
Abstract
Bipolar disorder (BPD) and major depressive disorder (MDD) are primary major mood disorders. Recent studies suggest that they share certain psychopathological features and common risk genes, but unraveling the full genetic architecture underlying the risk of major mood disorders remains an important scientific task. The public genome-wide association study (GWAS) data sets offer the opportunity to examine this topic by utilizing large amounts of combined genetic data, which should ultimately allow a better understanding of the onset and development of these illnesses. Genome-wide meta-analysis was performed by combining two GWAS data sets on BPD and MDD (19,637 cases and 18,083 controls), followed by replication analyses for the loci of interest in independent 12,364 cases and 76,633 controls from additional samples that were not included in the two GWAS data sets. The single-nucleotide polymorphism (SNP) rs10791889 at 11q13.2 was significant in both discovery and replication samples. When combining all samples, this SNP and multiple other SNPs at 2q11.2 (rs717454), 8q21.3 (rs10103191), and 11q13.2 (rs2167457) exhibited genome-wide significant association with major mood disorders. The SNPs in 2q11.2 and 8q21.3 were novel risk SNPs that were not previously reported, and SNPs at 11q13.2 were in high LD with potential BPD risk SNPs implicated in a previous GWAS. The genome-wide significant loci at 2q11.2 and 11q13.2 exhibited strong effects on the mRNA expression of certain nearby genes in cerebellum. In conclusion, we have identified several novel loci associated with major mood disorders, adding further support for shared genetic risk between BPD and MDD. Our study highlights the necessity and importance of mining public data sets to explore risk genes for complex diseases such as mood disorders.
Similar content being viewed by others


Introduction
Major mood disorders, including bipolar disorder (BPD) and major depressive disorder (MDD), are leading causes of disability worldwide that account for a substantial proportion of productivity loss, life quality impairment, poor physical health, and deaths by suicide1,2. Earlier family and twin studies indicated a pivotal role of genetic factors in the etiology of mood disorders3,4,5,6; however, the specific modes of inheritance have been found to be complex and polygenic7,8,9,10. In brief, while BPD and MDD differ from each other in age of onset, clinical presentation, and treatment response11, they still share several specific clinical features such as the presence of depressive episodes, mood instability and impaired cognition. Moreover, meta-analyses of family studies have found elevated rates of BPD in first-degree relatives of MDD patients and vice versa 12. Taken together, there are likely shared genetic components underlying the pathogenesis of BPD and MDD. However, molecular genetic studies have provided limited evidence for this contention so far and only a handful of common risk genes for major mood disorders have been reported to date13,14. For example, in a previous study, McMahon et al.15 performed a genome-wide association study (GWAS) in a major mood disorder sample of 13,600 individuals, and found a genomic locus at 3p21.1 showing genome-wide significant association, though the risk SNP was latter shown to have stronger association with BPD than with MDD16,17. Therefore, understanding the genetic mechanism and pathogenic basis of major mood disorders still remains an important task.
To date, there have been several GWAS conducted in BPD or MDD samples9,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38. Though the current GWAS of mood disorders have identified fewer genome-wide significant genes than expected, their genome-wide statistical results have been (completely or partially) released publicly and these are valuable resources for further larger-scale GWAS meta-analysis. This systematic utilization of these public GWAS resources offers a great opportunity to perform genome-wide screens of the underlying shared genetic factors and will provide valuable information that will benefit other studies of major mood disorders. Therefore, we conducted a genome-wide meta-analysis of GWAS statistics utilizing large samples of individuals diagnosed with BPD or MDD and control subjects, followed by replications of suggestive associations (p < 1.0 × 10–6) in multiple independent samples from diverse populations (including a total of 32,001 cases and 94,716 controls). This study highlights that there are undiscovered “treasures” underlying the current mood disorder GWAS data sets, and illustrates an example of utilizing available public resources to further dissect the genetic basis of mood disorders.
Methods
Research strategy and experimental design
We performed a meta-analysis of two GWAS data sets in a total of 19,637 cases and 18,083 controls of European ancestry. We then evaluated the SNPs with suggestive genome-wide associations (p < 1.0 × 10–6) in additional 12,364 cases and 76,633 controls from various ethnic groups (Supplementary Table S1). All the protocols and methods used in this study were approved by the institutional review board of the Kunming Institute of Zoology, Chinese Academy of Sciences.
GWAS data sets meta-analysis
In the discovery stage, we performed a meta-analysis using summary statistics from a BPD GWAS and a non-overlapping MDD GWAS9,18. The BPD GWAS sample9 comprised 10,410 cases and 10,700 controls that partially overlapped with the Psychiatric Genomics Consortium (PGC1) BPD GWAS sample20. Clinical information regarding lifetime history of psychiatric illnesses was collected via standardized semi-structured interviews, and lifetime diagnoses were based on operationalized criteria. All cases had experienced pathological episodes of elevated mood (mania or hypomania) and depression and met the criteria for BPD within the primary study classification system. Controls were individuals without BPD selected from the same ethnic groups within the same geographical area. For the MDD analyses, we collected data of 9227 patients and 7383 controls from the PGC1 MDD GWAS sample18. The cases were defined by having lifetime diagnoses of MDD according to DSM-IV criteria by trained interviewers, or based on clinician-administered DSM-IV checklists using structured diagnostic instruments. For most of these participants, cases were obtained from clinical sources, and controls were randomly selected from the population. Detailed description of the samples, data quality, genomic controls, and statistical analyses can be found in the original GWAS reports9,18.
Illumina OmniExpress, Omni2.5, HumanHap610K, HumanCNV360-Duo or Affymetrix 6.0 were used for the genotyping9,18. In both GWAS, strict quality control metrics were applied to ensure the quality of the results. Samples with poor call rates, gender discordance, and/or abnormal heterozygosity as well as those of non-European ancestry were excluded. SNPs with a call rate <95% or Hardy–Weinberg Equilibrium (HWE) p-value <1 × 10–6 were removed. Each GWAS was imputed separately using IMPUTE2, and SNPs which were poorly imputed (INFO score <0.3) or had minor allele frequency <5% were excluded. There were ~1.3 million SNPs left for the current meta-analysis. In each GWAS, the associations of clinical diagnosis with SNP dosage was tested using logistic regression under an additive model, and covariates such as sample grouping and principal components reflecting the ancestry were taken into consideration. Meta-analyses of GWAS summary statistics were conducted using an inverse variance method under an appropriate effect model according to the test of heterogeneity. Manhattan, quantile–quantile (QQ) and regional plots were made using R (qqman package)39 and LocusZoom40, respectively.
Replication and technical validation
Replication analyses of the candidate loci were undertaken in two stages in a total of 12,364 cases and 76,633 controls from nine additional samples. The purpose of stage 1 replication was to narrow down the list of candidate SNPs. Since we believe that true genetic risk factors for general populations should show consistent significant associations with mood disorders, we performed this initial replication using the results from either publicly released data sets or from our own samples. After the initial replication, the candidate SNPs underwent the stage 2 replication, in which we collected large samples from international collaborators, and the associations survived stage 1 replication were further tested to confirm their roles in mood disorders. Detailed information of individual samples—including diagnostic assessment, genotyping and quality control—are shown in the Supplemental Data and Supplementary Table S1. The subjects of the replication samples were recruited under relevant ethical and legal guidelines within their respective areas, and all subjects provided written informed consent prior to participation.
There were 40 SNPs reaching the significance threshold of p < 1.0 × 10–6 in the discovery GWAS meta-analysis and were taken forward for validation in the Replication sample I, which included a BPD-type I sample from Romania (Romanian, 451 cases and 318 controls)41 and a MDD sample from the CONVERGE consortium (Chinese, 5303 cases and 5337 controls)21. These two samples were mainly comprising more homogeneous patients that have been discussed previously21,41, ensuring that we can identify true signals using these samples. In addition, we believe that using trans-ethnic sample (CONVERGE) helps to screen for authentic risk signals across populations. The 40 SNPs were first analyzed in each of these two samples and in the total Replication sample I, and then meta-analyzed in the total Replication sample I combining the discovery GWAS under an inverse variance weighted fixed-effect model. p-Values for heterogeneity were calculated using the Cochran’s Q-test. Five SNPs showing p-value lower than 5.0 × 10–8 in the combined samples were considered genome-wide significant, and were then subject to analyses in Replication sample II to further validate the associations.
The data sources of Replication sample II were mainly international collaborators, and most of the data sets have been used in previous large-scale studies to identify risk loci for mood disorders13,14. In brief, the Replication sample II were comprised of 6610 cases and 70,978 controls in total from Australia (330 BPD cases and 1811 controls)14, Germany (181 BPD cases and 527 controls)13, Japan (2964 BPD cases and 61,887 controls)22, GAIN African American (362 BPD cases and 671 controls)34, the Netherlands (389 MDD cases and 2056 controls)38, Switzerland (the PsyCoLaus cohort of 1301 MDD cases and 1689 controls)13, and China (1083 MDD cases and 2337 controls). Association analyses for the risk SNPs were conducted first in each of the above samples alone using logistic regression, and then meta-analyzed in the pooled Replication sample II to examine the overall associations. All assays were performed blind to diagnosis and genotype.
Healthy subjects for expression quantitative trait loci analysis
To identify the impact of risk SNPs on mRNA expression, we utilized two well-characterized gene expression databases to explore the gene expression regulation in the human tissues, providing valuable resources for functional follow-up studies for certain disease-associated variants. We carried out both discovery and replication analyses respectively using each database. We first used the GTEx (Genotype-Tissue Expression project)42 data set for the discovery analyses. GTEx contains both genetic variation and RNA-seq gene expression data from a diverse set of human tissues. The dorsolateral prefrontal cortex (DLPFC), hippocampus and cerebellum tissues have been highlighted in the pathogenesis of major mood disorders, we therefore retrieved data of tissues from these brain regions from GTEx for the expression quantitative trait loci (eQTL) analyses. Genes within 200 kb away from each risk SNP were analyzed for eQTL effects of that particular SNP, and Bonferroni correction was applied according to the number of included genes and brain tissues. To replicate the discovery in an independent sample, we conducted the replication analyses using data from the Braineac43, the database containing genetic information and whole transcriptome microarray expression information from postmortem brain tissues of 134 normal Caucasians. More detailed information regarding sample collection, processing and analyses can be found in the original studies42,43.
Results
Discovery GWAS meta-analyses and replication sample I analyses
We first conducted a meta-analysis of a BPD GWAS and a MDD GWAS comprising of 19,637 cases and 18,083 controls of European ancestry. After quality control exclusions, ~1.3 million SNPs with minor allele frequency (MAF) > 5% were meta-analyzed in the discovery stage using an appropriate effect model selected according to the heterogeneity test results. We confirmed that the two data sets came from populations with a common distribution by generating the Manhattan and QQ plots (shown in Fig. 1 and Supplementary Fig. S1). The genomic inflation factor (λ GC) for the meta-analysis was then calculated to be at 1.019. This GWAS meta-analysis yielded a total of nine independent loci (defined by at least 1 Mb separation between each other) reaching suggestive genome-wide significance (p < 1.0 × 10–6, Supplementary Table S2).

Horizontal line indicates threshold for genome-wide significance (p < 5 × 10−8)
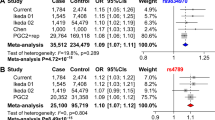
Those suggestive loci were then tested for their associations with major mood disorders in two additional samples (named Replication sample I as described above), including 5754 cases and 5655 controls in total, and the majority of the participants in Replication sample I were from China (5303 cases and 5337 controls, Supplementary Table S1). Notably, meta-analysis of populations with diverse ethnic backgrounds may increase the power to detect novel complex trait loci. This kind of study exhibits greater resolution for the fine-mapping of causal variants via leveraging differences in local linkage disequilibrium (LD) structure between ethnic groups44. Therefore, we believe that such trans-ethnic replication (European and Asian populations) will increase our confidence with the authenticity of the identified risk signal. Among the nine independent suggestive loci implicated in the GWAS meta-analysis, we found that three (2q11.2, 8q21.3, and 11q13.2) reached nominal significance in the Replication sample I (p < 0.05, Table 1 and S2). Meta-analysis in the sample pool combining discovery GWAS data sets and Replication sample I (including a total of 25,391 cases and 23,738 controls) showed that SNPs at 2q11.2, 8q21.3 and 11q13.2 were genome-wide significantly associated with major mood disorders (p < 5.0 × 10–8, Table 1 and Supplementary Table S2).
Although two SNPs in high LD (Europeans, r 2 = 0.97; East Asians, r 2 = 0.99) at 11q13.2 showed genome-wide significant associations with mood disorders in the discovery GWAS meta-analysis (rs7120256, p = 4.13 × 10–8; rs7119426, p = 4.33 × 10–8; Fig. 1 and Supplementary Table S2), they were not replicated in the Replication sample I (rs7120256, p = 0.479; rs7119426, p = 0.415; Supplementary Table S2). This phenomenon likely resulted from the genetic heterogeneity of these loci between populations. By contrast, there were other SNPs at 11q13.2 showing suggestive genome-wide significant associations in the GWAS meta-analysis (e.g., rs10791889, p = 4.40 × 10–7; rs2167457, p = 6.92 × 10–8; Table 1 and Supplementary Table S2), and were also associated (or marginally associated) with mood disorders in the Replication sample I (rs10791889, p = 0.012; rs2167457, p = 0.071; Table 1 and Supplementary Table S2). In the meta-analysis combining GWAS data sets and Replication sample I, we found that the latter two SNPs showed genome-wide significant associations (rs10791889, p = 1.70 × 10–8; rs2167457, p = 1.69 × 10–8; Table 1). A detailed LD examination revealed that rs10791889 and rs2167457 were in high LD (Europeans, r 2 = 0.92; Chinese, r 2 = 0.80); both were in strong LD with rs7120256 and rs7119426 in Europeans (r 2 ≥ 0.90), but in much weaker LD in Chinese population (r 2 < 0.60) (Supplementary Fig. S2). These data provide likely explanations for the divergent associations of the SNPs in the Replication sample I.
Several SNPs at 2q11.2 and 8q21.3 also achieved genome-wide significance when pooling the GWAS data sets and data of Replication sample I together (Supplementary Table S2), with the same direction of allelic effects in both European and Chinese populations. In contrast, other suggestive genome-wide significant SNPs within genomic regions of 1p31.1, 3p21.1, 3q11.2, 4q22.3, 17p12, and 20q13.12 in GWAS meta-analysis of European populations, were not significant in the Replication sample I (Supplementary Table S2).
Validations in replication sample II and joint meta-analyses
We further examined the genome-wide significant SNPs at 2q11.2 (rs17022433 and rs717454), 8q21.3 (rs10103191) and 11q13.2 (rs10791889 and rs2167457) in Replication sample II, which included a total of 6610 cases and 70,978 controls worldwide. In this replicative analysis, 11q13.2 SNPs were suggestively associated with major mood disorders (rs10791889, p = 1.48 × 10–2; Table 1), with the same direction of allelic effects as seen in the discovery GWAS meta-analysis. Notably, rs10791889 is the only SNP showing the genome-wide significance in the discovery and replication sample I, and was also nominally replicated in the independent replication sample II. The 2q11.2 and 8q21.3 SNPs, though not reaching nominal significance, showed the same trend of association with the discovery analysis (Table 1). We then combined samples from the GWAS datasets plus Replication sample I and II, which yielded a total of 32,001 cases and 94,716 controls, and conducted joint meta-analyses in this pooled sample. We found that SNPs at 2q11.2 (rs717454), 8q21.3 (rs10103191) and 11q13.2 (rs10791889 and rs2167457) were genome-wide significantly associated with major mood disorders (rs717454, p = 2.02 × 10–8; rs10103191, p = 2.55 × 10–8; rs10791889, p = 9.36 × 10–10; rs2167457, p = 4.92 × 10–9; Table 1). Regional plots of the risk loci are presented in Fig. 2. SNPs in 2q11.2 and 8q21.3 were novel mood disorders risk SNPs that have not been previously reported, and SNPs at 11q13.2 were in high LD with potential BPD risk SNPs implicated in previous GWAS analyses (rs10896135 was reported in those GWAS with p-value of 1.56 × 10–7, and it was in high LD with rs10791889 (r 2 > 0.9))20. The trans-ethnic replications increased our confidence with the authenticity of the risk loci, and were also consistent with our in prior hypotheses that most of the genome-wide significant SNPs would exhibit similar allelic frequencies between European and Asian populations (Europeans/Asians: rs717454-T, 0.570/0.688; rs10791889-T, 0.267/0.151; rs2167457-T, 0.272/0.133; for the only exception rs10103191, Europeans/Asians: rs10103191-A, 0.481/0.867).

40A physical map of the region is given and depicts known genes within the region, and the European population was used for the construction of LD structure
We then stratified the samples based on the case diagnostic status to examine the associations of the genome-wide significant variants with BPD (14,698 cases and 75,914 controls) or MDD (17,303 cases and 18,802 controls) alone. This stratified analysis revealed that the SNPs at 11q13.2 had larger effect sizes and stronger associations with BPD than with MDD, while SNPs at 2q11.2 and 8q21.3 showed similar effect sizes between BPD and MDD (Table 1). All the SNPs were associated with both disorders at nominal significance level.
Effects of the risk SNPs on nearby gene expression
The association between the risk SNPs with mood disorders in multiple independent samples lends statistical and biological support to the involvement of these genomic regions in risk for these disorders. However, these findings do not identify the underlying molecular mechanisms. Accumulating lines of evidence suggest that genetic risk factors likely contribute to the disease via affecting the expression of certain genes45,46,47,48. To explore whether transcriptional regulation explains the molecular mechanism underlying the risk SNPs identified in our meta-analyses, we carried out investigations using two existing eQTL databases42,43.
In the GTEx expression database42, the 2q11.2 SNPs (rs17022433 and rs717454) were significantly and selectively associated with LYG1 expression in the cerebellum (rs17022433, p = 0.0039; rs717454, p = 0.017; Fig. 3). More importantly, such associations in the cerebellum were replicated in the Braineac43 (rs17022433, p = 0.011; rs717454, p = 0.015; Supplementary Fig. S3). Rs10791889 and rs2167457 at 11q13.2 were significantly and specifically associated with the expression of C11orf80 in the cerebellum in GTEx database (rs10791889, p = 0.00051; rs2167457, p = 0.0017; Fig. 3). In Braineac, this association was replicated with marginal statistical significance in the cerebellum with the same allelic direction (rs10791889, p = 0.055; rs2167457, p = 0.079; Supplementary Fig. S3). While LYG1 and C11orf80 were the only significant genes in respective genomic risk regions, the eQTL associations survived multiple corrections and were repeatedly observed across independent data sets. Therefore, LYG1 and C11orf80 are likely reliable mood disorder related genes, and may contribute to the cerebellum associated pathogenic processes in the disease development. In fact, it has been reported that the cerebellum plays potential roles in psychiatric illnesses via its participation in the cortico-ponto-cerebello-thalamo-cortical circuit, by which it communicates with and modulates various congitive domains of the cerebral cortex; additionally, its role in motor coordination and procedural skill learning has been well established49,50,51. Konarski et al.50 have initially examined and synthesized the evidence from functional association studies of cerebellar stimulation, lesions, and brain imaging, through which they proposed the landmark hypothesis that abnormalities of the cerebellum play a crucial role in several psychiatric disorders including MDD and BPD.

Discussion
Major mood disorders are highly heritable traits, but the genetic association (e.g., GWAS) discoveries so far account for only a small portion of the inherited disease risk, which is probably primarily due to the polygenic nature and/or highly heterogeneous genetic architecture of the illnesses. However, GWAS remains an important approach, and it is widely accepted that the accumulation of such discoveries with growing sample sizes will serve as important steps toward the elucidation of biological pathways with etiologic relevance. As a result, mining the current available genetic data sets will help to identify previously undiscovered risk loci, deepening our knowledge of the pathogenesis of major mood disorders and providing valuable information about the genetic basis of these illnesses52,53,54,55.
The current study is the first report of genome-wide significant association between genetic variants on chromosome 2q11.2 and 8q21.3 and major mood disorders, although a limited number of previous studies have detected suggestive evidence of association in these regions15,20,31. We have also confirmed the associations of SNPs on chromosome 11 with mood disorders, which is consistent with a previous BPD GWAS20. These lines of evidence suggest potential involvement of those genomic regions in the illnesses, and eQTL analyses in brains have also highlighted certain genes. However, the functions of those genes in brain development and mood disorders are still unclear, so that further investigations are needed.
As previous major mood disorder GWAS have highlighted several genes, located elsewhere in the genome confer the risk of these illnesses20,30,31,33, we also tested those genes in our analyses. We detected nominal association for most of them (Supplementary Table S3), but our results cannot be considered as independent replications, as the samples we used partially overlapped with those in the previous studies20,30,31,33. A special mention deserves the intergenic SNP rs9834970 (TRANK1/LBA1 gene) that was not significant in the current meta-analysis of GWAS data sets (Supplementary Table S3) but was significant in the separate analysis of the Romanian BPD-type I sample41 and the Japanese BPD sample22 with the same direction of the effect as in the discovery study33. Notably, the significant results in our study may be different from those of the previous genome-wide analyses20,30,31,33, but this is not unexpected since each study has certain limitations in their power to detect the association and other unmeasurable factors such as sampling or technical differences between different studies may also contribute to this inconsistency. Therefore, even with such a large cohort, the aforementioned limitations might affect the power of this meta-analysis to capture all significant risk alleles at the genome-wide level.
Our genetic association results support the previous hypothesis that BPD and MDD share common genetic risk components12. However, these data are not sufficient to explain the phenomenon that only certain individuals among the risk variants carriers will develop BPD or MDD, while the others will remain healthy. Potential reasons may include the limited knowledge of the genetic risk variants for these illnesses, environmental influences, epistatic effects between variants, and perhaps unknown epigenetic factors.
This study also highlights the importance and necessity of utilizing public resources to dissect the genetic basis of complex diseases, such as major mood disorders. Many of the published GWAS have released (or partially released) their genome-wide statistical results18,19,20. Even though those GWAS have identified a limited number of genome-wide risk variants due to the sample size, they provided valuable data sets for further analyses and we believe that there are authentic undetected “treasures” underlying these public resources. In this case, we performed a meta-analysis of two public mood disorder GWAS data sets, followed by a set of independent replications, in which some of the replication samples were also available from public sources. Intriguingly, we identified some novel loci for major mood disorders with high confidence, suggesting that this approach is effective in studying major mood disorders, and may also be applicable in investigating other complex diseases.
Despite identifying several risk loci for major mood disorders, this study also raises the concern of potential bias of GWAS. Our current analyses largely rely on data of cases defined with wide diagnostic categories including all possible clinical presentations, and the fact that different cohorts were assembled by different researchers will thus introduce substantial variations regarding the clinical spectrum of cases. For example, systematic variation can occur in potentially important variables including disease symptoms, functional impairment, severity, comorbidity, response to interventions, familial loading, relevant environmental exposures, etc. As a result, the loci conferring risk across a broad phenotypic spectrum are most likely to emerge56. Therefore, future studies identifying the loci conferring risk of more specific symptoms are needed57, which will require different strategies for phenotypic refinement and the use of multiple large and well-characterized samples. A good example for this idea is the GWAS analyses of female individuals with recurrent MDD to reduce phenotypic heterogeneity, and successfully identified two genome-wide risk loci in a moderate sample size21. Besides, there are caveats remain to be addressed in the current study. For example, although we identified novel risk loci for major mood disorders, the lack of experimental investigation prevents us from further understanding their biological roles in the pathogenesis of the illnesses. Future studies on how these SNPs and genes confer risk of mood disorders are thus necessary.
In summary, using public resources, our GWAS meta-analysis followed by independent replications has identified several risk loci for categorically defined BPD and MDD. This is a clear demonstration of the genetic overlap between the major mood disorders that may explain susceptibility to such illnesses and conditions. These results not only contribute to our understanding of the pathogenesis of major mood illnesses, but also provide essential help in future reformation of psychiatric nosology, by contributing to a future scheme reflecting the underlying biology of psychiatric conditions rather than relying solely on the current diagnostic and classification system.
References
Cavanagh, J. T., Carson, A. J., Sharpe, M. & Lawrie, S. M. Psychological autopsy studies of suicide: a systematic review. Psychol. Med. 33, 395–405 (2003).
Lopez, A. D. & Murray, C. C. The global burden of disease, 1990-2020. Nat. Med. 4, 1241–1243 (1998).
Sullivan, P. F., Neale, M. C. & Kendler, K. S. Genetic epidemiology of major depression: review and meta-analysis. Am. J. Psychiatry 157, 1552–1562 (2000).
Craddock, N. & Forty, L. Genetics of affective (mood) disorders. Eur. J. Hum. Genet. 14, 660–668 (2006).
Craddock, N. & Jones, I. Genetics of bipolar disorder. J. Med. Genet. 36, 585–594 (1999).
Ding, Y. et al. Molecular and genetic characterization of depression: overlap with other psychiatric disorders and aging. Mol. Neuropsychiatry 1, 1–12 (2015).
Peterson, R. E. et al. The genetic architecture of major depressive disorder in Han Chinese women. JAMA Psychiatry 74, 162–168 (2017).
Milaneschi, Y. et al. Polygenic dissection of major depression clinical heterogeneity. Mol. Psychiatry 21, 516–522 (2016).
Ruderfer, D. M., et al. Polygenic dissection of diagnosis and clinical dimensions of bipolar disorder and schizophrenia. Mol. Psychiatry 19, 1017–1024 (2014).
Ross, J. et al. A rare variant in CACNA1D segregates with 7 bipolar I disorder cases in a large pedigree. Mol. Neuropsychiatry 2, 145–150 (2016).
Greenwood, T. A. Positive traits in the bipolar spectrum: the space between madness and genius. Mol. Neuropsychiatry 2, 198–212 (2017).
McGuffin, P. et al. The heritability of bipolar affective disorder and the genetic relationship to unipolar depression. Arch. Gen. Psychiatry 60, 497–502 (2003).
Chang H et al. The protocadherin 17 gene affects cognition, personality, amygdala structure and function, synapse development and risk of major mood disorders. Mol. Psychiatry https://doi.org/10.1038/mp.2016.231 (2017).
Li, M. et al. Allelic differences between Europeans and Chinese for CREB1 SNPs and their implications in gene expression regulation, hippocampal structure and function, and bipolar disorder susceptibility. Mol. Psychiatry 19, 452–461 (2014).
McMahon, F. J. et al. Meta-analysis of genome-wide association data identifies a risk locus for major mood disorders on 3p21.1. Nat. Genet. 42, 128–131 (2010).
Breen, G. et al. Replication of association of 3p21.1 with susceptibility to bipolar disorder but not major depression. Nat. Genet. 43, 3–5 (2011) author reply.
Vassos, E. et al. Replication study and meta-analysis in European samples supports association of the 3p21.1 locus with bipolar disorder. Biol. Psychiatry 72, 645–650 (2012).
Cross-Disorder Group of the Psychiatric Genomics Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 381, 1371–1379 (2013).
Major Depressive Disorder Working Group of the Psychiatric Gwas Consortium. et al. A mega-analysis of genome-wide association studies for major depressive disorder. Mol. Psychiatry 18, 497–511 (2013).
Psychiatric Gwas Consortium Bipolar Disorder Working Group. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat. Genet. 43, 977–983 (2011).
Converge consortium. Sparse whole-genome sequencing identifies two loci for major depressive disorder. Nature. 523, 588–591 (2015).
Ikeda M et al. A genome-wide association study identifies two novel susceptibility loci and trans population polygenicity associated with bipolar disorder. Mol. Psychiatry https://doi.org/10.1038/mp.2016.259 (2017).
Power, R. A. et al. Genome-wide association for major depression through age at onset stratification: major depressive disorder working group of the psychiatric genomics consortium. Biol. Psychiatry 81, 325–335 (2017).
Lewis, C. M. et al. Genome-wide association study of major recurrent depression in the U.K. population. Am. J. Psychiatry 167, 949–957 (2010).
Shi, J. et al. Genome-wide association study of recurrent early-onset major depressive disorder. Mol. Psychiatry 16, 193–201 (2011).
Muglia, P. et al. Genome-wide association study of recurrent major depressive disorder in two European case-control cohorts. Mol. Psychiatry 15, 589–601 (2010).
Hyde, C. L. et al. Identification of 15 genetic loci associated with risk of major depression in individuals of European descent. Nat. Genet. 48, 1031–1036 (2016).
Kohli, M. A. et al. The neuronal transporter gene SLC6A15 confers risk to major depression. Neuron 70, 252–265 (2011).
Hou, L. et al. Genome-wide association study of 40,000 individuals identifies two novel loci associated with bipolar disorder. Hum. Mol. Genet. 25, 3383–3394 (2016).
Muhleisen, T. W. et al. Genome-wide association study reveals two new risk loci for bipolar disorder. Nat. Commun. 5, 3339 (2014).
Ferreira, M. A. et al. Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat. Genet. 40, 1056–1058 (2008).
Cichon, S. et al. Genome-wide association study identifies genetic variation in neurocan as a susceptibility factor for bipolar disorder. Am. J. Hum. Genet. 88, 372–381 (2011).
Chen, D. T. et al. Genome-wide association study meta-analysis of European and Asian-ancestry samples identifies three novel loci associated with bipolar disorder. Mol. Psychiatry 18, 195–205 (2013).
Smith, E. N. et al. Genome-wide association study of bipolar disorder in European American and African American individuals. Mol. Psychiatry 14, 755–763 (2009).
Scott, L. J. et al. Genome-wide association and meta-analysis of bipolar disorder in individuals of European ancestry. Proc. Natl Acad. Sci. USA 106, 7501–7506 (2009).
Amin, N. et al. Nonsynonymous variation in NKPD1 increases depressive symptoms in European populations. Biol. Psychiatry 81, 702–707 (2017).
Direk N et al. An analysis of two genome-wide association meta-analyses identifies a new locus for broad depression phenotype. Biol. Psychiatry 82, 322–329 (2017).
Amin, N. et al. Exome-sequencing in a large population-based study reveals a rare Asn396Ser variant in the LIPG gene associated with depressive symptoms. Mol. Psychiatry 22, 537–543 (2017).
Turner, S. D. qqman: an R package for visualizing GWAS results using QQ and manhattan plots. Preprint at https://www.biorxiv.org/content/early/2014/05/14/005165 (2014).
Pruim, R. J. et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 26, 2336–2337 (2010).
Grigoroiu-Serbanescu, M., Diaconu, C. C., Heilmann-Heimbach, S., Neagu, A. I. & Becker, T. Association of age-of-onset groups with GWAS significant schizophrenia and bipolar disorder loci in Romanian bipolar I patients. Psychiatry Res. 230, 964–967 (2015).
GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 45, 580–585 (2013).
Ramasamy, A. et al. Genetic variability in the regulation of gene expression in ten regions of the human brain. Nat. Neurosci. 17, 1418–1428 (2014).
Morris, A. P. Transethnic meta-analysis of genomewide association studies. Genet. Epidemiol. 35, 809–822 (2011).
Luo, X.J. et al. Convergent lines of evidence support CAMKK2 as a schizophrenia susceptibility gene. Mol. Psychiatry 19, 774–783 (2014).
Li, M. et al. Allelic differences between Han Chinese and Europeans for functional variants in ZNF804A and their association with schizophrenia. Am. J. Psychiatry 168, 1318–1325 (2011).
Li, M. et al. Impact of a cis-associated gene expression SNP on chromosome 20q11.22 on bipolar disorder susceptibility, hippocampal structure and cognitive performance. Br. J. Psychiatry 208, 128–137 (2016).
Li, M. et al. A human-specific AS3MT isoform and BORCS7 are molecular risk factors in the 10q24.32 schizophrenia-associated locus. Nat. Med. 22, 649–656 (2016).
Andreasen, N. C. & Pierson, R. The role of the cerebellum in schizophrenia. Biol. Psychiatry 64, 81–88 (2008).
Konarski, J. Z., McIntyre, R. S., Grupp, L. A. & Kennedy, S. H. Is the cerebellum relevant in the circuitry of neuropsychiatric disorders? J. Psychiatry Neurosci. 30, 178–186 (2005).
Ito, M. Control of mental activities by internal models in the cerebellum. Nat. Rev. Neurosci. 9, 304–313 (2008).
Li, L., Chang, H., Peng, T., Li, M. & Xiao, X. Evidence of AS3MTd2d3-associated variants within 10q24.32-33 in the genetic risk of major affective disorders. Mol. Neuropsychiatry 2, 213–218 (2017).
Liu, Z., Huang, L., Luo, X. J., Wu, L. & Li, M. MAOA variants and genetic susceptibility to major psychiatric disorders. Mol. Neurobiol. 53, 4319–4327 (2016).
Li, M., Chang, H. & Xiao, X. BDNF Val66Met polymorphism and bipolar disorder in European populations: a risk association in case-control, family-based and GWAS studies. Neurosci. Biobehav. Rev. 68, 218–233 (2016).
Ou, J., Li, M., Xiao, X. The schizophrenia susceptibility gene ZNF804A confers risk of major mood disorders. World J. Biol. Psychiatry 18, 557–562 (2017).
Balaraman, Y., Lahiri, D. K. & Nurnberger, J. I. Variants in ion channel genes link phenotypic features of bipolar illness to specific neurobiological process domains. Mol. Neuropsychiatry 1, 23–35 (2015).
Zeledon, M. et al. Identification and functional studies of regulatory variants responsible for the association of NRG3 with a delusion phenotype in schizophrenia. Mol. Neuropsychiatry 1, 36–46 (2015).
Acknowledgements
This work was supported by grants from the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB13000000), and CAS Pioneer Hundred Talents Program (to M.L.). This work was also supported by the German Federal Ministry of Education and Research (BMBF) through the Integrated Network IntegraMent (Integrated Understanding of Causes and Mechanisms in Mental Disorders), under the auspices of the e:Med Programme (grant 01ZX1314A to S.C. and M.M.N., grant 01ZX1314G to M.R.). M.M.N. is a member of the DFG-funded Excellence-Cluster ImmunoSensation. The study was also supported by the German Research Foundation (DFG; grant FOR2107, RI908/11-1 to M.R., NO246/10-1 to M.M.N.). The Romanian sample recruitment and genotyping was funded by UEFISCDI, Bucharest, Romania, grant no. 89/2012 to M.G.S. and by the German Federal Ministry of Education and Research (BMBF) through the Integrated Network IntegraMent (grant 01ZX1314A to M.M.N. and S.C.). The Australian cohorts were supported by the Australian National Health and Medical Research Council Program Grant GNT1037196. The ERF study as a part of EUROSPAN (European Special Populations Research Network) was supported by European Commission FP6 STRP grant number 018947 (LSHG-CT-2006-01947) and also received funding from the European Community’s Seventh Framework Programme (FP7/2007-2013)/grant agreement HEALTH-F4-2007-201413 by the European Commission under the programme “Quality of Life and Management of the Living Resources” of 5th Framework Programme (no. QLG2-CT-2002-01254). High-throughput analysis of the ERF data was supported by joint grant from Netherlands Organization for Scientific Research and the Russian Foundation for Basic Research (NWO-RFBR 047.017.043). Exome sequencing was supported by the ZonMw grant (project 91111025). We are grateful to all study participants and their relatives, general practitioners, and neurologists for their contributions and to P. Veraart for her help in genealogy, J. Vergeer for the supervision of the laboratory work and P. Snijders for his help in data collection. N.A. is supported by the Hersenstichting Nederland (project number F2013(1)-28). The data in GAIN-AA sample used for the analyses described in this manuscript were obtained from dbGaP accession number phs000021.v3.p2. We are grateful to all the voluntary donors of DNA samples in this study. We thank members of the Psychiatric Genomic Consortium, who shared the PGC GWAS data.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing financial interests.
Additional information
Members of Advanced Collaborative Study of Mood Disorder (COSMO) Team
The members for the advanced Collaborative Study of Mood Disorder (COSMO) team are listed in the Supplemental Material.
MooDS Bipolar Consortium
Additional members in the MooDS Bipolar Consortium can be found in the Supplemental Material.
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The members of for the Advanced Collaborative Study of Mood Disorder (COSMO) Team are listed in the Supplemental Material.
Additional members in the MooDS Bipolar Consortium can be found in the Supplemental Material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xiao, X., Wang, L., Wang, C. et al. Common variants at 2q11.2, 8q21.3, and 11q13.2 are associated with major mood disorders. Transl Psychiatry 7, 1273 (2017). https://doi.org/10.1038/s41398-017-0019-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-017-0019-0
