
Abstract
Similarities and differences of phenotypes within local co-occurring species hold the key to inferring the contribution of stochastic or deterministic processes in community assembly. Developing both phylogenetic-based and trait-based quantitative methods to unravel these processes is a major aim in community ecology. We developed a trait-based approach that: (i) assesses if a community trait clustering pattern is related to increasing environmental constraints along a gradient; and (ii) determines quantitative thresholds for an environmental variable along a gradient to interpret changes in prevailing community assembly drivers. We used a regional set of natural shallow saline ponds covering a wide salinity gradient (0.1–40% w/v). We identify a consistent discrete salinity threshold (ca. 5%) for microbial community assembly drivers. Above 5% salinity a strong environmental filtering prevailed as an assembly force, whereas a combination of biotic and abiotic factors dominated at lower salinities. This method provides a conceptual approach to identify consistent environmental thresholds in community assembly and enables quantitative predictions for the ecological impact of environmental changes.
Similar content being viewed by others

Introduction
The main goal of community ecology is to understand the underlying forces that determine species numbers and identities, and their relative abundances, across spatio-temporal scales. Many recent efforts are aimed at developing robust methods to estimate and understand the relative importance of different community assembly processes [1]. Ecologists generally accept that community assembly must necessarily be driven by both dispersal-assembly and niche-assembly mechanisms [2,3,4]. Dispersal-assembly theories emphasize (1) stochastic colonization (driven largely by species pools) and (2) local extinctions (driven by random events affecting small populations sizes) [5, 6], while niche-assembly highlights (1) the importance of species differences and (2) their interactions in predicting community composition [7,8,9]. Niche-assembly mechanisms can affect either extinction or colonization. From this perspective, local extinctions result in general from negative interactions. They are often caused by competitive exclusion of sub-dominant species disappearing from the system due to a worse overall performance [10, 11], but strong environmental filtering (cf. species sorting [12]) can also prevent colonization and establishment of poorly adapted species.
In contrast, positive interactions (facilitation) decrease the probability of local extinctions, enhancing persistence and biodiversity [13,14,15]. Assessing the link between trait variation, the environment, and biotic interactions (either positive or negative) is not trivial [1, 16,17,18]. Further, there is an increasing recognition of the need for methods to make robust predictions that help understand responses of species assemblages to environmental shifts [19, 20].
In studying community assembly of macroorganisms, spatio-temporal scales are commonly huge, making long-term ecological research challenging. Microbes, in contrast, thrive in highly diverse communities and are characterized by short time scales. This makes microbial communities ideal study systems, providing robust insights into assembly processes [21, 22]. Species and individuals within assemblages respond to the environment according to their functional characteristics. In particular, microbes with stress tolerance traits are selected in specific environmental conditions [23]. Genomic traits allow the linkage of bacterial functions to their environmental preferences at different scales, from local habitats to biogeographical regions [24]. This approach could be especially helpful across habitats with strong environmental constraints, where a variety of functional adaptations can emerge along a gradient.
Moreover, there is an increasing recognition that variations in the distribution of functional traits along environmental gradients can be quantified [18, 25, 26]. Whenever niche-assembly mechanisms are overridden by strong random processes, the local community composition is expected to be akin to a random sample from the regional pool [27].
When this happens, local trait distributions should not differ significantly from the regional trait distribution. Conversely, differences between local and regional trait distributions are a fingerprint of the existence of specific drivers of community assembly. The classical framework by Webb [28] considers clustering in trait values as a signature of environmental filtering, while Mayfield and Levine [10] remarked that such clustering can result also from competitive exclusion.
Here, we use shifts in the distribution of functional traits along an environmental gradient to assess the ecological impact of a changing environment on community trait structure. Our methods are inspired by the data randomization/resampling null model approach [29]. Our trait-based approach consistently finds thresholds in an environmental variable along a gradient with the aim of interpreting meaningful changes in the type of drivers responsible for community assembly. We call this index-based method RTCC—Randomized Trait Community Clustering.
Materials and methods
Details related to sampling sites, molecular methods, bioinformatics processing, and additional statistics are provided in SI Materials and Methods, while null models of community assembly are fully described in this section. See also supplementary text and Fig. S1 for a summarized work-flow of the methods carried out in this study. In addition, source code for the RTCC Method and sample data files are available in GitHub. Briefly, samples were collected from the Monegros Desert area (NE Spain, 41°42′N, 0°20′W) which harbors among the largest number of inland saline lakes in Europe [30]. A total of 148 samples were taken from 14 different shallow lagoons (Fig. S4). We explored the spatio-temporal variation of a regional set of these local aquatic communities driven by a wide salinity gradient (0.1–40% of dissolved salts) (Fig. S5A). Temporal data of wind speeds were obtained from the Meteorological state agency reporting data of an automatic station in the area (Bujaraloz) (Fig. S5B). For DNA analyses, water samples were pre-filtered in situ through a 50 μm-pore-size net, to retain large zooplankton and algae, and 100–500 mL were subsequently filtered on 5 μm and then 0.2 μm pore-size polycarbonate membranes (47 mm diameter).
The membranes were enzymatically digested and phenol–chloroform extracted [30]. To obtain community profiles, we performed a NGS sequencing step targeting the V4 region of the bacterial 16S rRNA gene by means of a high-speed multiplexed Illumina MiSeq platform. Raw sequences were processed using the UPARSE pipeline [31]. After the processing steps, we only kept those samples that presented library sizes larger than 10,000 reads/sample. The final pool consisted of 136 samples with 9993 operational taxonomic units (OTUs). In order to associate OTUs to genome content, we used a 16S rRNA database [32] linked to the IMG genomic database [33]. OTUs were matched to 16S rRNA gene records available in the PATRIC genomic database (as of January 2016) [32]. The average percentage of abundance matched per sample was ca. 62%. We tested the representativeness of the subset matching genomes in our approach (see Figs.S1 and S2), i.e., we explored if the ordination pattern of the whole dataset was consistent with the ordination of the subset. In addition, a Mantel test (based on Spearman correlation) was also used to conclude on this decision-making step. See also the RTCC Method protocol in SI document. Functional predictions based on representative genomes allow for an inferring of genomic and metabolic potential of 16S rRNA sequences [34, 35]. For this we downloaded available genomic traits of potential interest from IMG [33]: genome size, gene count, % GC, % coding base, % CDS, % RNA, rRNA count, % transporter proteins, % signal peptide, and % transmembrane proteins. We selected 10 traits that summarize genomic structural/functional variability, some of them known to respond to ecological adaptations.
Null models of community assembly
In order to quantify the importance of environmental filtering in the assembly of ecological communities, we developed a measure of the degree of clustering of a set of samples (or sites). The pool of species was formed by all the species observed in the complete set of samples taken from the lagoons. First we calculated a measure of average dissimilarity in the trait distribution of a sample as the pair-wise mean trait difference between every species pair in the sample, the functional mean pairwise distance (MPD) [28]
where xi is the trait value of species i in the sample (with n reported species). Next we tested if the observed measure ∆x significantly differed from the expectation of the same measure across realizations of consistent null models with the same number of species as appearing in the empirical sample. We assumed that any species from the pool can take part in a null-model synthetic community. We build the null-model dissimilarity distribution over replicates to obtain a p-value, i.e., the quantile defined by the empirical difference within the distribution of simulated differences across realizations. We tested whether the empirical sample dissimilarity value was significantly large or small, indicating over-dispersion or clustering, respectively. If the observed difference of our empirical sample was under the lower 5% of the simulated distribution trait differences, we considered that sample as showing significant clustering (Fig. 1a, left). We repeated this test for every sample in our data set, which yielded the p-value distributions for each target trait (Fig. 1a, right). Let N be the number of samples in the data set, and Nc the number of samples that showed significant clustering when tested against the null model. We defined a clustering index,
as the proportion of samples showing significant clustering over all the samples considered. Note that the clustering index is a global measure for the whole set of samples. Error bars for the clustering index were calculated by averaging this quantity over 100 repetitions of the distribution of p-values according to the null model (Fig. 1b).

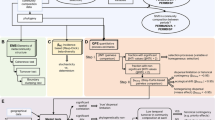
Methodological framework for the Randomized Trait Community Clustering (RTCC) method. a For each sample from different water bodies over a given region we test the hypothesis that observed average dissimilarity (see Eq. (1)) is compatible with a given null model. These non-parametric randomizations assign a p-value for each sample. For all samples in the dataset we represent the p-value distributions obtained by the null model for a given functional trait. Significantly low p-values are related to clustering (p < 0.05), and significantly high p-values to over-dispersion. b For the whole sample set, which we define here as a metacommunity, we calculate the fraction of samples that yield significant clustering, i.e., its clustering index (see Eq. (2)). We average several realizations of the null model in order to get average clustering index and error bars. c Samples are sequentially removed along decreasing values of the environmental variable to define nested metacommunities; for each of them we calculate the clustering index (left). The significance of this curve is assessed by building an ensemble of curves corresponding to removing samples in random orderings, which are then used to define the shaded area (right)
The null models developed differ in the definition of the species pools potentially available for each of the samples, that is, in the way species were drawn from the species pool:
-
i.
Random assembly: For all samples, all species are potentially available. They were chosen at random from the pool with equal probability.
-
ii.
Abundance-based assembly: For all samples, all species are potentially available, but had a probability of being chosen proportional to their abundance in the entire species pool.
-
iii.
Environmental range-based assembly: For a given sample, only those species whose ranges for the environmental factor encompass the value observed in that sample are eligible to be drawn and form the corresponding randomly generated sample. Potentially eligible species are drawn with uniform probability. Then, in this constrained way, we build a set of random synthetic samples, each characterized by a given value of the environmental factor and the same number of species as observed in each corresponding observed sample. For each species, the range of the environmental factor is defined by the minimum and maximum values of the variable across the samples in which the species is observed.
-
iv.
Environmental range and abundance-based assembly: As (iii), but, additionally, species whose ranges for the environmental factor contain the value of the empirical sample are randomly drawn proportionally to their abundances in the species pool.
For the purpose of discerning if significant clustering (or the lack of it) was influenced by the environmental gradient, we iteratively excluded the sample from the set with the highest value of the environmental variable of interest, also excluding from the pool, the corresponding species that appeared only in that sample, and recalculated the clustering index with the remaining samples (Fig. 1c, left). Additionally, we tested the significance of clustering patterns according to the four null models by applying the same sequential removal of samples, but first randomizing their salinity order (see Supplementary Materials for further details). These randomizations provided a 95% confidence interval for the clustering index (Fig. 1c, right) that allows you to decide if the observed clustering pattern as the environmental variable decreases is related to the environmental factor or not.
Statistics
Significant breakpoints of observed patterns along environmental gradients, either from ordination analyses or clustering indices, were estimated by means of the maximum F statistic derived from sequential Chow-tests (sctest function from strucchange package) [36] (see Fig. S3).
Results
We developed a detailed theoretical and methodological framework for the RTCC method (see Fig. 1, the methods section for more details, and Fig. S1 for a summarized workflow of the methodology). To test the potential of this trait-based approach for gaining new insights about different community assembly mechanisms, we explored microbial communities along an environmental (salinity) gradient in a set of shallow saline ponds. We first evaluated 10 genomic traits that were available in public databases and that summarized genomic structural/functional variability, some of them known to respond to ecological adaptations (Fig. 2 and see Supplementary Material, section "Matching genomes"). Among them, the percentage of DNA dedicated to signal peptide synthesis exhibited the strongest clustering signal. Therefore, this trait, characterizing each species within a sample, was primarily used to further conduct the RTCC analyses. The pairwise differences for this trait were significantly smaller than those expected in synthetic communities randomly assembled from the pool according to the first null model (see “"Methods” section below).

Clustering distribution patterns for different functional traits. The p-value distributions obtained by the random assembly null model for 10 functional traits are shown. Colored stripes show the ranges of significant over-dispersion (p ≥ 0.95) and significant clustering (p ≤ 0.05). Signal peptide exhibit a strong signal of clustering
To assess the role of environmental factors in sorting species from empirical communities along the gradient, we analyzed the relationship between clustering patterns and two environmental variables with a battery of assembly null models (see the “Methods” section). Salinity and wind velocity should have different impacts on community assembly and, therefore, result in distinct patterns of clustering index values. We use wind as a control variable because it should represent a stochastic continuous perturbation, while salinity would provide a deterministic influence. We represented clustering index values by sequentially removing samples in decreasing order of the environmental variable (see Fig. 3a, b). We then tested the significance of the environmental variable by comparing the declining patterns with replicates of the same methodology based on random orderings (95% confidence intervals are shown as shaded areas). The rightmost point of the curves corresponded to the clustering index for all samples in the data set. The random assembly null model yielded a clustering index value h~0.9, which means that around a 90% of samples were not compatible with random assemblages, that is, showed an average trait dissimilarity significantly different from null model expectations. In other words, the vast majority of empirical samples exhibited a high degree of clustering in signal peptide compared to purely random assemblages. Such level of clustering declined when synthetic communities were built by selecting species with a probability proportional to relative abundances, and when ranges of salinity were taken into account in the assembly of simulated communities. As null model complexity increased, the fraction of samples compatible with the null hypothesis increased (see Fig. 3b). For the most constrained model, which takes into account both species abundances and salinity ranges, only at most ~30% of samples were not compatible with the null hypothesis underlying this model (see Fig. 3, lower curve). All null models showed a plateau in clustering index as the environmental variable decreased, but, at some point, a sharp decline occurred. The curves for salinity lay outside the confidence interval expected for randomized orderings, hence the null model was not able to explain the variation of clustering as salinity decreased. This result is consistent with a deterministic role of the salinity environmental gradient in community assembly. We repeated the same methodology using wind speed as a control environmental variable. Wind velocity showed a different pattern from salinity, that is, white curves laying within the shaded area in both tests (Fig. 3a). Since clustering values for ordered sample removal lie within null model confidence intervals for randomized orderings, wind speed is not a deterministic driver of community assembly, but, as expected, plays a stochastic role (Fig. 3a). Although the OTU-based ordination approach revealed a weak positive correlation signal, this marginal correlation is well below the strong correlation observed for salinity (compare D and B panels in Fig. S6).

Null models for community assembly. a Randomized signal peptide community clustering in salinity and maximum wind speed according to null models (i) and (ii), random assembly and abundance-based assembly, respectively. The clustering index for the signal peptide trait is plotted versus the maximum value of the environmental variable among all samples still present after every removal step. Samples are sequentially removed one-by-one in decreasing order of the variable. Note that wind velocities have been normalized so that their maximum values correspond to the maximum salinity observed. The shaded gray areas correspond to confidence intervals for the different null models when the random removal of samples is repeated multiple times. b Same as a for null models (iii) and (iv), environmental range-based assembly, and environmental range and abundance-based assembly, respectively, which are meaningful only for salinity. c Clustering index curves are plotted again along with their thresholds, all of them lying within the rectangle (shaded in gray) yielded by the OTU-based approach, whose limits corresponds to the significant breakpoints of the observed pattern along the salinity gradient (Fig. S6)
Chow tests were used to determine a threshold value in salinity over which environmental constraints emerge as a leading driver. We found percentages of salinity ranging from 3.2% for the random assembly null model and 4.8% for the ranges and abundances null model. These values lay within the range of salinity that presented significant breakpoints on the changing rate of communities in the ordination analysis (Fig. 3c, see also Fig. S6). We observed that traits other than signal peptide could be used to determine thresholds along the gradient (for example, the percentage of GC pairs in sequences; see Fig. S7). However, this was not necessarily true for any trait (for example, the percentage of DNA associated to transporter proteins, see also Fig. S7). Finally, signal peptide average values across genomes per sample (relative to the metacommunity average) were plotted along the salinity gradient. We observed that average trait values decreased as salinity increased (Fig. S8).
Discussion
Competitive exclusion increasing local extinctions of sub-adapted species and strict environmental filtering preventing effective colonization of non-tolerant species along an environmental gradient, lie at two extremes of a continuum. Accurately disentangling these two main drivers is challenging. We developed a method to evaluate the effect of an environmental gradient on community trait structure, and provide a conceptual framework (Fig. 4) to interpret consistent discrete environmental thresholds that helps separate an assembly regime driven by a rich combination of several biotic and abiotic factors from a regime characterized by strong environmental constraints. The observed threshold unveils the switch point above which a structuring force starts dominating along the gradient (see the highest values of the clustering index along the plateau). Conversely, the sharp decline of the clustering index below the threshold value can be regarded as a signature of the rapid decay of the predominant role of a given environmental variable in driving local community assembly (Fig. 4). Thus, the lower the environmental pressure the lower the proportion of samples exhibiting significant clustering (i.e., the lower clustering index) due to the increased dispersion of trait values within local samples.

Conceptual framework. A conceptual model for the interpretation of results based on the Randomized Trait Community Clustering (RTCC) method. We indicate the type of dominant deterministic forces acting along a gradient of a meaningful environmental variable. The picture shows the pattern observed along the salinity gradient by using null model (iv). The shaded gray area corresponds to confidence intervals for the same null model but randomizing sequential sample removal. A significant correlation of the trait clustering pattern with an environmental gradient exists if clustering index values for a given trait (under ordered sequential removal) lie outside the shaded area, this is, the null model fails to explain the variation of clustering along the gradient. The vertical line is the threshold salinity value separating the two community assembly regimes
Trait-based approaches have been widely used by ecologists to identify general community-level patterns of macro-organisms based on phenotypic characters [25, 37,38,39]. Some authors have made considerable efforts to introduce this framework in the study of microbial communities [22, 40,41,42], among others. Burke et al. [43] suggests that gene function, rather than the RNA taxonomy approach, holds the key to properly studying bacterial community assembly. Community composition may vary through several processes, while community functionality may remain stable due to functional redundancy of taxa [44, 45]. If stochastic forces are not too strong, selected species traits are expected to be differently favored and cluster around certain optimal values along a structuring environmental gradient. The distribution of trait values across species along a given gradient will be the result of a balance between environmental filtering and biotic interactions. Some traits can be unambiguously related to either stress-tolerance or competitive abilities. In this case, they can be used to distinguish between environmental filtering vs. competitive exclusion [17, 23] in spite of these two process producing the same trait clustering patterns [10].
Our findings indicate that the relative amount of genomic DNA coding for signal peptide shows a significant clustering pattern along the salinity gradient. Signal peptides are short peptides (5–30 aminoacids) present at the N-terminus of newly synthesized proteins that control the final destiny of these proteins, this is, whether they end up anchored to the cell membrane or are extracellularly secreted [46]. The diversity of these signaling systems decreases along salinity gradients in favor of the signal peptide-dependent Tat system, which dominates under high salinity conditions [47,48,49]. The observed decrease in average signal peptide values per sample along the salinity gradient (see Fig. S8) suggests that this trait could be involved in salinity stress tolerance, although confirming this requires further experimental research. Likewise, as salinity increases, we also observed higher similarity in GC content across locally co-occurring species (see Fig. S7). Several studies have suggested that hypersaline inhabitants are characterized by high GC content although there are some exceptions to these general patterns [50]. Moreover, it has been suggested that the stress effect of extreme environments is reflected in the nucleotide composition [40]. Other functional traits are not critically influenced by this gradient. In other words, some traits with relevant ecological roles may be related to other functional strategies not linked to the main environmental gradient, such as, for instance, the rRNA operon copy number, known to reflect bacterial growth rates [51], which are related to competitive ability [41].
Salinity has been described as one of the major environmental drivers of microbial community composition at a global scale [52,53,54]. Important ecological changes have been described along salinity gradients from multipond solar salterns [55], with decreasing biodiversity as salinity increases [56], with an accompanying loss of metabolic processes [57]. Here, the specific trait pattern reveals the ecological functional adaptation of bacterial communities to the salinity gradient. Structural changes shown under a taxonomic view followed a similar pattern along the gradient (Fig. S6B). An increase in salinity beyond a threshold close to 5% triggers both a rapid change in community composition, and a decrease in mean relative signal peptide values (Fig. S8, upper panel). Maximum trait clustering was also reached at the same threshold. Above 5% salinity, community trait structure tends to stabilize, which means that there is functional redundancy in taxa while community composition still changes, but showing a slower turnover.
Between ca. 10% and 40% salinity range, community composition remained more stable. This shows that the RTCC method provided information not only about functional adaptation of communities—complementing results from their taxonomic turnover—but also about the accurate position of a salinity threshold (5%) over which environmental constraints strongly shape community assembly. Since traits are selected based on the degree of clustering signal rather than on previous trait knowledge, our method opens new possibilities to objectively determine both whether or not and where a functional adaptation occurs along an environmental gradient.
It is reasonable to think that, in principle, if an environmental variable changes very smoothly, community composition, and, consequently, community trait structure, should also change smoothly. However, the opposite seems to be true in microbial communities along a salinity gradient. The RTCC accurately finds the position of a salinity threshold.
Ecological theory predicts that the end points of community assembly can be multiple [58]. The possibility that several stable species assemblages can exist under the same environmental conditions opens the door to observing abrupt transitions in community composition as a single environmental variable slowly changes. The study of the origins and underlying causes of these break points in community composition along environmental gradients deserves further empirical and theoretical work.
To conclude, we provide a new conceptual approach with the RTCC method that could be useful in guiding hypothesis-driven studies coping with the high complexity of biological systems. Although we used a data set from microbial communities, we highlight the potential of this method to be applied more generally to quantitative phenotypic or genotypic trait data measured for macro-organismal communities as well. A combination of theoretical modeling through trait-based analyses allows us to go beyond description and deepen our mechanistic understanding of community assembly. Quantifying how community assembly is shaped by the environment is critical to predicting the ecological impact of environmental changes, and our approach provides a powerful tool to advance this analysis.
References
van der Plas F, Janzen T, Ordóñez A, Fokema W, Reinders J, Etienne RS, et al. A new modeling approach estimates the relative importance of different community assembly processes. Ecology. 2015;96:1502–15.
Vellend M. Conceptual synthesis in community ecology. Q Rev Biol. 2010;85:183–206.
Vellend M. The theory of ecological communities (MPB-57). Princeton University Press; 2016.
Gravel D, Canham CD, Beaudet M, Messier C. Reconciling niche and neutrality: the continuum hypothesis. Ecol Lett. 2006;9:399–409.
MacArthur RH. Species packing and what interspecific competition minimizes. Proc Natl Acad Sci USA. 1969;64:1369–71.
Hubbell SP. The unified theory of biodiversity and biogeography. Princeton: Princeton University Press; 2001.
Tilman D. Resource competition and community structure. Princeton, NJ: Princeton University Press; 1982.
Law R, Morton RD. Permanence and the assembly of ecological communities. Ecology. 1996;77:762–75.
Law R, Weatherby AJ, Warren PH. On the invasibility of persistent protist communities. Oikos. 2000;88:319–26.
Mayfield MM, Levine JM. Opposing effects of competitive exclusion on the phylogenetic structure of communities. Ecol Lett. 2010;13:1085–93.
Chesson PL. Mechanisms of maintenance of species diversity. Annu Rev Ecol Syst. 2000;31:343–66.
Liebhold AM, Chase J. Metacommunity ecology. Princeton: Princeton University Press; 2018.
Bertness M, Callaway RM. Positive interactions in communities. Trends Ecol Evol. 1994;9:191–3.
Maestre FT, Callaway RM, Valladares F, Lortie CJ. Refining the stress-gradient hypothesis for competition and facilitation in plant communities. J Ecol. 2009;97:199–205.
Bastolla U, Fortuna MA, Pascual-García A, Ferrera A, Luque B, Bascompte J. The architecture of 290 mutualistic networks minimizes competition and increases biodiversity. Nature. 2009;458:1018–20.
Helmus MR, Savage K, Diebel MW, Maxted JT, Ives AR. Separating the determinants of phylogenetic community structure. Ecol Lett. 2007;10:917–25.
Kraft NJB, Adler PB, Godoy O, James EC, Fuller S, Levine JM. Community assembly, coexistence and the environmental filtering metaphor. Funct Ecol. 2014;29:592–9.
Peres-Neto PR, Dray S, Braak CJ. Linking trait variation to the environment: critical issues with community-weighted mean correlation resolved by the fourth-corner approach. Ecography. 2017;40:806–16. arXiv:arXiv:0902.0132v1
Wisz MS, Pottier J, Kissling WD, Pellissier L, Lenoir J, Damgaard CF, et al. The role of biotic interactions in shaping distributions and realised assemblages of species: implications for species distribution modelling. Biol Rev. 2013;88:15–30.
Townsend Peterson A, Soberón J, Pearson RG, Anderson RP, Martínez-Meyer E, Nakamura M, et al. Ecological niches and geographic distributions. Oxford: Princeton University Press; 2011.
Barberán A, Casamayor EO, Fierer N. The microbial contribution to macroecology. Front Microbiol. 2014;5:203.
Dini-Andreote F, Stegen JC, van Elsas JD, Salles JF. Disentangling mechanisms that mediate the balance between stochastic and deterministic processes in microbial succession. Proc Natl Acad Sci USA. 2015;112:E1326–E1332.
Goberna M, Navarro-Cano JA, Valiente-Banuet A, García C, Verdú M. Abiotic stress tolerance and competition-related traits underlie phylogenetic clustering in soil bacterial communities. Ecol Lett. 2014;17:1191–201.
Barberán A, Ramirez KS, Leff JW, Bradford MA, Wall DH, Fierer N. Why are some microbes more ubiquitous than others? Predicting the habitat breadth of soil bacteria. Ecol Lett. 2014;17:794–802.
Ter Braak CJ, Cormont A, Dray S. In the fourth-corner problem reports. Ecology. 2012;93:1525–6.
Edwards KF, Litchman E, Klausmeier CA. Functional traits explain phytoplankton responses to environ mental gradients across lakes of the United States. Ecology. 2013;94:1626–35.
Etienne RS, Alonso D. A dispersal-limited sampling theory for species and alleles. Ecol Lett. 2005;8:1147–56. arXiv.
Webb CO, Ackerly DD, McPeek MA, Donoghue MJ. Phylogenies and community ecology. Annu Rev Ecol Systemat. 2002;33:475–505.
Gotelli NJ, Graves GR. Null models in ecology. Washington: Smithsonian Institution Press; 1996.
Casamayor EO, Triadó-Margarit X, Castañeda C. Microbial biodiversity in saline shallow lakes of the Monegros Desert, Spain. FEMS Microbiol Ecol. 2013;85:503–18.
Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10:996–8.
Wattam AR, Abraham D, Dalay O, Disz TL, Driscoll T, Gabbard JL, et al. PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 2014;42:581–91.
Markowitz VM, Chen I-MA, Palaniappan K, Chu K, Szeto E, Pillay M, et al. IMG 4 version of the integrated microbial genomes comparative analysis system. Nucleic Acids Res. 2014;42:D560–7.
Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nature Biotechnol. 2013;31:814–21.
Nemergut DR, Knelman JE, Ferrenberg S, Bilinski T, Melbourne B, Jiang L, et al. Decreases in average bacterial community rRNA operon copy number during succession. ISME J. 2015;10:1147–56.
Zeileis A, Kleiber C, Krämer W, Hornik K. Testing and dating of structural changes in practice. Comput Stat Data Anal. 2003;44:109–23.
McGill BJ, Enquist BJ, Weiher E, Westoby M. Rebuilding community ecology from functional traits. Trends Ecol Evol. 2006;21:178–85.
Adler PB, Fajardo A, Kleinhesselink AR, Kraft NJB. Trait-based tests of coexistence mechanisms. Ecol Lett. 2013;16:1294–306.
Thakur MP, Wright AJ. Environmental filtering, niche construction, and trait variability: the missing discussion. Trends Ecol Evol. 2017;32:884–6.
Barberán A, Fernández-Guerra A, Bohannan BJM, Casamayor EO. Exploration of community traits as ecological markers in microbial metagenomes. Mol Ecol. 2012;21:1909–17.
Krause S, Le Roux X, Niklaus PA, Van Bodegom PM, Lennon JT, Bertilsson S, et al. Trait-based approaches for understanding microbial biodiversity and ecosystem functioning. Front Microbiol. 2014;5:251.
Ortiz-Álvarez R, Fierer N, de los Ríos A, Casamayor EO, Barberán A. Consistent changes in the taxonomic structure and functional attributes of bacterial communities during primary succession. ISME J. 2018;12:1658–67.
Burke C, Steinberg P, Rusch D, Kjelleberg S, Thomas T. Bacterial community assembly based on functional genes rather than species. Proc Natl Acad Sci. 2011;108:14288–93.
Grossmann L, Beisser D, Bock C, Chatzinotas A, Jensen M, Preisfeld A, et al. Trade-off between taxon diversity and functional diversity in European lake ecosystems. Mol Ecol. 2016;25:5876–88.
Louca S, Polz MF, Mazel F, Albright MBN, Huber JA, O’Connor MI, et al. Function and functional redundancy in microbial systems. Nat Ecol Evol. 2018;2:936–43.
von Heijne G. Signal sequences: the limits of variation. J Mol Biol. 1985;184:99–105.
Wesley RR, Brüser T, Kissinger JC, Pohlschröder M. Adaptation of protein secretion to extremely high-salt conditions by extensive use of the twin-arginine translocation pathway. Mol Microbiol. 2002;45:943–50.
van der Ploeg R, Mäder U, Homuth G, Schaffer M, Denham EL, Monteferrante CG, et al. Environmental salinity determines the specificity and need for tat-dependent secretion of the YwbN protein in Bacillus subtilis. PLoS One. 2011;6:1–10.
van der Ploeg R, Monteferrante CG, Piersma S, Barnett JP, Kouwen TRHM, Robinson C, et al. High-salinity growth conditions promote tat-independent secretion of tat substrates in Bacillus subtilis. Appl Environ Microbiol. 2012;78:7733–44.
Paul S, Bag SK, Das S, Harvill ET, Dutta C. Molecular signature of hypersaline adaptation: insights from genome and proteome composition of halophilic prokaryotes. Genome Biol. 2008;9:R70.
Klappenbach JA, Dunbar JM, Schmidt TM. rRNA operon copy number reflects ecological strategies of bacteria. Appl Environ Microbiol. 2000;66:1328–33.
Lozupone CA, Knight R. Global patterns in bacterial diversity. Proc Natl Acad Sci USA. 2007;104:11436–40.
Auguet J-C, Barberán A, Casamayor EO. Global ecological patterns in uncultured Archaea. ISME J. 2010;4:182–90.
Barberán A, Casamayor EO. Global phylogenetic community structure and beta-diversity patterns in surface bacterioplankton metacommunities. Aquat Microb Ecol. 2010;59:1–10.
Gasol JM, Casamayor EO, Joint I, Garde K, Gustavson K, Benlloch S, et al. Control of heterotrophic prokaryotic abundance and growth rate in hypersaline planktonic environments. Aquat Microb Ecol. 2004;34:193–206.
Casamayor EO, Triadó-Margarit X. Microbial diversity and novelty along salinity gradients. New York: Springer; 2013. p. 1–8.
Oren A. Bioenergetic aspects of halophilism. Microbiol Mol Biol Rev. 1999;63:334–48.
Bunin G. Ecological communities with Lotka–Volterra dynamics. Phys Rev E. 2017;95:1–8.
Acknowledgements
This work was funded by the Spanish "Ministerio de Economía y Competitividad" under the projects BRIDGES (CGL2015-69043-P, to EOC and DA), and the Ramón y Cajal Fellowship program (DA). Both VJO and MM-S have been supported by Ph.D. contracts funded by the Spanish "Ministerio de Economía y Competitividad" under the projects BRIDGES and SITES (CGL2015-69043-P, CGL2012-39964 to EOC and DA). We thank Joan Cáliz and Gerard Funosas for help in bioinformatic analyses and fruitful discussions and the facilities and warm and creative atmosphere provided by the "white room" in the Computational Biology Lab (CBL) of the Center for Advanced Studies of Blanes.
Author information
Authors and Affiliations
Contributions
EOC, JAC, and DA conceived the study. EOC and XT-M carried out the sampling and field work. JAC, VJO and DA designed the null models. RO-A, MM-S, and XT-M conducted bioinformatic analyses. MM-S, VJO, JAC, and XT-M contributed with scripts and analyses. JAC and XT-M led the analyses of the data. XT-M, JAC, and MM-S assembled and organized the supplementary materials. XT-M, JAC, and DA led the writing of the manuscript. All authors contributed to developing the concepts along several “Bridges” workshops, discussed and contributed critically to the drafts and gave final approval for publication.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Triadó-Margarit, X., Capitán, J.A., Menéndez-Serra, M. et al. A Randomized Trait Community Clustering approach to unveil consistent environmental thresholds in community assembly. ISME J 13, 2681–2689 (2019). https://doi.org/10.1038/s41396-019-0454-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-019-0454-4
This article is cited by
-
Ecological and Metabolic Thresholds in the Bacterial, Protist, and Fungal Microbiome of Ephemeral Saline Lakes (Monegros Desert, Spain)
Microbial Ecology (2021)
-
Towards sustainable agriculture: rhizosphere microbiome engineering
Applied Microbiology and Biotechnology (2021)
