as having BPD by a consensus of at least two experienced psychiatrists. Diagnoses were further confirmed with an Extensive Clinical Interview and a Structured Clinical Interview for DSM-IV Axis/Disorders, Patient Version (SCID-P), as previously described26,27. Cases were excluded if they had either a pre-existing history of schizophrenia, mental retardation, or drug/alcohol addiction. Subjects with concurrent diagnosis of other mental illnesses were also excluded to minimize potential compounding conditions in our analysis. The control subjects were recruited from Mainland China with their medical and family history information records. Subjects with any history of major neuropsychiatric or neurological disorders (e.g., BPD, depression, schizophrenia, attention deficit hyperactivity disorder, and mental retardation), or with a family history of severe mental illnesses, were excluded from this study.
SNP selection
A total of 21 SNPs were selected based on the results of four previous BPD GWAS studies in European and Japanese populations14,15,17,18. Among them, 13 SNPs (rs17183814, rs2302417, rs3804640, rs11724116, rs10455979, rs10821745, rs7122539, rs12575685, rs10744560, rs4447398, rs11647445, rs112114764, and rs6130764) were selected from the BPD PGC2 GWAS (29,764 cases and 169,118 controls of European ancestry) because of their genome-wide significant associations with BPD14. Two genome-wide significant SNPs (rs4236274 and rs12553324) were chosen from Hou et al. GWAS (9784 cases and 30,471 controls of European origin)15. One genome-wide risk SNP (rs12290811) was obtained from Muhleisen et al.18 GWAS (9747 cases and 14,278 controls of European ancestry), and five additional SNPs (rs4332037, rs78089757, rs174576, rs329674, and rs76317718) were chosen from a previous GWAS study by Ikeda et al.17 (2964 cases and 61,887 controls of Japanese origin, and 7481 cases and 9250 controls of European ancestry). In this trans-ethnic analysis performed by Ikeda et al.17, two (rs4332037 and rs174576) of the five selected SNPs showed genome-wide significant associations with BPD in both Europeans and Japanese; the other three SNPs (rs78089757, rs329674, and rs76317718), though not highlighted in cross-population analysis, all showed marginal genome-wide significance in Japanese (p < 5.00E−07). Given the similar genetic background between Japanese and Han Chinese populations, we also examined whether these three SNPs (rs78089757, rs329674, and rs76317718) were associated with BPD in Han Chinese, and whether the associations could reach genome-wide level of significance when all the East Asian samples were combined. The statistics (p values and odds ratios (ORs)) of the 21 SNPs were retrieved from the original studies and summarized in Table 1.
SNP genotyping
Peripheral venous blood samples were collected and were stored at −80 °C. Genomic DNA was extracted from peripheral blood leukocytes using high-salt extraction procedures according to the manufacturer’s protocol. Each sample was genotyped at a multiplex level per well on a custom array using the Sequenom MassARRAY system following the manufacturer’s instructions, which is similar to a previous study28. The software MassARRAY TYPER 4.0 was used to analyze the mass spectrograms and to call the SNP genotypes. Raw genotype data (cluster plots) of the SNPs were visually assessed by two investigators. All assays were performed blind to diagnosis and genotype.
Statistical analysis in Chinese sample
Hardy–Weinberg equilibrium test was performed for all SNPs in controls using Pearson χ2-test with one degree of freedom. PLINK (v1.07) was used to analyze the statistical association between single SNPs and illness conditions, i.e., to calculate the allelic p-values, ORs and 95% confidence intervals (CIs)29. In the direct replication analysis of previously reported SNPs, we also applied one-tailed test in our Han Chinese sample, while in the meta-analysis of all available samples, two-tailed tests were used. Haplotype analysis was performed using the online SHEsis website (http://analysis.bio-x.cn/)30,31, and only haplotypes with frequencies higher than 0.001 in both cases and controls were considered. The regional association results were plotted using LocusZoom (http://locuszoom.sph.umich.edu/locuszoom/)32.
Meta-analysis across different samples
After calculating the statistics of the 21 SNPs in our Han Chinese sample, we then conducted meta-analysis of data from this Chinese sample and previous published GWAS studies. Specifically, we performed meta-analysis of the 13 SNPs (rs17183814, rs2302417, rs3804640, rs11724116, rs10455979, rs10821745, rs7122539, rs12575685, rs10744560, rs4447398, rs11647445, rs112114764, and rs6130764) chosen based on the PGC2 GWAS (European ancestry)14 using data from our Chinese sample and the PGC2 GWAS (including both discovery and replication stages, a total of 29,764 cases and 169,118 controls, data were from Table S2 in their study14); for the two SNPs (rs4236274 and rs12553324) retrieved from Hou et al. GWAS15, the meta-analysis was performed using statistics from our Chinese sample and the original Hou et al. GWAS (including both discovery and replication stages, a total of 9784 cases and 30,471 controls, data were from Table 2 in their study15); the SNP rs12290811 chosen from the GWAS by Muhleisen et al.18 underwent meta-analysis using data obtained from our Chinese sample and their original study (including both discovery and replication stages, a total of 9747 cases and 14,278 controls, data were from Table 2 in their study18). We also examined five additional SNPs (rs4332037, rs78089757, rs174576, rs329674, and rs76317718) reported in the Ikeda et al. GWAS17. Notable, meta-analysis of rs174576, the SNP significantly linked to BPD in both Japanese and European individuals, was conducted using data from our Chinese sample, two Japanese samples, and a European study22 (including 20,129 cases and 21,524 controls, the sample size was different from PGC121 or PGC214 GWAS). Rs78089757 was not polymorphic in Europeans, and meta-analysis of the SNP was therefore conducted in solely East Asian populations, including our primary Chinese sample, three additional Chinese control sample sets, as well as two Japanese samples17. The meta-analysis of other three SNPs (rs4332037, rs329674, and rs76317718) was conducted using data from our Chinese sample and the data shown in Ikeda et al.17 (from either Table 1 or Table 2 of their study). The detailed information about the sample size and source of data for each SNP during meta-analysis is shown in Table S1. The metafor package in R was used to obtain pooled estimates of ORs across different samples, and to test for heterogeneity of the ORs33.
Expression quantitative trait loci (eQTL) analyses in public datasets
We explored the impact of BPD risk SNPs on gene expression levels in human brains. For this analysis, three public gene expression datasets of genome-wide genotype data as well as RNA-seq data were used. These datasets are (1) BrainSeq34, (2) Genotype-Tissue Expression project (GTEx)35, and (3) Brain xQTL36.
BrainSeq (http://eqtl.brainseq.org/phase1/eqtl/) contains data of polyA + RNA-seq expression quantitative trait loci (eQTL) associations in the dorsolateral prefrontal cortex (DLPFC) tissues collected from 175 schizophrenia patients and 273 controls34. These individuals were all aged >13 and were mainly Caucasians and African Americans. As described in the website, the RNA-seq data was analyzed under the additive genetic effect model for the eQTL results, and was adjusted for sex, ancestry, and expression heterogeneity (principal components). We herein retrieved the results of eQTL associations from the 273 unaffected controls only. Detailed information about the BrainSeq can be found in the original publication34.
The GTEx (https://www.gtexportal.org/) contains information of both genetic variation and RNA-seq mRNA expression from diverse human tissues. So far, data of more than 3797 tissues from 150 postmortem donors are available for research purpose. We have thus retrieved the results of eQTL associations from the 118 frontal cortex (BA9) of healthy controls for the current study. Detailed information can be found in the original report35.
The Brain xQTL Serve (http://mostafavilab.stat.ubc.ca/xQTLServe/) allows assessment of the impact of genetic variation on multiple types of molecular traits derived from the human brain cortex36. Their samples were mainly collected using autopsies from participants of the Religious Orders Study and the Rush Memory and Aging Project. As described in this early study36, postmortem frozen samples of the DLPFC were obtained from these individuals, and polyA + RNA-seq gene expression (n = 494) analyses were performed using these samples. The effects of known (e.g., RNA integrity number, sex, age, diagnostic status, pH, PMI, and batch effects) and hidden (e.g., RNA quality and variation in cell type proportions) confounding factors were removed from the molecular phenotype data using linear regression. The authors employed Spearman’s rank correlation to estimate the association between each SNP and gene expression. This dataset provides statistical results, and the visual plots are not available in their website at the time we accessed. Detailed information can be found in the original study36.
Replication of eQTL effects in Chinese brain amygdala sample
In order to replicate the observed eQTL associations of BPD risk SNPs in Chinese population, we also collected amygdala tissues of 65 nonpsychiatric controls from the Chinese Brain Bank Center. It is recognized that patients with BDP normally showed mood dysregulation as well as impaired cognitive, social, and autonomic functions3,4,37. Amygdala is primarily related to emotion, mood, and cognition38, and is therefore believed to be an important brain region in the pathogenesis of BPD. Indeed, decrease of brain amygdala volume was frequently observed in BPD patients compared to healthy controls37,39,40,41,42,43, and functional magnetic resonance imaging studies have also revealed aberrant amygdala activation in BPD patients compared with normal subjects37,44,45,46,47,48. We thus believe that this amygdala sample will provide pivotal information regarding the molecular mechanisms underlying BPD pathogenesis in Chinese population.
Total RNAs from the brain amygdala tissues were isolated by TRIzol reagent (Life technologies, USA) according to the manufacture’s protocol. The complementary DNA (cDNA) was synthesized by using a first strand cDNA synthesis kit (K1612, ThermoFisher Scientific, USA). The reverse transcription reaction mixture included 2 μg total RNA, 1.0 μL random hexamer primer, 4.0 μL 5× reaction buffer, 1.0 μL dNTP mix, 2.0 μL RiboLock RNase inhibitor, 2.0μL M-MuLV reverse transcriptase and nuclease-free water in a final volume of 20 μL. The reverse transcription reaction was incubated for 5 min at 25 °C and followed by 60 min at 42 °C. After the reverse transcription reaction, 80 μL nuclease-free water was added to make the final volume of 100 μL, for further transcription analysis. For quantitative real-time polymerase chain reaction (qRT-PCR), the reaction mixture contained 10.0 μL 2× SYBR master mix (Roche, USA), 2.0 μL primers (10 μM), 1.0 μL cDNA and 7.0 μL nuclease-free water. The qRT-PCR was performed on a 7900HT Fast Real-Time PCR System (Applied Biosystems, USA) following the program that firstly 95 °C for 10 min, followed by 40 repeated cycles of 95 °C for 15 s and 60 °C for 30 s. The FADS1 expression was determined by relative quantitative analysis and RPS13 was employed as an internal control gene. The relative gene expression was presented as the means of –ΔCt for a statistical test against genotypic groups49, and the p values were calculated using one-way ANOVA. The sequences of primers used for amplification of RPS13 were 5′-CCCCACTTGGTTGAAGTTGA-3′ (forward) and 5′-CTTGTGCAACACCATGTGAA-3′ (reverse), and sequences of primers for FADS1 were 5′-TATATGCCGTACAACCACCAGC-3′ (forward) and 5′-GAAGCGGACGTAGAAGGTAATCA-3′ (reverse).
Cross-disorder analysis using statistical data from CONVERGE GWAS
Previous studies have suggested substantial genetic overlap between BPD and major depressive disorder (MDD)50,51,52,53. To investigate if the aforementioned BPD risk SNPs were also associated with risk of MDD in Han Chinese population, we utilized data of Han Chinese individuals from the previously published CONVERGE MDD GWAS10. In brief, the CONVERGE GWAS consisted of 5303 MDD cases and 5337 mentally healthy controls, in which the MDD cases were diagnosed according to the DSM-IV criteria10. As described earlier, controls were recruited either from local communities or from the patients who underwent minor surgical procedures at the general hospitals. Detailed information about the sample composition, quality control, and statistical methods can be found in the original study10.
Results
Of the 3102 individuals genotyped, 1794 were male and 1308 were female. The final genotyping rate was 0.9855. The allele and genotype frequencies of the 21 SNPs are shown in Table 1 and Table S2. Sixteen of the 21 (76.2%) SNPs exhibited the same direction of allelic effects regarding their roles in BPD susceptibility in our Han Chinese sample compared with previous GWAS. Six SNPs had nominal significant associations with the illness (p < 0.05, Table 1) at one-tailed test in our Chinese samples, and two additional SNPs showed marginal significance (p < 0.10, Table 1). Meta-analysis combining our Chinese samples with data of previous studies yielded genome-wide significant associations for 15 these tested SNPs (two-tailed p < 5.00E−08, Table S1).
FADS1 is a risk gene for bipolar disorder
Our study, rather than providing solely the above replication results of previously reported SNPs, has also highlighted several SNPs for further analyses. First, rs174576 in FADS1/2 region, the SNP showing genome-wide significant association in the meta-analysis of Japanese and European PGC1 GWAS samples (two-tailed p = 1.34E−10, OR = 1.130)17, is also significantly associated with BPD in our Han Chinese sample (one-tailed p = 0.0229, OR = 1.110, Table 1). When our Han Chinese samples were combined with data from previous GWAS samples of Japanese17 (2964 cases and 61,887 controls) and Europeans22 (20,129 cases and 21,524 controls) for further meta-analysis, a stronger genome-wide association between rs174576 and BPD was seen under fixed effect model (two-tailed p = 9.33E−13, OR = 1.098, Table 2).
The allele frequencies of rs174576 in European and East Asian populations are similar (Figure S1), and in both populations there are multiple SNPs in high linkage disequilibrium (LD) with rs174576 (r2 > 0.8, Figure S2). Although it is difficult to determine the potential causative variant for this locus, functional prediction in GWAVA dataset (http://www.sanger.ac.uk/sanger/StatGen_Gwava)54 and HaploReg (http://archive.broadinstitute.org/mammals/haploreg/haploreg.php)55 showed several SNPs deserving further investigation (Table S3 and Figure S3).
To gain more insights into this issue, we conducted eQTL analysis in brain tissues as it is proven to be effective in discovering the underlying molecular mechanism of risk association56,57,58. This approach has little dependence on the causative variant, and LD SNPs of the causative variant could also show eQTL associations with the relevant gene expression24,25. Briefly, eQTL analysis of rs174576 with nearby gene expression in brain samples was performed using BrainSeq as the discovery sample34. In this dataset, we observed that rs174576 was significantly associated with the gene expression of FADS1 (two-tailed p = 2.65E−10, Fig. 1), with the BPD risk allele [A] predicting lower levels of FADS1 mRNA. We then examined the eQTL association between rs174576 and FADS1 using the GTEx and Brain xQTL datasets35,36, and the above finding was reproduced (two-tailed p = 1.60E−05 in GTEx and two-tailed p = 6.77E−14 in Brain xQTL, Fig. 1 and Table S4, respectively), with the eQTL associations in the three datasets all in the same directions. We then conducted eQTL analysis between rs174576 and FADS1 expression in our Han Chinese amygdala samples (n = 65) to validate if this eQTL association exists across different ethnic populations, and more intriguingly, we found that rs174576 was also significantly associated with FADS1 expression in Han Chinese (two-tailed p = 0.0357, Fig. 1). Together, these data strongly support the important role of rs174576 and FADS1 in BPD pathogenesis, and the causal factors among rs174576 and its linked SNPs remains to be determined. We also tested the association between rs174576 and the expression of FADS2 in the above mRNA datasets, but did not see significant associations (Tables S4 and S5). In the Brain xQTL dataset, rs174576 was also significantly associated with TMEM258 expression (Table S4), but this association was not replicated in GTEx or BrainSeq database (Table S5), and was therefore not followed up in our Chinese amygdala sample.

Association of rs174576 with FADS1 mRNA expression in BrainSeq (a), GTEx (b), and our own Chinese sample (c). DLPFC dorsolateral prefrontal cortex, FC frontal cortex
MAD1L1 SNPs and haplotypes are associated with bipolar disorder
Besides rs174576, SNPs (rs4236274 and rs4332037) in MAD1L1 also worth further investigations. Rs4236274 and rs4332037 are two SNPs in close proximity (54.4 kb between each other, their positions are shown in Figure S4) in the intron regions of MAD1L1. The two SNPs are in weak LD in both European and East Asian populations from 1000 Genomes Project59 (both r2 = 0.085), which was further seen in our own Han Chinese samples (r2 = 0.123). More importantly, the two (partially) independent SNPs have been reported to be genome-wide significantly associated with BPD in different GWAS studies15,17. In Hou et al. GWAS comprising of all European individuals15, rs4236274 showed genome-wide significant associations (two-tailed p = 8.49E−12, OR = 1.149, Table 1 and Figure S4), while rs4332037 only displayed nominal associations (two-tailed p = 0.00392, Figure S4). In Ikeda et al.17 GWAS including Japanese and European populations, rs4332037 was genome-wide significantly associated with BPD (two-tailed p = 1.91E−09, OR = 1.170, Table 1). In the current Han Chinese sample, both SNPs exhibited nominal significant associations with BPD (rs4236274, one-tailed p = 0.00731, OR = 1.140; rs4332037, one-tailed p = 0.0104, OR = 1.218; Table 1) in the same allelic directions and similar effect sizes compared with the previous GWAS studies15,17.
Given that rs4236274 and rs4332037 are in low LD, we also conducted the haplotype analysis of these two SNPs in our Han Chinese sample, and found that a common protective haplotype (G–C) showed significantly lower frequency in BPD cases (0.518) compared with controls (0.553) (haplotype p = 0.00845, OR = 0.868, Fisher’s exact test, Table 3). Global test also confirmed the haplotype analysis results (Global p = 0.00786, Fisher’s exact test). These results suggest that there are likely (at least partially) independent risk association signals in the MAD1L1 region, and there might be other causative variants remains to be identified. Overall, our single SNP and haplotype analyses support that MAD1L1 is a risk locus for BPD in Chinese population. However, neither of these MAD1L1 SNPs are associated with any gene expression in brains in the published eQTL datasets (data not shown)34,35,36, further studies elucidating their functional impacts are thus needed.
A rare variant rs78089757 is genome-wide significantly associated with bipolar disorder in East Asians
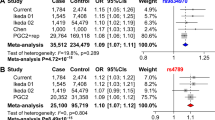
In addition, we have also replicated the nominal or marginal significant associations at several other loci, such as rs2302417 in ITIH3 and rs17183814 in SCN2A (one-tailed p value < 0.10, Table 1). Among these, there is a SNP rs78089757 located in 10q26.13 (intergenic region), which has been previously reported in a Japanese GWAS17, did not achieve genome-wide significance (two-tailed p = 3.99E−07, OR = 1.410, Table 1) probably due to the limited sample size. According to previous studies17, this SNP is rare in Japanese population (minor allele frequency < 0.05), and is almost monomorphic in European populations (not included in the current GWAS of BPD in European populations probably due to its low frequency). We thus checked the data from 1000 Genomes Project59 and confirmed that rs78089757 is also a rare SNP in Chinese population. We then performed further tests to understand its role in the genetic risk of BPD in Chinese, and found that rs78089757 was also nominally associated with BPD in 1146 cases and 1956 controls (one-tailed p = 0.0440, OR = 1.358, Table 1), with similar effect size as that in the Japanese GWAS17. To maximize the statistical power of our analyses, we then collected all available genotypic data through combining Han Chinese control subjects from 1000 Genomes Project59 (n = 208 individuals), CONVERGE Consortium10 (n = 5222 subjects), and an additional control cohort (n = 1908 individuals) with our own Han Chinese sample, and the association became stronger in the combined analysis (one-tailed p = 0.00176, OR = 1.526 in a total of 1146 cases and 9294 controls, Table 4). A meta-analysis combining Han Chinese data with Japanese GWAS17 showed that rs78089757 was genome-wide significantly associated with BPD (two-tailed p = 5.22E−09, OR = 1.429, Table 4), indicating that it is likely a risk SNP in East Asian populations. While rs78089757 could be an East Asian specific risk locus, but whether it is involved in BPD susceptibility in European populations remains unclear. Current European GWAS studies have not covered this SNP, and its surrounding genetic variations were not associated with BPD in European populations (the statistical data was from Hou et al. GWAS15, a total of 3095 SNPs in this region were included, and the regional-wide corrected p value should be 0.05/3095 = 1.62E−05, Figure S5).
SCN2A and SHANK2 are associated with major depressive disorder in Han Chinese
Given the substantial genetic overlap between BPD and MDD50,51,52,53, we also examined the associations of the 21 BPD risk SNPs with MDD in a published Chinese GWAS conducted by CONVERGE Consortium (5303 cases and 5337 controls)10. Among the 21 tested SNPs, 18 European BPD risk alleles were also highlighted in Chinese MDD cases compared with controls. Importantly, two SNPs (rs17183814 and rs12575685) have even achieved nominal significance (Table S6). Rs17183814 locates in the gene SCN2A. This SNP was previously reported to confer genome-wide significant risk of BPD in Europeans (two-tailed p = 2.02E−09, OR = 1.142, Table 1)14, and is also associated with BPD in our Han Chinese sample with the similar effect size (one-tailed p = 0.0311, OR = 1.153, Table 1). We reveal that the BPD risk allele [G] of the SNP also increases risk of MDD in a similar effect size (two-tailed p = 0.00875, OR = 1.116, Table S6). The second SNP rs12575685 locates in the gene SHANK2, and is also significantly associated with MDD in Chinese (two-tailed p = 0.00558, OR = 1.095, Table S6). However, it did not show evidence of association with BPD in our Chinese sample (one-tailed p = 0.143, OR = 1.066, Table 1). The considerable OR for this SNP suggests that this negative result is probably due to the limited sample size.
Discussions
In the current studies, we have performed replications of previous BPD GWAS findings in an independent case and control sample set of Chinese origin. By combining our data with the previous BPD GWAS datasets, we have further confirmed that several genes (such as FADS1, MAD1L1, SCN2A, and ITIH3) are likely involved in the genetic risk of BPD. We also observed a genome-wide significant locus at 10q26.13 in East Asian populations.
This study has provided essential information as we have used a newly collected BPD sample from Mainland China, which has not been explored before the year of 2018. While we have tested whether the risk associations observed in other populations could be replicated in Chinese, further expanding analyses are also warranted to uncover Chinese specific genetic risk factors for BPD. Additionally, we have also utilized the brain amygdala samples from Chinese population. Although the sample size is relatively moderate compared to European brain eQTL studies34,36,56, it is still one of the largest brain samples ever reported in Chinese population. These data are therefore valuable in helping elucidate the molecular mechanisms underlying genetic risk factors of BPD and other psychiatric illnesses.
We have reported the intriguing finding of the association between rs174576 and FADS1 expression. Indeed, this genomic region has been recently highlighted in several BPD GWAS14,17. However, this genomic region contains numerous high-LD SNPs and spans many genes (Figure S2), making it difficult to pinpoint the causative gene(s). Our analyses, together with previous studies, suggest that FADS1 is likely a BPD-related gene, and decreased expression of FADS1 might be a potential risk factor. However, this gene was not highlighted in recent studies of genome-wide RNA-seq analyses in the brains of BPD patients60,61,62,63, and in a candidate gene study, mRNA expression of FADS1 was slightly higher in patients with BPD compared with nonpsychiatric controls64. Therefore, the mechanism by which FADS1 involved in BPD pathogenesis is rather more complicated and remains to be elucidated. The FADS1 gene encodes a member of the fatty acid desaturase (FADS) protein family. As described previously, FADS1 desaturates omega-3 and omega-6 polyunsaturated fatty acids at the delta-5 position and thereby catalyzes the final step of eicosapentaenoic acid (EPA) and arachidonic acid formation65. Interestingly, the SNP rs174576 has been reported to correlate with whole-body fat oxidation66, desaturase indices, and the impact of hormonal contraceptives on plasma docosahexaenoic acid concentrations67,68. While long-chain (>20 C-atoms) polyunsaturated fatty acids (LC PUFAs) are known to be important for the functional integrity of brain development, cognitive abilities, and mood69,70, the association between rs174576 and psychiatric disorders gains further support. These lines of evidence are in line with a previous study17, in which the authors speculated that alterations in lipid signaling pathways and metabolism might be involved in the pathophysiology of BPD. In fact, epidemiological studies found that increased incidences of hyperlipidemia and metabolic syndrome appeared in BPD patients compared with general populations71,72,73. In addition, a previous study found that rs174576 was associated with white matter abnormality in 83 preterm infants (quantified in vivo using diffusion tensor imaging), providing possible evidence that susceptibilities to BPD later in life may be linked to the in uterus exposure to environmental stress74.
Another gene implicated in the current study, MAD1L1, encodes the mitotic spindle assembly checkpoint protein MAD1 that facilitates appropriate onset of anaphase after proper alignment of all chromosomes at the metaphase plate75. MAD1L1 functions as a homodimer. By interacting with MAD2L1, HDAC1, and histone deacetylase 276,77, this protein likely plays a role in cell cycle control and tumor suppression78. The MAD1L1 gene is also known as human accelerated region 3 (HAR3; as mentioned in previous studies79,80, HARs are a set of 49 human genomic segments that are conserved throughout vertebrate evolution but are sharply different in human beings), and thus may have played a key role in human evolution79,80. Intriguingly, MAD1L1 has also been implicated in the genetic susceptibility to schizophrenia9,11,81,82, another psychiatric disease sharing substantial risk factors with BPD. It was also found that MAD1L1 was linked to the reward systems functioning in healthy adults (an intermediate phenotype for BPD and schizophrenia)83, adding further evidence for its involvement in psychiatric abnormalities. Although the exact function of MAD1L1 in brain development and even these illnesses remains unclear, the above evidence strongly suggests that the acceleration of MAD1L1 region in humans could play pivotal roles in the arising of these human-specific or human-dominant diseases (i.e., BPD and schizophrenia). Further studies dissecting the underlying mechanisms are therefore necessary.
We have also replicated the nominal or marginal associations between variants at ITIH3, SCN2A, and CD47 and the risk of BPD in our Han Chinese sample. ITIH3 is located at chromosome 3p21, the genomic region repeatedly showing significant associations with BPD14,15,17,20,84,85 and other mental illnesses9,11,86,87. This chromosome 3p21 region contains multiple high-LD variants in a tight cluster of genes, and thus a causative gene is yet to be determined. SCN2A encodes sodium voltage-gated channel alpha subunit 2 (Nav1.2). Although this gene has not been highlighted in earlier BPD studies, de novo mutations in SCN2A were found associated with autisms88,89,90,91, and common variations within or near this gene were also associated with cognitive performance in schizophrenia patients92. Besides, the gene CACNA1C also deserved further attention. In the current study, the CACNA1C SNP rs10744560 did not achieve nominal significance in our Chinese sample (p = 0.143), however, its OR in our sample is consistent with that in European GWAS14 (1.130 in Chinese versus 1.076 in Europeans), suggesting that the size of our sample is likely too small to reveal the true significant association between this SNP and BPD. The genome-wide significant association in the meta-analysis of our Chinese sample and previous studies also support this conclusion. CACNA1C is a promising BPD candidate gene that encodes the Cav1.2 subunit of voltage-gated calcium channels93,94; it is one of the best replicated genetic susceptibility genes for BPD14,18,19,21 and other psychiatric disorders9,11,52,95,96,97 across distinct populations, and the risk gene also affects the disease relevant phenotypes in humans98,99,100 and in animal models101,102,103 as implicated by multiple studies. Our results thus strongly supports the involvement of these genes in the genetic risk of BPD, but further replication studies are also necessary.
There are, however, several limitations in the present study and we are cautious about the interpretation of these results. First, we replicated the eQTL association between rs174576 and FADS1 expression in our Han Chinese amygdala sample using qRT-PCR techniques, while in the three European eQTL datasets, they all utilized RNA-seq methods. It is therefore interesting to examine the variation of gene expression quantification results using different methods, and future replication of the eQTL associations using RNA-seq in our Chinese amygdala sample is needed. Second, although we have excluded individuals with histories of major neuropsychiatric or neurological disorders in the control samples, there are the possibilities that some control individuals might also have other types of mental illness, such as cyclothymic disorder (this illness was not individually included in our BPD patients sample)104, although the prevalence of those illnesses in general populations is relatively low and might not significantly alter the association results.
In summary, we have provided additional support for prior association findings in FADS1, MAD1L1, SCN2A, and ITIH3, and have confirmed a risk locus at 10q26.13 that is associated with BPD. While we have presented the genetic association study of BPD using the Han Chinese samples recruited in Mainland China, and have explored relevant mechanisms using the cohort of brain amygdala samples of Chinese origin, the samples sizes are yet to be enlarged for better resolution and statistical power.
References
Craddock, N. & Jones, I. Genetics of bipolar disorder. J. Med. Genet. 36, 585–594 (1999).
Craddock, N. & Sklar, P. Genetics of bipolar disorder. Lancet 381, 1654–1662 (2013).
Grande, I., Berk, M., Birmaher, B. & Vieta, E. Bipolar disorder. Lancet 387, 1561–1572 (2016).
Muller-Oerlinghausen, B., Berghofer, A. & Bauer, M. Bipolar disorder. Lancet 359, 241–247 (2002).
Penzes, P., Cahill, M. E., Jones, K. A., VanLeeuwen, J. E. & Woolfrey, K. M. Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 14, 285–293 (2011).
Forrest, M. P., Parnell, E. & Penzes, P. Dendritic structural plasticity and neuropsychiatric disease. Nat. Rev. Neurosci. 19, 215–234 (2018).
Harrison, P. J., Geddes, J. R. & Tunbridge, E. M. The emerging neurobiology of bipolar disorder. Trends Neurosci. 41, 18–30 (2018).
Li, Z. et al. Genome-wide association analysis identifies 30 new susceptibility loci for schizophrenia. Nat. Genet. 49, 1576–1583 (2017).
Pardinas, A. F. et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat. Genet. 50, 381–389 (2018).
Converge consortium. Sparse whole-genome sequencing identifies two loci for major depressive disorder. Nature 523, 588–591 (2015).
Schizophrenia Working Group of the Psychiatric Genomics C. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014).
Ryu, E. et al. A genome-wide search for bipolar disorder risk loci modified by mitochondrial genome variation. Mol. Neuropsychiatry 3, 125–134 (2017).
Cichon, S. et al. Genome-wide association study identifies genetic variation in neurocan as a susceptibility factor for bipolar disorder. Am. J. Hum. Genet. 88, 372–381 (2011).
Stahl E., et al. Genomewide association study identifies 30 loci associated with bipolar disorder. bioRxiv 2017: 173062.
Hou, L. et al. Genome-wide association study of 40,000 individuals identifies two novel loci associated with bipolar disorder. Hum. Mol. Genet. 25, 3383–3394 (2016).
Chen, D. T. et al. Genome-wide association study meta-analysis of European and Asian-ancestry samples identifies three novel loci associated with bipolar disorder. Mol. Psychiatry 18, 195–205 (2013).
Ikeda, M. et al. A genome-wide association study identifies two novel susceptibility loci and trans population polygenicity associated with bipolar disorder. Mol. Psychiatry 23, 639–647 (2018).
Muhleisen, T. W. et al. Genome-wide association study reveals two new risk loci for bipolar disorder. Nat. Commun. 5, 3339 (2014).
Ferreira, M. A. et al. Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat. Genet. 40, 1056–1058 (2008).
McMahon, F. J. et al. Meta-analysis of genome-wide association data identifies a risk locus for major mood disorders on 3p21.1. Nat. Genet. 42, 128–131 (2010).
Psychiatric Gwas Consortium Bipolar Disorder Working Group. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat. Genet. 43, 977–983 (2011).
Bipolar Disorder and Schizophrenia Working Group of the Psychiatric Genomics Consortium. Genomic dissection of bipolar disorder and schizophrenia, including 28 subphenotypes. Cell 173, 1705–1715 e16 (2018).
Lee, M. T. et al. Genome-wide association study of bipolar I disorder in the Han Chinese population. Mol. Psychiatry 16, 548–556 (2011).
Edwards, S. L., Beesley, J., French, J. D. & Dunning, A. M. Beyond GWASs: illuminating the dark road from association to function. Am. J. Hum. Genet. 93, 779–797 (2013).
Bray, N. J. & Hill, M. J. Translating genetic risk loci into molecular risk mechanisms for schizophrenia. Schizophr. Bull. 42, 5–8 (2016).
Zhang, X. et al. Association of genetic variation in CACNA1C with bipolar disorder in Han Chinese. J. Affect. Disord. 150, 261–265 (2013).
Zhang, C. et al. ZNF804A genetic variation confers risk to bipolar disorder. Mol. Neurobiol. 53, 2936–2943 (2016).
Guan, L. et al. Common variants on 17q25 and gene–gene interactions conferring risk of schizophrenia in Han Chinese population and regulating gene expressions in human brain. Mol. Psychiatry 21, 1244–1250 (2016).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Li, Z.et al. A partition-ligation-combination-subdivision EM algorithm for haplotype inference with multiallelic markers: update of the SHEsis http://analysis.bio-x.cn. Cell Res. 19, 519–523 (2009).
Shi, Y. Y. & He, L. SHEsis, a powerful software platform for analyses of linkage disequilibrium, haplotype construction, and genetic association at polymorphism loci. Cell Res. 15, 97–98 (2005).
Pruim, R. J. et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 26, 2336–2337 (2010).
Viechtbauer, W. Conducting meta-analyses in R with the metafor package. J. Stat. Softw. 36, 1–48 (2010).
Jaffe, A. E. et al. Developmental and genetic regulation of the human cortex transcriptome illuminate schizophrenia pathogenesis. Nat. Neurosci. 21, 1117–1125 (2018).
GTEx Consortium. The genotype-tissue expression (GTEx) project. Nat. Genet. 45, 580–585 (2013).
Ng, B. et al. An xQTL map integrates the genetic architecture of the human brain’s transcriptome and epigenome. Nat. Neurosci. 20, 1418–1426 (2017).
Garrett, A. & Chang, K. The role of the amygdala in bipolar disorder development. Dev. Psychopathol. 20, 1285–1296 (2008).
Phillips, M. L., Drevets, W. C., Rauch, S. L. & Lane, R. Neurobiology of emotion perception I: The neural basis of normal emotion perception. Biol. Psychiatry 54, 504–514 (2003).
Blumberg, H. P. et al. Amygdala and hippocampal volumes in adolescents and adults with bipolar disorder. Arch. Gen. Psychiatry 60, 1201–1208 (2003).
Dickstein, D. P. et al. Frontotemporal alterations in pediatric bipolar disorder: results of a voxel-based morphometry study. Arch. Gen. Psychiatry 62, 734–741 (2005).
Chang, K. et al. Reduced amygdalar gray matter volume in familial pediatric bipolar disorder. J. Am. Acad. Child Adolesc. Psychiatry 44, 565–573 (2005).
Pfeifer, J. C., Welge, J., Strakowski, S. M., Adler, C. M. & DelBello, M. P. Meta-analysis of amygdala volumes in children and adolescents with bipolar disorder. J. Am. Acad. Child Adolesc. Psychiatry 47, 1289–1298 (2008).
Hibar, D. P. et al. Subcortical volumetric abnormalities in bipolar disorder. Mol. Psychiatry 21, 1710–1716 (2016).
Altshuler, L. et al. Increased amygdala activation during mania: a functional magnetic resonance imaging study. Am J Psychiatry 162, (1211–1213 (2005).
Pavuluri, M. N., O’Connor, M. M., Harral, E. & Sweeney, J. A. Affective neural circuitry during facial emotion processing in pediatric bipolar disorder. Biol. Psychiatry 62, 158–167 (2007).
Yurgelun-Todd, D. A. et al. fMRI during affect discrimination in bipolar affective disorder. Bipolar Disord. 2(3 Pt 2), 237–248 (2000).
Rich, B. A. et al. Limbic hyperactivation during processing of neutral facial expressions in children with bipolar disorder. Proc. Natl Acad. Sci. USA 103, 8900–8905 (2006).
Almeida, J. R., Versace, A., Hassel, S., Kupfer, D. J. & Phillips, M. L. Elevated amygdala activity to sad facial expressions: a state marker of bipolar but not unipolar depression. Biol. Psychiatry 67, 414–421 (2010).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25, 402–408 (2001).
Levinson, D. F. The genetics of depression: a review. Biol. Psychiatry 60, 84–92 (2006).
Ding, Y. et al. Molecular and genetic characterization of depression: overlap with other psychiatric disorders and aging. Mol. Neuropsychiatry 1, 1–12 (2015).
Green, E. K. et al. The bipolar disorder risk allele at CACNA1C also confers risk of recurrent major depression and of schizophrenia. Mol. Psychiatry 15, 1016–1022 (2010).
Schulze, T. G. et al. Molecular genetic overlap in bipolar disorder, schizophrenia, and major depressive disorder. World J. Biol. Psychiatry 15, 200–208 (2014).
Ritchie, G. R., Dunham, I., Zeggini, E. & Flicek, P. Functional annotation of noncoding sequence variants. Nat. Methods 11, 294–296 (2014).
Ward, L. D. & Kellis, M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 40(Database issue), D930–D934 (2012).
Fromer, M. et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat. Neurosci. 19, 1442–1453 (2016).
Sekar, A. et al. Schizophrenia risk from complex variation of complement component 4. Nature 530, 177–183 (2016).
Li, M. et al. A human-specific AS3MT isoform and BORCS7 are molecular risk factors in the 10q24.32 schizophrenia-associated locus. Nat. Med. 22, 649–656 (2016).
Genomes Project Consortium. et al. An integrated map of genetic variation from 1092 human genomes. Nature 491, 56–65 (2012).
Cruceanu, C. et al. Transcriptome sequencing of the anterior cingulate in bipolar disorder: dysregulation of G protein-coupled receptors. Am. J. Psychiatry 172, 1131–1140 (2015).
Akula, N. et al. RNA-sequencing of the brain transcriptome implicates dysregulation of neuroplasticity, circadian rhythms and GTPase binding in bipolar disorder. Mol. Psychiatry 19, 1179–1185 (2014).
Pacifico, R. & Davis, R. L. Transcriptome sequencing implicates dorsal striatum-specific gene network, immune response and energy metabolism pathways in bipolar disorder. Mol. Psychiatry 22, 441–449 (2017).
Zhao, Z. et al. Transcriptome sequencing and genome-wide association analyses reveal lysosomal function and actin cytoskeleton remodeling in schizophrenia and bipolar disorder. Mol. Psychiatry 20, 563–572 (2015).
Liu, Y. & McNamara, R. K. Elevated Delta-6 desaturase (FADS2) gene expression in the prefrontal cortex of patients with bipolar disorder. J. Psychiatr. Res. 45, 269–272 (2011).
Park, W. J., Kothapalli, K. S., Lawrence, P., Tyburczy, C. & Brenna, J. T. An alternate pathway to long-chain polyunsaturates: the FADS2 gene product Delta8-desaturates 20:2n−6 and 20:3n-3. J. Lipid Res. 50, 1195–1202 (2009).
Roke, K., Jannas-Vela, S., Spriet, L. L. & Mutch, D. M. FADS2 genotype influences whole-body resting fat oxidation in young adult men. Appl. Physiol. Nutr. Metab. 41, 791–794 (2016).
Abdelmagid, S. A. et al. Ethnicity, sex, FADS genetic variation, and hormonal contraceptive use influence delta-5- and delta-6-desaturase indices and plasma docosahexaenoic acid concentration in young Canadian adults: a cross-sectional study. Nutr. Metab. 12, 14 (2015).
Roke, K. & Mutch, D. M. The role of FADS1/2 polymorphisms on cardiometabolic markers and fatty acid profiles in young adults consuming fish oil supplements. Nutrients 6, 2290–2304 (2014).
Schuchardt, J. P. et al. Genetic variants of the FADS gene cluster are associated with erythrocyte membrane LC PUFA levels in patients with mild cognitive impairment. J. Nutr. Health Aging 20, 611–620 (2016).
Lattka, E., Illig, T., Heinrich, J. & Koletzko, B. FADS gene cluster polymorphisms: important modulators of fatty acid levels and their impact on atopic diseases. J. Nutr. Nutr. 2, 119–128 (2009).
Hsu, J. H., Chien, I. C. & Lin, C. H. Increased risk of hyperlipidemia in patients with bipolar disorder: a population-based study. Gen. Hosp. Psychiatry 37, 294–298 (2015).
Bai, Y. M., Su, T. P., Chen, M. H., Chen, T. J. & Chang, W. H. Risk of developing diabetes mellitus and hyperlipidemia among patients with bipolar disorder, major depressive disorder, and schizophrenia: a 10-year nationwide population-based prospective cohort study. J. Affect Disord. 150, 57–62 (2013).
McIntyre, R. S. et al. Bipolar disorder and metabolic syndrome: an international perspective. J. Affect Disord. 126, 366–387 (2010).
Boardman, J. P. et al. Common genetic variants and risk of brain injury after preterm birth. Pediatrics 133, e1655–e1663 (2014).
Jin, D. Y., Spencer, F. & Jeang, K. T. Human T cell leukemia virus type 1 oncoprotein Tax targets the human mitotic checkpoint protein MAD1. Cell 93, 81–91 (1998).
Sironi, L. et al. Mad2 binding to Mad1 and Cdc20, rather than oligomerization, is required for the spindle checkpoint. EMBO J. 20, 6371–6382 (2001).
Yoon, Y. M. et al. WD repeat-containing mitotic checkpoint proteins act as transcriptional repressors during interphase. FEBS Lett. 575, 23–29 (2004).
Musacchio, A. & Salmon, E. D. The spindle-assembly checkpoint in space and time. Nat. Rev. Mol. Cell Biol. 8, 379–393 (2007).
Pollard, K. S. et al. An RNA gene expressed during cortical development evolved rapidly in humans. Nature 443, 167–172 (2006).
Pollard, K. S. et al. Forces shaping the fastest evolving regions in the human genome. PLoS. Genet. 2, e168 (2006).
Ruderfer, D. M. et al. Polygenic dissection of diagnosis and clinical dimensions of bipolar disorder and schizophrenia. Mol. Psychiatry 19, 1017–1024 (2014).
Su, L. et al. Genetic association of GWAS-supported MAD1L1 gene polymorphism rs12666575 with schizophrenia susceptibility in a Chinese population. Neurosci. Lett. 610, 98–103 (2016).
Trost, S. et al. Investigating the impact of a genome-wide supported bipolar risk variant of MAD1L1 on the human reward system. Neuropsychopharmacology 41, 2679–2687 (2016).
Vassos, E. et al. Replication study and meta-analysis in European samples supports association of the 3p21.1 locus with bipolar disorder. Biol. Psychiatry 72, 645–650 (2012).
Breen, G. et al. Replication of association of 3p21.1 with susceptibility to bipolar disorder but not major depression. Nat. Genet. 43, 3–5 (2011). author reply.
Hamshere, M. L. et al. Genome-wide significant associations in schizophrenia to ITIH3/4, CACNA1C and SDCCAG8, and extensive replication of associations reported by the Schizophrenia PGC. Mol. Psychiatry 18, 708–712 (2013).
Li, Z. et al. Loci with genome-wide associations with schizophrenia in the Han Chinese population. Br. J. Psychiatry 207, 490–494 (2015).
Li, J. et al. Genes with de novo mutations are shared by four neuropsychiatric disorders discovered from NPdenovo database. Mol. Psychiatry 21, 290–297 (2016).
Li, J. et al. Targeted sequencing and functional analysis reveal brain-size-related genes and their networks in autism spectrum disorders. Mol. Psychiatry 22, 1282–1290 (2017).
Sanders, S. J. et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485, 237–241 (2012).
D’Gama, A. M. et al. Targeted DNA sequencing from autism spectrum disorder brains implicates multiple genetic mechanisms. Neuron 88, 910–917 (2015).
Dickinson, D. et al. Differential effects of common variants in SCN2A on general cognitive ability, brain physiology, and messenger RNA expression in schizophrenia cases and control individuals. JAMA Psychiatry 71, 647–656 (2014).
Bhat, S. et al. CACNA1C (Cav1.2) in the pathophysiology of psychiatric disease. Prog. Neurobiol. 99, 1–14 (2012).
Moon, A. L., Haan, N., Wilkinson, L. S. & Thomas, K. L., & Hall, J. Association with psychiatric disorders, behavior, and neurogenesis. Schizophr. Bull. 44, 958–965 (2018).
Ripke, S. et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat. Genet. 45, 1150–1159 (2013).
Zheng, F. et al. Further evidence for genetic association of CACNA1C and schizophrenia: new risk loci in a Han Chinese population and a meta-analysis. Schizophr. Res. 152, 105–110 (2014).
He, K. et al. CACNA1C, schizophrenia and major depressive disorder in the Han Chinese population. Br. J. Psychiatry 204, 36–39 (2014).
Bigos, K. L. et al. Genetic variation in CACNA1C affects brain circuitries related to mental illness. Arch. Gen. Psychiatry 67, 939–945 (2010).
Erk, S. et al. Brain function in carriers of a genome-wide supported bipolar disorder variant. Arch. Gen. Psychiatry 67, 803–811 (2010).
Cosgrove, D. et al. Cognitive characterization of schizophrenia risk variants involved in synaptic transmission: evidence of CACNA1C’s role in working memory. Neuropsychopharmacology 42, 2612–2622 (2017).
Dao, D. T. et al. Mood disorder susceptibility gene CACNA1C modifies mood-related behaviors in mice and interacts with sex to influence behavior in mice and diagnosis in humans. Biol. Psychiatry 68, 801–810 (2010).
Dedic, N. et al. Cross-disorder risk gene CACNA1C differentially modulates susceptibility to psychiatric disorders during development and adulthood. Mol. Psychiatry 23, 533–543 (2018).
Lee, A. S. et al. Selective genetic deletion of cacna1c in the mouse prefrontal cortex. Mol. Psychiatry 17, 1051 (2012).
Van Meter, A. R., Youngstrom, E. A. & Findling, R. L. Cyclothymic disorder: a critical review. Clin. Psychol. Rev. 32, 229–243 (2012).
Acknowledgments
We acknowledge with appreciation all the individuals with bipolar disorders and mentally healthy controls whose contributions made this work possible. We are deeply grateful to all the participants as well as to the physicians working on this project. This work was supported by grants from the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant no., XDB13000000); National Natural Science Foundation of China (81722019, 31701133, 31701088, 81471358, and 81771450); the medical and health science and technology project in Zhejiang (2018KY721); Shanghai Municipal Education Commission—Gaofeng Clinical Medicine Grant Support (20152530); the Shanghai Municipal Commission of Health and Family Planning Foundation, Key Developing Disciplines (2015ZB0405); Yunnan Provincial Science and Technology Department—Kunming Medical University Joint Applied Basic Research Project (2015FB031) and Health Science and Technology Plan projects in Yunnan Province (2017NS028) to Weiqing Liu; and research foundation from West China Psychiatric Association to Fang Liu. Xiao Xiao was supported by the Chinese Academy of Sciences Western Light Program, and Youth Innovation Promotion Association, CAS. Ming Li was supported by CAS Pioneer Hundred Talents Program and the 1000 Young Talents Program. The authors wish to acknowledge Prof. Jonathan Flint and the CONVERGE Consortium for providing the genotype data in control subjects of Han Chinese populations.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, L., Chang, H., Zhou, DS. et al. Replicated associations of FADS1, MAD1L1, and a rare variant at 10q26.13 with bipolar disorder in Chinese population. Transl Psychiatry 8, 270 (2018). https://doi.org/10.1038/s41398-018-0337-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-018-0337-x
This article is cited by
-
Clinical and genetic contributions to medical comorbidity in bipolar disorder: a study using electronic health records-linked biobank data
Molecular Psychiatry (2024)
-
CEGA: a method for inferring natural selection by comparative population genomic analysis across species
Genome Biology (2023)
-
CYP2C19-rs4986893 confers risk to major depressive disorder and bipolar disorder in the Han Chinese population whereas ABCB1-rs1045642 acts as a protective factor
BMC Psychiatry (2023)
-
MAD1L1 and TSNARE gene polymorphisms are associated with schizophrenia susceptibility in the Han Chinese population
BMC Medical Genomics (2021)
-
Further confirmation of netrin 1 receptor (DCC) as a depression risk gene via integrations of multi-omics data
Translational Psychiatry (2020)
