
Abstract
Sequenced archaeal genomes are mostly smaller and more streamlined than typical bacterial genomes; however, members of the Methanosarcina genus within the Euryarchaeaota are a significant exception, with M. acetivorans being the largest archaeal genome (5.8 Mbp) sequenced thus far. This finding is partially explained by extensive gene duplication within Methanosarcina spp. Significantly, the evolutionary pressures leading to gene duplication and subsequent genome expansion have not been well investigated, especially with respect to biological methane production (methanogenesis), which is the key biological trait of these environmentally important organisms. In this study, we address this question by specifically probing the functional evolution of two methylamine-specific methyltransferase paralogs in members of the Methanosarcina genus. Using the genetically tractable strain, M. acetivorans, we first show that the two paralogs have distinct cellular functions: one being required for methanogenesis from methylamine, the other for use of methylamine as a nitrogen source. Subsequently, through comparative sequence analyses, we show that functional divergence of paralogs is primarily mediated by divergent evolution of the 5′ regulatory region, despite frequent gene conversion within the coding sequence. This unique evolutionary paradigm for functional divergence of genes post-duplication underscores a divergent role for an enzyme singularly associated with methanogenic metabolism in other aspects of cell physiology.
Similar content being viewed by others


Introduction
Gene duplication provides the raw material for functional innovation: duplicated genes comprise anywhere between 15–65% of an organism’s genome [1, 2]. The evolutionary fate of duplicate genes is well documented and three alternative outcomes are commonly proposed [1, 3,4,5]. Functional redundancy arising immediately post-duplication leads to relaxed selection. As a result, mutations accumulate in one or both paralogs [1, 3, 5]. Fixation of deleterious mutations in one paralog might lead to non-functionalization: formation of a pseudogene and eventual loss, which is considered to be the most likely outcome of a gene duplication event [3, 4]. In contrast, paralogs may be stably maintained over time if each copy acquires a distinct function either through neo-functionalization or sub-functionalization [1, 3]. Neo-functionalization implies that one paralog acquires a novel, beneficial function, while the other paralog maintains the original function; whereas sub-functionalization implies that each copy retains a complementary portion of the original function(s) of the ancestral gene [1, 3,4,5].
Most empirical models suggest that functional divergence is imperative for the stable maintenance of duplicated genes in a genome over time [3,4,5]. Accordingly, sequence divergence is propagated and maintained by a combination of neutral and selective forces [3, 5]. In this regard, gene conversion (a sequence overwriting mechanism that operates by unidirectional transfer of DNA from a donor sequence to a homologous acceptor sequence and results in the homogenization of both sequence into the donor form) is considered to be detrimental [4,5,6]. In fact, it is often conjectured that hitherto unknown mechanisms must exist to allow paralogs to escape the homogenizing ‘tether’ of gene conversion [6]. Although, under certain circumstances, i.e., for genes like rrn (encoding rRNA) [7] and tuf (encoding the elongation factor Ef-Tu) [8], concerted evolution (non-independent evolution of paralogs due to gene conversion) ensures structural and functional integrity of housekeeping gene products. Gene conversion is also frequently observed in metabolic genes across Bacteria and Archaea [9,10,11,12,13] where, often without any experimental validation, it is naively assumed that functional homogenization of paralogs catalyzing critical metabolic reactions enhances metabolic robustness [14]. Here, on the path to uncovering the evolutionary trajectory of paralogous operons involved in methanogenesis from methylamine (also referred to as monomethylamine), we stumbled upon a contrasting paradigm wherein isozymes perform distinct cellular functions despite signatures of gene conversion in the coding sequence.
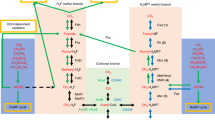
Methanogenesis, biological methane production coupled to growth and energy conservation, is restricted to, and widespread in, members of the archaeal domain [15, 16]. Distributed across a wide range of anoxic environments, from the human gut to hydrothermal vents, methanogens produce ca. two-third of the annual emissions of this potent greenhouse gas, the remainder coming from geochemical sources [17, 18]. Unlike most methanogens, which reduce CO2 with H2 during methanogenesis, Methanosarcina spp. have a wider metabolic breadth, exemplified by their ability to grow on reduced one-carbon (C1) compounds such as methanol, methylated sulfides, and methylated amines (known as methylotrophic methanogenesis), as well as acetate (known as aceticlastic methanogenesis) [18, 19]. Methylotrophic methanogenesis occurs via a disproportionation reaction in which one molecule of a reduced C1 compound is oxidized to CO2 for every three molecules that are reduced to methane (Fig. 1 and Supplementary Fig. 1) [20]. The process is initiated by a substrate-specific methyltransferase (methyltransferase 1 or MT1) that differs depending on the growth substrate. Most MT1 enzymes are heterodimers comprised of a methyltransferase protein and a cognate corrinoid-binding protein (MtmB and MtmC, respectively, in the case of methylamine). These proteins are typically encoded in an operon [21]. The corrinoid-bound methyl group is then transferred to coenzyme M (mercaptoethanesulfonic acid) by a second methyltransferase (MtbA) to form methyl-coenzyme M (CH3-CoM), which is subsequently reduced to methane by the enzyme methyl coenzyme M reductase (MCR) [19, 21]. Curiously, most Methanosarcina spp. encode at least two copies of the mtmCB operon (Fig. 1) [22, 23]. Whether this redundancy enhances metabolic robustness or indicates unique functional roles for each paralog is unclear.

Methylamine-mediated methanogenic metabolism in Methanosarcina acetivorans. a A methylamine-specific methyltransferase catalyzes the first step in the methyl-transfer reactions that leads to generation of methyl-coenzyme M (CH3-CoM). For every four molecules of CH3-CoM generated, one is oxidized to CO2 in a step-wise fashion and the other three are reduced to methane (CH4) with the sulfhydrl group of coenzyme B (CoB) as the electron donor by the enzyme methyl-coenzyme M reductase (MCR). The heterodisulfide (CoM-S-S-CoB) produced by MCR is reduced to regenerate CoM and CoB by heterodisulfide reductase (HDR) using reducing equivalents obtained from the oxidation of CH3-CoM to CO2. b The methyl-transfer reaction from methylamine to CH3-CoM involves two methyltransferases. The first methyltransferase (MT1) is substrate-specific and comprises of a heterodimer of a methyltransferase protein (MtmB) and a corrinoid-binding protein (MtmC). MT1 catalyzes the transfer of the methyl group from methylamine to the corrinoid protein (MtmC). This reaction releases free ammonium ions (NH4+). The second methyltransferase (MT2) catalyzes the methyl-transfer reaction from the corrinoid protein (MtmC) to CoM to generate CH3-CoM. c Methanosarcina acetivorans encodes two copies of the methylamine-specific MT1 operon in its chromosome. The operon in green (mtmC1B1) is found in the vicinity of the methylamine-specific MT2 gene (mtbA1) and a methylated amine-specific permease gene (mttP). The operon in orange (mtmC2B2) is encoded more than 2 Mb away on the chromosome next to genes encoding components of the NH4+ assimilation pathway
In this work, we investigate the functional role of each of the mtmCB paralogs in the genetically tractable methanogenic archaeon, Methanosarcina acetivorans. Our results indicate that MtmC1B1 (encoded by MA0144 and MA0145) allows growth and methanogenesis from methylamine, whereas MtmC2B2 (encoded by MA2971 and MA2972) enables optimal utilization of methylamine as a nitrogen source. Phylogenetic analyses of the paralogs across the Methanosarcina genus suggests that the coding sequences of the two paralogs have undergone frequent gene conversion, with functional divergence likely having been mediated by divergent evolution of the 5′ regulatory region. Overall, this study reevaluates the role of gene conversion post-duplication and emphasizes the role of regulatory sequences in the functional evolution of paralogs.
Results
Methylamine growth requires only one of the two methylamine methyltransferases in M. acetivorans
Methanosarcina acetivorans encodes two copies of the methylamine-specific methyltransferase (mtmB) and corrinoid-binding protein (mtmC) (Fig. 1). To characterize the functional roles of each mtmCB operon in M. acetivorans, we generated unmarked single-deletion mutants lacking either mtmC1B1 or mtmC2B2, as well as a double mutant lacking both operons, using a recently developed Cas9-mediated genome editing technique for M. acetivorans [24]. Subsequently, we quantified the growth rate and growth yield (maximum optical density at 600 nm) of the parental strain and each mutant on all methylated amines that can serve as growth substrates for M. acetivorans [18, 20, 25] (Fig. 1, Supplementary Fig. 1). No significant difference in growth rate or growth yield was observed for the ∆mtmC2B2 mutant relative to the parent strain (referred to as WT henceforth) under any of the conditions tested (Fig. 2; Supplementary Fig. 1). In contrast, the ∆mtmC1B1 mutant had 50% and 70% lower growth yields on trimethylamine (TMA) (p < 0.001; two-sided t-test with the mean of three biological replicates) and dimethylamine (DMA) (p < 0.001), respectively (Fig. 2b). The ∆mtmC1B1 mutant was incapable of growth on methylamine (Fig. 2). The growth phenotype of the ∆mtmC1B1∆mtmC2B2 double mutant resembled that of the ∆mtmC1B1 mutant (Fig. 2).

Growth of mtmCB mutants on methylated amines. a Mean growth rate (h−1) and b mean maximum optical density (OD) (measured at 600 nm) of the parent strain (referred to as WT; in gray), the ∆mtmC1B1 single mutant (green), the ∆mtmC2B2 single mutant (orange), and the ∆mtmC1B1∆mtmC2B2 double mutant (blue) in high-salt (HS) medium with 50 mM trimethylamine hydrochloride (TMA), 50 mM dimethylamine hydrochloride (DMA), and 50 mM monomethylamine (MMA) as the sole methanogenic substrate. The error bars indicate the 95% confidence interval of the mean of three independent biological replicates. NG indicates no growth after three months of incubation at 37 °C
MtmCB2 enables optimal utilization of methylamine as a nitrogen source in M. acetivorans
Because loss of the mtmC2B2 operon does not affect methanogenesis and growth using methylamine, we explored additional growth conditions. Previous studies showed that the expression of mtmC2B2 increases under nitrogen-limiting environments in M. mazei, suggesting the possibility that this operon might be required for use of methylamine as a nitrogen source [26, 27]. However, this hypothesis has never been experimentally tested.
Prior to testing our hypothesis regarding the role of MtmCB2, we optimized the growth medium to exclude any nitrogenous compounds that might confound the interpretation of our results. HS (high-salt) medium used for cultivation of Methanosarcina spp. from marine environments contains 19 mM NH4Cl and 2.8 mM cysteine, with N2/CO2 (80:20) gas in the headspace [28]. Methanosarcina strains (including M. acetivorans) encode functional nitrogenases and can use N2 gas as a nitrogen source [22, 29]; whether the nitrogen from cysteine could also be assimilated was untested prior to this study [30]. We observed that the WT strain could grow in modified HS medium (with 125 mM methanol as the growth substrate) lacking NH4Cl and containing Argon/CO2 (80:20) in the headspace (Supplementary Fig. 2A), suggesting that cysteine can serve as a nitrogen source for this organism. Consistent with this idea, no growth was observed when cysteine was also eliminated from the medium (Supplementary Fig. 2A). Cysteine primarily serves as a reducing agent in HS medium; to compensate for its absence, we increased the concentration of sodium sulfide (Na2S.9H2O), another reductant, in the modified nitrogen-free (N-free) HS medium (Supplementary Fig. 2B)
To test whether MtmC2B2 allowed use of methylamine as a nitrogen source, we examined growth in N-free HS medium with 125 mM methanol as the methanogenic substrate and 5 mM methylamine as the sole nitrogen source (note: no growth was observed in this medium if either methanol or methylamine were absent). Relative to the WT, the ∆mtmC1B1 mutant had a modest albeit statistically significant decrease in growth (Fig. 3). In contrast, the ∆mtmC2B2 mutant grew 30% slower (p = 0.001) with lower cell yields at as well (34%; p < 0.001) (Fig. 3). No growth was detected for the ∆mtmC1B1∆mtmC2B2 double mutant. Complementation of the ∆mtmC1B1∆mtmC2B2 double mutant by chromosomal insertion of either the mtmC1B1 operon or the mtmC2B2 operon at the ssuC locus [24] rescued growth (Fig. 3). Notably, the growth rate was significantly higher when the double mutant was complemented with the mtmC2B2 operon compared with the mtmC1B1 operon (Fig. 3).

Nitrogen assimilation phenotype of mtmCB mutants. a Mean growth rate (h−1) and b mean maximum optical density (OD) (measured at 600 nm) of the parent strain (WT) and mutants in nitrogen-free high-salt (HS) medium with 125 mM methanol as the primary carbon and energy source for methanogenesis along with 5 mM monomethylamine (MMA; orange) or nitrogen gas (blue) as the sole nitrogen source. The error bars indicate the 95% confidence interval of the mean of three independent biological replicates. An * indicates p < 0.05 and ** indicates p < 0.01 using a two-tailed t-test. NG indicates no growth after three months of incubation at 37 °C and ND indicates that growth data were not determined
Methylamine-specific methyltransferases are not involved in nitrogen fixation
Global regulators like NrpR and NrpA play a critical role in mediating the expression of genes involved in the uptake and assimilation of nitrogen sources in methanogenic archaea [31, 32]. A characteristic palindromic DNA motif near the promoter of mtmC2B2 suggests that it might be a part of the NrpR regulon (Supplementary Fig. 3B) [27, 31]. To test whether deletion of the mtmC2B2 operon indirectly impacts the assimilation of nitrogen from methylamine by interfering with the NrpR regulon, we tested the ability of the ∆mtmC1B1, ∆mtmC2B2 single mutants to fix N2. When compared with WT, no significant difference in growth rate or yield was observed for either mutant during growth in N-free HS medium with 125 mM methanol as the methanogenic substrate and N2 as the sole nitrogen source (Fig. 3).
Phylogenetic analyses indicate that gene conversion is common and frequent within the mtmCB coding sequence
Our data clearly indicate that the mtmCB paralogs have distinct cellular functions. To trace the evolutionary trajectory underlying functional divergence, we profiled mtmCB homologs in thirty Methanosarcina strains with completely sequenced genomes. Approximately 65% of the sampled strains encode two copies of mtmCB, another 23% have a third copy, and only four strains encoded just one copy of the operon (Supplementary Table 1). With the exception of the third copy, which is always found in the immediate vicinity of the mtmC1B1 operon (Supplementary Fig. 4), the two paralogs are at least 1 Mbp apart on the chromosomes of their respective hosts (Supplementary Table 1). The genomic neighborhood of the mtmC1B1 operon includes genes involved in transport and metabolism of methylated amines and the synteny of this locus is conserved across all sequenced Methanosarcina spp. (Supplementary Fig. 4 and Supplementary Table 2). On the other hand, genes associated with nitrogen metabolism, such as glutamine amidotransferase (GATase; MA2966) as well as an NAD(P)/FAD oxidoreductase (MA2968) and a putative ferredoxin (MA2969), likely providing reducing equivalents for the GS-GOGAT cycle, are frequently found near the mtmC2B2 locus (Supplementary Fig. 4 and Supplementary Table 2). The observed synteny of the two loci is consistent with the hypothesis that the mtmCB duplication event preceded speciation of Methanosarcina spp.
Strikingly, phylogenetic analyses of mtmC and mtmB show that the nucleotide sequence of paralogs from the same strain are often more closely related to each other than to the copy that would be expected based on the genomic neighborhood (Fig. 4). We suspect that this phenomenon stems from frequent gene conversion between paralogs rather than HGT events.

Phylogenetic analysis of mtmCB paralogs in Methanosarcina spp. A maximum-likelihood phylogenetic tree of the a nucleotide sequence of the mtmC ORF and b nucleotide sequence of the mtmB ORF from thirty Methanosarcina strains with completely sequenced genomes. Sequences in green, orange, and blue represent mtmC1, mtmC2, and mtmC3 or mtmB1, mtmB2, and mtmB3, respectively, as determined by the chromosomal synteny and conserved motifs in the 5′ region upstream of the coding sequence (see ‘Annotation of mtmCB paralogs’ in the materials and methods section for further details). The node labels indicate bootstrap support values
Promoter sequences mediate functional divergence of methylamine-specific methyltransferases
To understand how each mtmCB paralog acquired a distinct function, despite gene conversion, we examined the regulatory region upstream of mtmCB. The transcription start site (TSS), TATA box and B recognition element (BRE) of the mtmC1B1 operon was experimentally determined in M. barkeri MS [25] and similar analyses have also been conducted for the mtmC2B2 operon in M. mazei Gö1 [27]. The promoter of the third copy of mtmCB is yet to be determined therefore was excluded from these analyses. Notably, the promoter element of mtm1CB1 and mtmC2B2 are highly conserved in Methanosarcina spp. (Supplementary Fig. 3 and Supplementary Fig. 5). Therefore, we generated a ‘promoter’ tree with sequences corresponding to TATA box ±50 bp for the two paralogs (to account for putative binding sites for cis regulatory elements) (Supplementary Fig. 6 and Supplementary Table 3). Unlike the gene trees (Fig. 5), the promoter tree has a distinct topology with two well-supported clades corresponding to each paralog (Fig. 5). A similar topology was also observed for a phylogenetic tree constructed with 500 nucleotides upstream of the mtmC1 and mtmC2 start codons (Supplementary Fig. 7).

Contrasting the coding sequence phylogeny and the promoter phylogeny of mtmCB paralogs in Methanosarcina. A tanglegram comparing the maximum-likelihood phylogenetic tree of the concatenated nucleotide sequence of the mtmC and mtmB ORF (left) with the promoter tree of corresponding operons (right) in thirty Methanosarcina strains with completely sequence genomes. Sequences in green and orange, represent the coding sequence or promoter of mtmC1/mtmB1 or mtmC2/mtmB2, respectively. All mtmC3B3 sequences were eliminated from these analyses since the promoter of this operon is yet to be determined
Discussion
The genome architecture of Methanosarcina strains deviates from the majority of sequenced archaeal strains that have compact and highly streamlined genomes: significant genome expansion may have resulted from extensive gene duplication [14, 33, 34]. Here, we delve into the ramifications of gene duplication of methylamine-specific methyltransferases (encoded by mtmCB) on the physiology and ecology of Methanosarcina spp.
Our mutant analyses indicate that MtmCB paralogs have distinct metabolic roles in methanogenic growth and nitrogen assimilation (Figs. 2 and 3). These findings are in accord with global transcriptional studies in the closely related strain, M. mazei, which have shown that the mtmC1B1 operon is specifically upregulated during growth on methylated amines, whereas the expression of the mtmC2B2 operon is elevated during nitrogen limitation albeit under slightly confounding conditions where methylated amines were also the sole methanogenic substrate [26, 27].
In marine environments, methylamine is a significant source of carbon and nitrogen for microbial growth [35, 36]. Temporal or spatial concentration gradients of methylamine are bound to exist; encoding two functionally specialized copies of MtmCB would enable methanogens like M. acetivorans to adapt to distinct ecological niches. Under oligotrophic conditions, where nitrogen is often a limiting resource, MtmC2B2 would facilitate these organisms to scavenge nitrogen from methylamine. In contrast, when methylamine is abundant in the environment, or produced intracellularly during growth on other methylated compounds like trimethylamine (TMA) or dimethylamine (DMA) (Supplementary Fig. 1), MtmC1B1 would enhance growth rate and cell yield. Significantly, similar tradeoffs between the use of methylamine for carbon versus nitrogen have led to the maintenance of degenerate (functionally redundant but evolutionarily unrelated) pathways for methylamine oxidation in the genomes of methylotrophic bacteria [37].
MtmC1B1 still retains some capacity to assimilate nitrogen from methylamine (Fig. 2). Thus, one might wonder why a single enzyme is incapable of performing both functions? The free energy yield (∆G°′) for methanogenic growth on methylamine (−57.5 kJ/mol substrate) is much lower in comparison to other methylated compounds like dimethylsulfide (−73.8 kJ/mol substrate), methanol (−77.6 kJ/mol substrate), DMA (−108.0 kJ/mol substrate), or TMA (−164 kJ/mol substrate) [38, 39]. Therefore, from a thermodynamic perspective, methylamine is an inferior substrate and would only be ideal for growth when concentrations are high (Supplementary Fig. 1). Accordingly, cells would require a methylamine-specific methyltransferase that is highly expressed and specifically upregulated when present at levels that support growth. In contrast, under conditions where methylamine is the most favorable nitrogen source, but where superior carbon sources are available, the organism would benefit from a methylamine-specific methyltranferase expressed at a level consistent with the cellular demand for nitrogen. Given than methanogens use ca. 20% of their growth substrate for biomass [19, 40], with the underlying assumption that the cellular concentration of methylamine is not rate-limiting and the composition of biomass is in accord with the Redfield ratio [41], then the expression of this nitrogen assimilating methylamine-specific methyltransferase would have to be at least 50-fold lower than the paralog used for growth on methylamine as a methanogenic substrate. Ideally, the expression of this enzyme should be mediated by a nitrogen-sensing regulon, as noted by the presence of a NrpR binding site upstream of the mtmC2B2 CDS (Supplementary Fig. 3 and Supplementary Fig. 5) [27]. Therefore, we posit that adaptive constraints arising from incompatible regulons sensing methanogenic substrates and nitrogen sources might prevent the evolution of a single ortholog that is optimized for both functions.
Strikingly, when we sought to recapitulate the evolutionary trajectory of MtmCB paralogs across members of the Methanosarcina genus, the outcome was inconsistent with our expectations based on aforementioned genetic analyses in M. acetivorans. Based on the topology of the trees for the coding sequence of mtmC and mtmB (Fig. 4), one might infer that the duplication event has occurred multiple times, often very recently, or that horizontal gene transfer is rampant within and across closely related strains, which contradicts the notion that the paralogs may have evolved unique functions. However, neither the conserved chromosomal synteny, especially in the neighborhood of mtmC1B1 (Supplementary Fig. 4 and Supplementary Table 2), nor the topology of the promoter tree (Supplementary Fig. 6), lends support to these hypotheses. Significantly, the topology of the promoter tree (Supplementary Fig. 6) is consistent with the species tree for Methanosarcina spp. [42]. Taken together, these data support the inference that, despite the homogenizing effect of gene conversion in the coding sequence of mtmC and mtmB, divergent evolution of the 5′ regulatory region is likely to have conferred the distinct functional roles uncovered in our genetic studies.
Whether gene conversion among paralogs is common and frequent in Methanosarcina spp. remains to be tested. However, in contrast to our results, previous studies indicate that the isozymes encoded by the three methanol-specific methyltransferases (mtaCB) in M. acetivorans do not seem to undergo gene conversion and are functionally redundant [43, 44]. These key differences in the evolutionary trajectory of methanol- and methylamine-specific methyltransferases indicate that the basal frequency of gene conversion in these methanogenic archaea might not be unusually high; further suggesting that gene conversion between mtmCB paralogs might be under selection. The molecular and evolutionary processes that lead to gene conversion within the coding sequence but not the flanking regulatory regions are beyond the scope of this work but deem further investigation. In conclusion, our study demonstrates, both, a unique physiological and evolutionary paradigm for functional evolution post-duplication of an enzyme to expand its metabolic scope beyond methanogenesis.
Materials and methods
Strains, media, and growth conditions
All M. acetivorans strains were grown in single-cell morphology [28] in bicarbonate-buffered high-salt (HS) liquid medium in sealed tubes with N2/CO2 (80/20) at 8–10 psi in the headspace unless specified. For growth on methylated amines as the primary methanogenic substrate, trimethylamine, dimethylamine, and methylamine were added to a final concentration of 50 mM. NH4Cl and cysteine hydrochloride were excluded from nitrogen-free HS medium and Na2S.9H2O was added to a final concentration of 1.0 mM (instead of 0.4 mM in unaltered HS medium). Ar/CO2 (80/20) at 8–10 psi was provided in the headspace for growth with 5 mM methylamine as the sole nitrogen source whereas N2/CO2 (80/20) at 8–10 psi in the headspace for growth with N2 gas as the sole nitrogen source. Methanol served as the primary methanogenic for growth in nitrogen-free HS medium with alternate nitrogen sources and was added to a final concentration of 125 mM. All substrates were added to the medium prior to sterilization in an autoclave. Growth rate measurements were conducted with three independent cultures (biological replicates) pre-acclimated to the growth medium. A 1:10 dilution of a late-exponential phase culture was used as the inoculum for growth rate measurement. Plating on HS medium containing 50 mM TMA solidified with 1.7% agar was conducted in an anaerobic glove chamber (Coy Laboratory Products, Grass Lake, MI) as described previously [45]. Solid media plates were incubated in an intra-chamber anaerobic incubator maintained at 37 °C with N2/CO2/H2S (79.9/20/0.1) in the headspace as described previously [45]. Puromycin (CalBiochem, San Diego, CA) was added to a final concentration of 2 μg/mL from a sterile, anaerobic stock solution to select for transformants containing the pac (puromycin transacetylase) cassette. The purine analog 8-aza-2,6-diaminopurine (8ADP) (R. I. Chemicals, Orange, CA) was added to a final concentration of 20 µg/mL from a sterile, anaerobic stock solution to select against the hpt (phosphoribosyltransferase) cassette encoded on pC2A-based plasmids. E. coli strains were grown in LB broth at 37 °C with standard antibiotic concentrations. WM4489, a DH10β derivative engineered to control copy number of oriV-based plasmids [46], was used as the host strain for all plasmids generated in this study (Supplementary Table 4). Plasmid copy number was increased dramatically by supplementing the growth medium with sterile rhamnose to a final concentration of 10 mM.
Plasmids
All plasmids used in this study are listed in Supplementary Table 4. Plasmids for Cas9-mediated genome editing were designed as described previously [24]. Standard techniques were used for the isolation and manipulation of plasmid DNA. WM4489 was transformed by electroporation at 1.8 kV using an E. coli Gene Pulser (Bio-Rad, Hercules, CA). All pDN201-derived plasmids were verified by Sanger sequencing at the Roy J. Carver Biotechnology Center, University of Illinois at Urbana-Champaign and all pAMG40 cointegrates were verified by restriction endonuclease analysis.
Transformation of M. acetivorans
All M. acetivorans strains used in this study are listed in Supplementary Table 5. Liposome-mediated transformation was used for M. acetivorans as described previously [47] using 10 mL of late-exponential phase culture of M. acetivorans and 2 µg of plasmid DNA for each transformation.
Annotation of mtmCB paralogs
Based on previous studies [22, 25, 27], the mtmCB operon proximal to mttP and mtbA1 was designated as mtmC1B1 (Supplementary Table 2 and Supplementary Fig. 4) and the operon containing a highly conserved region in the promoter with a conserved sequence of GGTA-N6-TTCC, likely corresponding to the NrpR binding site [27, 31], was designated as mtmC2B2 (Supplementary Fig. 3B). For those genomes that contain a third copy mtmCB operon, the sequence identity of the 5′ upstream region was used to distinguish mtmC1B1 and mtmC3B3 (Supplementary Fig. 8).
Phylogenetic analyses
The nucleotide sequences of mtmC and mtmB, and the 5′ upstream region were aligned using the MUSCLE plug-in [48] with default parameters in Geneious version R9 [49]. All maximum-likelihood (ML) trees were generated with RaXML version 7.2.8 [50] using the generalized time-reversible (GTR)+GAMMA nucleotide substitution model and the rapid bootstrapping algorithm to search for the best scoring ML tree. Tanglegrams were generated using Dendroscope version 3.5.9 [51]. Trees were displayed using Fig Tree v1.4.3 (http://tree.bio.ed.ac.uk/software/figtree/).
References
Lynch M, Conery JS. The evolutionary fate and consequences of duplicate genes. Science. 2000;290:1151–5.
Teichmann SA, Babu MM. Gene regulatory network growth by duplication. Nat Genet. 2004;36:492–6.
Lynch M, Katju V. The altered evolutionary trajectories of gene duplicates. Trends Genet. 2004;20:544–9.
Zhang J. Evolution by gene duplication: an update. Trends Ecol Evol. 2003;18:292–8.
Innan H, Kondrashov F. The evolution of gene duplications: classifying and distinguishing between models. Nat Rev Genet. 2010;11:97–108.
Katju V. In with the old, in with the new: the promiscuity of the duplication process engenders diverse pathways for novel gene creation. Int J Evol Biol. 2012;2012:1–24.
Liao D. Gene conversion drives within genic sequences: concerted evolution of ribosomal RNA genes in bacteria and archaea. J Mol Evol. 2000;51:305–17.
Lathe WC, Bork P. Evolution of tuf genes: ancient duplication, differential loss and gene conversion. FEBS Lett. 2001;502:113–6.
Yáñez-cuna FO, Castellanos M, Romero D. Biased gene conversion in Rhizobium etli is caused by preferential double-strand breaks on one of the recombining homologs. J Bacteriol. 2016;198:591–9.
Rodriguez C, Romero D. Multiple recombination events maintain sequence identity among members of the nitrogenase multigene family in Rhizobium etli. Genetics. 1998;149:785–94.
Zucko J, Long PF, Hranueli D, Cullum J. Horizontal gene transfer and gene conversion drive evolution of modular polyketide synthases. J Ind Microbiol Biotechnol. 2012;39:1541–7.
Bergholz TM, Tarr CL, Christensen LM, Betting DJ, Whittam TS. Recent gene conversions between duplicated glutamate decarboxylase genes (gadA and gadB) in pathogenic Escherichia coli. Mol Biol Evol. 2007;24:2323–33.
Mao D, Grogan DW. How a genetically stable extremophile evolves: modes of genome diversification in the archaeon Sulfolobus acidocaldarius. J Bacteriol. 2017;199:1–18.
Mahadevan R, Lovley DR. The degree of redundancy in metabolic genes is linked to mode of metabolism. Biophys J. 2008;94:1216–20.
Borrel G, Adam PS, McKay LJ, Chen L-X, Sierra-García IN, Sieber CMK, et al. Wide diversity of methane and short-chain alkane metabolisms in uncultured archaea. Nat Microbiol. 2019;4:603–13.
Vanwonterghem I, Evans PN, Parks DH, Jensen PD, Woodcroft BJ, Hugenholtz P, et al. Methylotrophic methanogenesis discovered in the archaeal phylum Verstraetearchaeota. Nat Microbiol. 2016;1:16170.
Conrad R. Microbial Ecology of methanogens and methanotrophs. Adv Agron. 2007;96:1–63.
Liu Y, Whitman WB. Metabolic, phylogenetic, and ecological diversity of the methanogenic archaea. 2008. Ann N Y Acad Sci. 2008;1125:171–89.
Thauer RK, Kaster A, Seedorf H, Buckel W, Hedderich R. Methanogenic archaea:ecologically relevant differences in energy conservation. Nat Rev Microbiol. 2008;6:579–92.
Hippe H, Caspari D, Fiebig K, Gottschalk G. Utilization of trimethylamine and other N-methyl compounds for growth and methane formation by Methanosarcina barkeri. Proc Natl Acad Sci USA. 1979;76:494–8.
Burke SA, Krzycki JA. Reconstitution of monomethylamine: coenzyme M methyl transfer with a corrinoid protein and two methyltransferases purified from Methanosarcina barkeri. J Biol Chem. 1997;272:16570–7.
Galagan JE, Nusbaum C, Roy A, Endrizzi MG, Macdonald P, Fitzhugh W, et al. The genome of M. acetivorans reveals extensive metabolic and physiological diversity. Genome Res. 2002;12:532–42.
Maeder DL, Anderson I, Brettin TS, Bruce DC, Gilna P, Han CS, et al. The Methanosarcina barkeri genome: comparative analysis with Methanosarcina acetivorans and Methanosarcina mazei reveals extensive rearrangement within Methanosarcinal Genomes. J Bacteriol. 2006;188:7922–31.
Nayak DD, Metcalf WW. Cas9-mediated genome editing in the methanogenic archaeon Methanosarcina acetivorans. Proc Natl Acad Sci USA. 2017;114:2976–81.
Burke SA, Lo SL, Krzycki JA. Clustered genes encoding the methyltransferases of methanogenesis from monomethylamine. J Bacteriol. 1998;180:3432–40.
Veit K, Ehlers C, Schmitz RA. Effects of nitrogen and carbon sources on transcription of soluble methyltransferases in Methanosarcina mazei strain Gö1. J Bacteriol. 2005;187:6147–54.
Veit K, Ehlers C, Ehrenreich A, Salmon K, Hovey R, Gunsalus RP, et al. Global transcriptional analysis of Methanosarcina mazei strain Gö1 under different nitrogen availabilities. Mol Gen Genomics. 2006;276:41–55.
Sowers KR, Boone JE, Gunsalus RP. Disaggregation of Methanosarcina spp. and growth as single cells at elevated osmolarity. Appl Environ Microbiol. 1993;59:3832–9.
Leigh JA. Nitrogen fixation in methanogens: the archaeal perspective. Mol Biol. 2000;2:125–31.
Mazumder TK, Nishio N, Fukuzaki S, Nagai S. Effect of sulfur-containing compounds on growth of Methanosarcina barkeri in defined medium. Appl Environ Microbiol. 1986;52:617–22.
Weidenbach K, Ehlers C, Kock J, Ehrenreich A, Schmitz RA. Insights into the NrpR regulon in Methanosarcina mazei Gö1. Arc Microbiol. 2008;190:319–32.
Weidenbach K, Ehlers C, Schmitz RA. The transcriptional activator NrpA is crucial for inducing nitrogen fixation in Methanosarcina mazei Gö1 under nitrogen-limited conditions. FEBS J. 2014;281:3507–22.
Kellner S, Spang A, Offre P, Szöllősi GJ, Petitjean C, Williams TA. Genome size evolution in the Archaea. Emerg Top Life Sci. 2018;2:595–605.
Koonin EV, Wolf YI. Genomics of bacteria and archaea: the emerging dynamic view of the prokaryotic world. Nucleic Acids Res. 2008;36:6688–719.
Taubert M, Grob C, Howat AM, Burns OJ, Pratscher J, Jehmlich N. et al. Methylamine as a nitrogen source for microorganisms from a coastal marine environment. Environ Microbiol.2017;19:2246–57.
Stein LY. Methylamine: a vital nitrogen (and carbon) source for marine microbes. Environ Microbiol.2017;19:2117–8.
Nayak DD, Agashe D, Lee M, Marx CJ. Selection maintains apparently degenerate metabolic pathways due to tradeoffs in using methylamine for carbon versus nitrogen. Curr Biol. 2016;26:1416–26.
Patterson JA, Hespell RB. Trimethylamine and methylamine as growth substrates for rumen bacteria and Methanosarcina barkeri. Curr Microbiol. 1979;3:79–83.
Finster K, Tanimoto Y, Bak F. Fermentation of methanethiol and dimethylsulfide by a newly isolated methanogenic bacterium. Arch Microbiol. 1992;157:425–30.
Weimer P, Zeikus J. One carbon metabolism in methanogenic bacteria. Arch Microbiol.1978;119:49–57.
Cleveland CC, Liptzin D. C: N: P stoichiometry in soil: is there a “Redfield ratio” for the microbial biomass? Biogeochemistry. 2007;85:235–52.
Borrel G, O’Toole PW, Harris HMB, Peyret P, Brugère JF, Gribaldo S. Phylogenomic data support a seventh order of methylotrophic methanogens and provide insights into the evolution of methanogenesis. Genome Biol Evol. 2013;5:1769–80.
Pritchett MA, Metcalf WW. Genetic, physiological and biochemical characterization of multiple methanol methyltransferase isozymes in Methanosarcina acetivorans C2A. Mol Microbiol. 2005;56:1183–94.
Bose A, Pritchett MA, Rother M, Metcalf WW. Differential regulation of the three methanol methyltransferase isozymes in Methanosarcina acetivorans C2A. J Bacteriol. 2006;188:7274–83.
Metcalf WW, Zhang JK, Wolfe RS. An anaerobic, intrachamber incubator for growth of Methanosarcina spp. on methanol-containing solid media. Appl Environ Microbiol. 1998;64:768–70.
Kim SY, Ju KS, Metcalf WW, Evans BS, Kuzuyama T, Van Der Donk WA. Different biosynthetic pathways to fosfomycin in Pseudomonas syringae and Streptomyces species. Antimicrob Agents Chemother. 2012;56:4175–83.
Metcalf WW, Zhang JK, Apolinario E, Sowers KR, Wolfe RS. A genetic system for archaea of the genus Methanosarcina: liposome-mediated transformation and construction of shuttle vectors. Proc Natl Acad Sci USA. 1997;94:2626–31.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7.
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–9.
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3.
Huson DH, Richter DC, Rausch C, Dezulian T, Franz M, Rupp R. Dendroscope: an interactive viewer for large phylogenetic trees. BMC Bioinformatics. 2007;8:460.
Acknowledgements
The authors acknowledge the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences of the U.S. Department of Energy through Grant DE-FG02-02ER15296 (to WWM), the Carl R. Woese Institute for Genomic Biology postdoctoral fellowship (to DDN), and the Simons Foundation Life Sciences Research Foundation postdoctoral fellowship (to DDN). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Author information
Authors and Affiliations
Contributions
DDN and WWM contributed to conceptualization, data curation, formal analysis, supervision, funding acquisition, project administration, methodology, and writing. DDN performed all the experiments outlined in this work.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Nayak, D.D., Metcalf, W.W. Methylamine-specific methyltransferase paralogs in Methanosarcina are functionally distinct despite frequent gene conversion. ISME J 13, 2173–2182 (2019). https://doi.org/10.1038/s41396-019-0428-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-019-0428-6
This article is cited by
-
Unique genes carried by abundant species enhance CH4 emissions during the growing season at the Tibetan Plateau
Soil Ecology Letters (2021)
