
Abstract
Adequate nutrition during the pre- and early-postnatal periods plays a critical role in programming early neurodevelopment. Disruption of neurodevelopment by nutritional deficiencies can result not only in lasting functional deficits, but increased risk of neuropsychiatric disease in adulthood. Historical periods of famine such as the Dutch Hunger Winter and the Chinese Famine have provided foundational evidence for the long-term effects of developmental malnutrition on neuropsychiatric outcomes. Because neurodevelopment is a complex process that consists of many nutrient- and brain-region-specific critical periods, subsequent clinical and pre-clinical studies have aimed to elucidate the specific roles of individual macro- and micronutrient deficiencies in neurodevelopment and neuropsychiatric pathologies. This review will discuss developmental iron deficiency (ID), the most common micronutrient deficiency worldwide, as a paradigm for understanding the role of early-life nutrition in neurodevelopment and risk of neuropsychiatric disease. We will review the epidemiologic data linking ID to neuropsychiatric dysfunction, as well as the underlying structural, cellular, and molecular mechanisms that are thought to underlie these lasting effects. Understanding the mechanisms driving lasting dysfunction and disease risk is critical for development and implementation of nutritional policies aimed at preventing nutritional deficiencies and their long-term sequelae.
Similar content being viewed by others
Early life nutrition and risk of neuropsychiatric disorders
Neurodevelopment is a complex process that involves not only establishment and expansion of neuronal and glial cell populations, but maturation of complex cell morphologies and establishment of appropriate connectivity among cells and among brain regions. The successful construction of these neural circuits during development is critical for the ability of the individual to perform complex behaviors. Increasing evidence supports the hypothesis that disruption of these processes during early development can result in neurocognitive and neuropsychiatric dysfunctions that appear later in life and can last across the lifespan.1 Although much of neurodevelopment proceeds according to a relatively hard-wired, time-locked, experience-independent genetic program, environmental experiences, and exposures exert significant influences on neurodevelopment through the complex interplay between genes and the environment.
Nutrition is one of the most important environmental factors in early neurodevelopment. Although almost all nutrients are needed for normal brain development, a subset of nutrients plays a particularly significant role because they support the high rate of brain metabolism during late fetal and early postnatal life.2 These include macronutrients such as protein, long-chain polyunsaturated fatty acids (LC-PUFAs), and glucose, as well as select micronutrients such as iron, zinc, and vitamins, which are involved in a variety of critical neurodevelopmental processes across brain regions (reviewed in detail in refs 3,4,5) (Table 1). Moreover, while it has been well established that fetal nutrition plays a role acutely in fetal development and pregnancy outcomes,6 it is increasingly understood that fetal nutrition also exerts long-term effects on offspring health and disease risk—including neurocognitive health and disease—into adulthood and throughout the lifespan. This growing body of both clinical and preclinical literature supports the hypothesis that fetal and early postnatal macro- and micro-nutritional status is linked to the risk of psychopathology later in life.7,8,9
Cohort studies of historical periods of famine, such as the Dutch Hunger Winter and the Chinese Famine, have provided foundational evidence for the role of fetal and early postnatal undernutrition in neuropsychiatric disease. These studies demonstrate that inadequate nutrition during fetal development is associated with an increased risk of schizophrenia and affective disorders, among individuals prenatally exposed to famine.10,11,12 Studies of preclinical models have helped to elucidate the specific roles and mechanisms of individual macro- and micronutrient deficiencies that are particularly associated with the development of neuropsychiatric pathologies.
A comprehensive review of all nutrients is beyond the scope of this article. Thus, the following sections focus on iron deficiency (ID) as a paradigm for the effects of nutritional deficiency on neurodevelopment and neuropsychiatric disease risk, due to its extensive clinical and preclinical literature. ID has been implicated in multiple studies as a risk factor for developmentally based psychopathologies in humans13,14,15 and iron’s biology of action has been well studied. Thus, this review discusses fetal-neonatal ID from the epidemiological evidence linking it to neuropsychiatric disorders, to the structural, cellular, and molecular mechanisms that underlie its lasting effects. Additionally, the importance and potential application of understanding the underlying mechanisms in shaping therapies is discussed.
The Developmental Origins of Health and Disease (DOHaD) hypothesis
The Developmental Origins of Health and Disease, or DOHaD, hypothesis posits that the early-life environment, particularly the fetal and early postnatal environment, influences later-life health outcomes and disease risks. The mechanism by which this effect occurs is the subject of considerable research effort. Originally the DOHaD hypothesis was applied to risk for adult cardiovascular disease.16,17 However, it is now understood that a wide range of early-life exposures and conditions, including malnutrition, can affect lifetime health and disease risk across multiple organ systems. One of the classic examples with respect to this nutritional deprivation hypothesis comes from cohort studies of individuals who were exposed to gestational malnutrition during the Dutch Famine of 1944–1945. During this 5-month period, daily maternal rations fell to below 1000 calories. As a result, pregnant women and their fetuses were exposed to famine during early, mid, or late gestation.18 These cohort studies demonstrated that gestational exposure to famine was associated with a variety of adverse health outcomes in adulthood, including obstructive airway disease,19 impaired glucose tolerance,20 dyslipidemia,21 coronary heart disease,22 and schizophrenia.10,11 Interestingly, famine during different periods of gestation was associated with distinct health risks, suggesting that discrete critical periods exist during the fetal period for different organ systems and health outcomes. These observations also suggested that disruption of nutritional status during critical periods results in permanent compromise of organ systems and related health outcomes.
Nutrition and critical periods of neurodevelopment
A critical period in organ system development is characterized as a window of time in which there is a high degree of plasticity and thus increased malleability in response to environmental exposures, including nutrition.23 With respect to nutrition, critical periods demarcate time windows in which a tissue is most sensitive to the presence or absence of a specific nutrient. Generally, a critical period coincides with the time when a given nutrient is in highest demand—often during a period of rapid tissue growth and development. Because tissue growth and development are highly metabolic processes, and sufficient nutrient supply is critical for energy and metabolism, tissues are most likely to suffer detrimental effects from insufficient supply during this window. Each tissue develops on a different time scale and has different nutritional requirements for appropriate development,23,24 and so an exposure at a given time point may affect one tissue or function but not another. As such, there is no single critical period during development; rather, there are a series of nutrient- and tissue-specific critical periods throughout the broad time-span of development.23
Relative to other primates, humans have larger and more highly metabolic brains than would be predicted based on average body size. The human brain has a higher energy demand per unit weight than any other tissue, and in the adult, the brain accounts for 20–25% of basal energy supply despite comprising only 2% of total body mass.25 The developing fetal and early postnatal brain has an even higher metabolic demand than the adult brain, accounting for as much as 60% of the total metabolic rate.25 As such, the developing brain is exquisitely sensitive to nutrient supply. The brain does not develop as a uniform tissue. Rather, each region develops on a unique timeline, with unique timing of nutrient demands, resulting in a series of brain region-specific critical periods. Thus, the effects of insufficient nutrient supply on the developing brain vary depending on the nutrient, the duration of the deficiency and the neurodevelopmental timing of the deficiency relative to the various regional critical periods.26 Failure to construct a brain region during its critical period can lead to residual structural and connectivity defects, persistent neurochemical and electrophysiological abnormalities, or permanent dysregulation of gene expression.
Disruption of neurodevelopmental processes by nutritional deficiency can result in irreversible changes to tissue structure, metabolic regulation, physiology and function, which ultimately manifests as functional deficits and increased risk for neuropsychiatric disease, although the specific manifestation of the deficiency is dependent upon the nutrient and the timing of the deficiency (Table 2).
Clinical studies linking early-life iron deficiency and risk of psychiatric diseases
ID is the most common micronutrient deficiency worldwide, affecting an estimated 2 billion people and 40–50% of pregnant women and preschool-aged children.27,28 While ID is most prevalent in developing countries, it is notable among nutrient deficiencies for its prevalence in industrialized countries, including the United States, where 18% of pregnant women and 14% of 1–2 year olds are affected.29,30 Though ID is ubiquitous across age groups, ID in pregnant women and young children is of particular interest due to the well-established relationship between developmental ID and later-life neurologic dysfunction.
Longitudinal cohort studies of formerly ID (FID) individuals who experienced iron deficiency during infancy have provided detailed information about the specific cognitive and socioemotional functions that are impaired by developmental iron deficiency. At 5 years of age, FID children exhibited slower perceptual speed and poorer understanding of quantitative concepts31 and impaired language abilities.32 In early adolescence, FID individuals were characterized as having slower perceptual speed, poorer spatial memory, and developmental delays in a selective attention task.31 These impairments may contribute to the lower scores of FID adolescents on tests of reading and arithmetic as well as the increased likelihood to repeat a grade or receive referrals for tutoring.33 As young adults, FID individuals performed worse on measures of recognition memory and strategy shifting.34 Finally, outcome measures of employment and education at 25 years suggested that the cognitive deficits experienced throughout infancy, childhood, and adolescence had a significant impact on the lives of FID adults.35 Compared with non-ID individuals, a larger proportion of FID adults did not complete secondary school and were not actively pursuing further training or education.35
Likewise, ID infants and FID children and adults exhibit long-lasting socioemotional and affective deficits. At ages 4–5, FID children exhibited reduced positive affect during an interactive task with their mother36 or a stranger37 and were found to be more passive and unengaged than iron sufficient children.37,38 FID children also displayed poorer self-control on a delayed-gratification task.37 As early adolescents, FID individuals received higher ratings from parents or teachers on measures of anxiety and depression, social problems, delinquent behavior, and aggression.33 Finally, FID adults self-reported more feelings of detachment and negative emotions and had lower ratings for emotional health.35
Separate cohort studies have used measures of maternal iron intake and maternal iron deficiency during pregnancy to assess the effects of iron deficiency during fetal development on long-term neurodevelopmental outcomes of offspring. These studies have found a significant association between maternal iron deficiency and increased risk of schizophrenia spectrum disorders in adulthood,13,15 as well as an association between low maternal iron intake and increased risk of autism spectrum disorders.14
Collectively, these human cohort studies point to a specific set of neurocognitive and neuropsychiatric deficits that arise from fetal and early postnatal iron deficiency, implicating a critical role for iron in the normal development of these functions.
Biological bases for the long-term neuropsychiatric effects of early-life nutritional perturbations
Understanding the potential mechanisms by which early-life nutritional perturbations influence neurodevelopment and subsequent risk for neuropsychiatric disease is critical for developing effective prevention and treatment strategies. The two central hypotheses for these effects are residual structural defects and dysregulation of genes involved in neural function. In the residual structural defects hypothesis, an early-life nutritional exposure, such as malnutrition, during a critical period of development results in aberrant structural development ranging from gross structural abnormalities to fine ultrastructural changes.23,39,40 In the gene dysregulation hypothesis, the early-life nutritional environment aberrantly programs gene expression, which in turn stably alters the subsequent phenotype.41,42,43,44,45 Though these can be posed as two separate hypotheses, in fact, it is likely a combination of both that drives aberrant neurodevelopment and lifetime risk for neuropsychiatric dysfunction.
Residual structural defects
Rodent and porcine models of fetal-neonatal ID have both demonstrated gross morphological changes to both gray and white matter in the brains of developmentally iron deficient individuals. In a porcine model, developmental ID during the first 30 days of life (approximately equivalent to the first 4 months of human postnatal development) results in a decreased total brain volume, with specific decreases in several brain regions including cortex and hippocampus.46 This is accompanied by decreased total white matter volume and integrity.47 Rodent models of early life ID corroborate these effects on both hippocampal volume48,49 and white matter volume/integrity.50 Importantly, these changes are partially reversible: iron restoration starting at 30 days of life is sufficient to normalize gross brain volume after only 30 days of iron repletion in the porcine model. However, disorganization of brain structure persists despite normalization of total brain volume, with remaining deficits in hippocampal volume and white matter organization/integrity.46 Together, these results indicate that while iron repletion may correct gross morphological measures such as total brain volume, ultrastructural changes to gray and white matter may persist beyond the period of iron deficiency and into adulthood.
Rodent models, as well as in vitro primary culture models of ID have been critical in determining the ultrastructural effects of developmental iron deficiency, particularly on neurons. The neuronal ultrastructural effects of developmental ID were first characterized using a rat model of fetal-neonatal ID in which animals experience iron deficiency throughout gestation and early postnatal life. At postnatal day (P) 15—the peak of hippocampal dendritogenesis—ID rats had truncated apical dendrites in hippocampal area CA1.39 Additionally, branches in the dendritic arbor of hippocampal neurons occurred more proximally to the soma and dendritic spine heads were smaller.51 These hippocampal neuronal findings have been corroborated using mouse models of non-anemic ID40,52 and primary neuronal culture models of ID.53 In the rat model of ID, these hippocampal neuronal ultrastructural deficits persisted into adulthood despite early-life iron repletion.39,51 Further refinement of iron repletion experiments determined that there was a critical period for iron in hippocampal neurodevelopment. If iron was restored within the critical period, than adult hippocampal neuronal structure was normalized; however, if iron repletion occurred outside of the critical period, then hippocampal neuronal structure remained permanently altered.40
Increasing evidence shows that cortical neurons are also affected by developmental ID. Ultrastructural changes to both apical and basal dendritic arbors have been characterized in cortical neurons in rodent models of ID, primarily manifesting as decreased branching of dendrites proximal to the cell body.54 Correspondingly, in a study of infants of mothers with low iron intake during pregnancy, cortical gray matter fractional anisotropy (FA) was increased in infants with low maternal iron intake.55 In cortical tissue, FA decreases as neurodevelopment progresses and dendritic arborization increases.56,57 Thus, the increased FA seen in infants of ID mothers supports the preclinical model findings of compromised cortical neuron structure.
Notably, neuronal development is a highly metabolically demanding process that is dependent on adequate availability of energy substrates for development of complex cell morphology.58,59 Production of the metabolic substrate ATP by mitochondria is iron-dependent, as several key enzymes in the electron transport chain are mechanistically dependent on iron for their enzymatic activity.60 Preliminary evidence suggests that mitochondrial function may thus be compromised by developmental iron deficiency,53 contributing to the above-described neuronal ultrastructural defects associated with developmental ID.
Rodent models have also been critical in determining the ultrastructural effects of developmental ID on white matter. Oligodendrocytes, the myelinating cells of the central nervous system, stain more densely for intracellular iron than other cell types in the central nervous system,61 and the peak of iron uptake in the developing brain corresponds to the peak period of myelination,50,62 suggesting that adequate iron supply is important for their development and function. In both in vitro and in vivo models, oligodendrocyte precursor cell (OPC) differentiation to oligodendrocytes is impaired when iron uptake in impaired.63,64 Additionally, the diameter of myelinated axons is significantly decreased in developmentally ID rats, even in adulthood, suggesting that iron is critical for establishment of myelination.54 This has been noted to correspond functionally with decreased conduction velocity.65
In order to understand the relationship between developmental ID and complex psychopathologies, it is important to understand not only its effects on individual brain regions and cell types, but also its effects of the developmentally driven interconnection of multiple brain regions and cell types. Because each brain area develops on a different time scale, critical periods differ between and even within brain areas,24,26 and an exposure at a given time point in development may affect one brain region or physiologic function but not another. This non-uniform effect on the developing brain can disrupt the connectivity among brain regions that work together in complex circuits that are critical for higher-order behaviors in adulthood.
For example, proper functional control of the ventral tegmental area (VTA) loop depends on the balance between tonic hippocampal input and intermittent frontal lobe input. Disruption of this balance is postulated to result in schizophrenia,66 a psychopathology that presents later in life but that has distinct developmental origins. Within the loop, the hippocampus develops earlier and more rapidly than the frontal cortex during late fetal/early neonatal life, and thus is far more vulnerable to the adverse effects of fetal deficiencies of nutrients such as iron that support rapid growth. This selective vulnerability of the hippocampus (relative to frontal cortex) to developmental iron deficiency has the potential to disrupt VTA circuit structure and function by unbalancing the inputs. Further contributing to the unbalance of VTA loop connectivity, the dopaminergic system, a key neurotransmitter system in VTA loop, is disrupted by ID due to the iron-dependence of enzymes in the dopamine synthesis pathway.67 Pre-clinical models of fetal/neonatal iron deficiency support this hypothesis.68,69
Emerging evidence in human cohorts also supports the hypothesis that functional connectivity of brain regions is disrupted by ID. A functional magnetic resonance imaging (fMRI) study of developmentally ID individuals in adulthood found significant changes in connectivity of the Default Mode Network (DMN), a major network of interconnected regions in the brain.70 Connectivity of the posterior cingulate cortex within the DMN was particularly affected by ID.70 Similar disruptions to the DMN are seen in a variety of neurocognitive and neuropsychiatric disorders.71
Gene dysregulation
Widespread hippocampal gene dysregulation has been shown in both rodent and porcine models of developmental ID, both acutely72,73 and in adulthood after complete resolution of ID.44,45 The dysregulated genes map to functions critical for neurodevelopment as well as neuropsychiatric disorders including mood disorders, pervasive developmental disorders, autism, and schizophrenia.44,45,72,73 These widespread and stable long-term alterations to gene expression implicate an underlying transcriptional regulatory mechanism that functions on a genome-wide scale, such as epigenetic modification.
Epigenetics is the study of how supragenomic modifications to DNA and chromatin can alter gene expression and resultant phenotype, independent of alterations to the genome itself. Two major classes of epigenetic modification include cytosine modifications (DNA methylation and DNA hydroxymethylation) and histone modifications. Each of these classes of epigenetic modifications is associated with distinct effects on transcriptional regulation. Additionally, all have been demonstrated to be environmentally labile, meaning that their prevalence and distribution in the genome can be altered, sometimes permanently, in response to environmental conditions and exposures such as stress,74,75 toxicants,76,77,78 and nutrition.41,42,43,79,80 Thus, epigenetic modification presents a plausible mechanism by which the environment, including nutritional status, can exert an effect on central nervous system gene expression and subsequent neurodevelopmental and neuropsychiatric outcomes.
DNA methylation is an epigenetic modification generated by covalent addition of a methyl group to cytosine nucleotides, primarily at CpG dinucleotides. On a genome-wide scale, the hippocampal DNA methylation pattern is significantly altered by developmental ID in a porcine model, with 853 differentially methylated CpG sites identified in the developmentally iron deficient hippocampus compared to controls.73 However, relatively few of these differentially methylated CpG sites map to differentially expressed genes,73 leaving open the question of the functional significance of these changes. DNA methylation has also been assessed at the gene-specific level at the brain derived neurotrophic factor (Bdnf) promoter. Bdnf, a gene critical for neuronal development, synaptic formation and plasticity, learning, and memory is downregulated in the hippocampus both acutely during the period of developmental ID,81 and in adulthood after complete resolution of ID.82 In adult, formerly iron deficient rats, total Bdnf-IV promoter methylation rate is significantly decreased, with 2 of 7 specific CpG sites significantly hypomethylated.80 Given that promoter hypomethylation is canonically associated with transcriptional activation,83 the functional significance of these changes at the downregulated Bdnf-IV promoter is again uncertain.
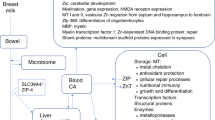
Despite the evidence that developmental ID alters DNA methylation, there is no clear biological mechanism through which iron directly influences the establishment of DNA methylation. In contrast, active DNA demethylation relies on the iron-dependent family of TET methylcysosine dioxygenases which require iron for their enzymatic conversion of methylcytosine to hydroxymethylcytosine and its derivatives (Fig. 1).84 Hydroxymethylcytosine, or DNA hydroxymethylation, itself is a stable epigenetic modification that may alter gene expression.85,86 However, it has not yet been assessed in the context of developmental iron deficiency.

Epigenetic modifications of DNA and chromatin are sensitive to nutritional status. Two major categories of epigenetic modification, DNA methylation (left) and histone modification, specifically histone methylation (right), are both modified by iron deficiency. DNA methylation consists of cytosine bases with a covalently added methyl group. Active DNA demethylation is performed by TET methylcytosine dioxygenases, which require iron for their enzymatic conversion of methylcytosine to hydroxymethylcytosine and its derivatives. Lysine residues of histone tails can undergo mono-, di-, or trimethylation. JARID histone demethylases enzymatically remove methyl groups from di- or trimethylated lysine residues, and also require iron for their enzymatic activity. C Cytosine, hME Hydroxymethyl, K Lysine, Me Methyl
Histone modifications are covalent modifications to the amino acid residues of histone tails, and include histone acetylation, methylation, phosphorylation, and ubiquitination. The histone code is complex and has not been studied in depth in the context of developmental ID. However, one family of histone modifications, lysine methylation, has been of particular interest due to its mechanistic dependence on iron. Removal of methyl groups from lysine residues is catalyzed by the JmjC ARID-domain containing histone demethylase (JARID) family proteins, which have an absolute requirement for iron for their enzymatic activity (Fig. 1).87,88 Accordingly, in the adult, formerly iron deficient rat hippocampus, enrichment of histone methylation is significantly altered at the dysregulated Bdnf-IV promoter.80 K27me3 and K4me3, histone modifications associated with silenced genes, were found to be enriched at the Bdnf-IV promoter, while K4me3, a modification associated with active gene expression, was significantly depleted.80 Interestingly, properly timed dietary supplementation with the methyl donor choline mitigates the effects of ID on histone methylation in the hippocampus of preclinical models.80 This finding underscores the potential for dietary epigenetic “workarounds” in humans in the near future.
Clinical implications
Emerging literature suggests that early life folate, LC-PUFA, Vitamin A and Vitamin D status may also influence the risk of later psychopathology.8,9,89 Folic acid (folate) is of particular interest because it fits into a more general category of one-carbon metabolites along with its dietary counterparts choline, betaine, methionine, and their co-factors vitamins B12 and B6. These nutrients are found in methyl diets, which have been shown to promote epigenetic modifications in the developing brain. A meta-analysis identified low maternal folate and high homocysteine levels as risk factors for schizophrenia in the offspring.90 Low maternal periconceptional folic acid intake, especially in the context of a pesticide exposure as a “second hit” increases the risk of autism in the offspring by up to 2.5-fold.91 It will be important to consider the proper timing, dose, and duration of dietary methyl donors with respect to brain development in light of the finding that these diets have potential for epigenetic modification of brain genes.42,92 Clinical trials of choline for vulnerable pediatric populations, such as children with fetal alcohol syndrome, have already begun and show positive effects on learning and memory behavior.93
Due to the high worldwide prevalence of malnutrition, particularly among pregnant women and young children, understanding the effects of nutrient deficiencies on long-term neurocognitive outcomes and risk for neuropsychiatric disorders is critical because sound nutritional policy and practice can potentially mitigate risk. Prevention of nutrient perturbations during fetal and early postnatal life is an accomplishable goal with long-term societal implications. Women of child-bearing age should be in optimal nutritional health entering into pregnancy, particularly with respect to critical nutrients such as those listed in Tables 1 and 2. Prevention strategies to optimize fetal brain nutritional status throughout pregnancy include not only maintaining maternal nutritional sufficiency but also reducing conditions in nutritionally sufficient women that nevertheless alter fetal nutritional status. These conditions include maternal hypertension, the most common cause of intrauterine growth restriction (IUGR) in developed countries,94 gestational diabetes mellitus,95 maternal smoking,96 obesity/excessive gestational weight gain,97,98 and maternal stress.99 Postnatally, provision of human milk and maintenance of the status of nutrients listed in Table 1 are key prevention strategies. While the promising pre-clinical data on the role of nutrient-mediated epigenetic modification of brain development suggests a potential for therapeutic intervention in humans, it is too soon to propose methyl diets as routine therapeutic interventions until more is known about the optimal timing, dose, and duration of these agents.100
References
Cuthbert, B. N. & Insel, T. R. Toward the future of psychiatric diagnosis: the seven pillars of RDoC. BMC Med. 11, 126 (2013).
Georgieff, M. K., Brunette, K. E. & Tran, P. V. Early life nutrition and neural plasticity. Dev. Psychopathol. 27, 411–423 (2015).
González, H. F. & Visentin, S. Micronutrients and neurodevelopment: an update. Arch. Argent. Pediatr. 114, 570–575 (2016).
Cusick, S. E. & Georgieff, M. K. The role of nutrition in brain development: the golden opportunity of the “First 1000 Days”. J. Pediatr. 175, 16–21 (2016).
Krebs, N. F., Lozoff, B. & Georgieff, M. K. Neurodevelopment: the impact of nutrition and inflammation during infancy in low-resource settings. Pediatrics 139, S50–S58 (2017).
Abu-Saad, K. & Fraser, D. Maternal nutrition and birth outcomes. Epidemiol. Rev. 32, 5–25 (2010).
Bale, T. L. et al. Early life programming and neurodevelopmental disorders. Biol. Psychiatry 68, 314–319 (2010).
Davis, J. et al. A review of vulnerability and risks for schizophrenia: beyond the two hit hypothesis. Neurosci. Biobehav. Rev. 65, 185–194 (2016).
Brown, A. S. & Susser, E. S. Prenatal nutritional deficiency and risk of adult schizophrenia. Schizophr. Bull. 34, 1054–1063 (2008).
Susser, E. S. & Lin, S. P. Schizophrenia after prenatal exposure to the Dutch Hunger Winter of 1944-1945. Arch. Gen. Psychiatry 49, 983–988 (1992).
Susser, E. et al. Schizophrenia after prenatal famine. Further evidence. Arch. Gen. Psychiatry 53, 25–31 (1996).
St Clair, D. et al. Rates of adult schizophrenia following prenatal exposure to the Chinese famine of 1959-1961. J. Am. Med. Assoc. 294, 557–562 (2005).
Insel, B. J., Schaefer, C. A., McKeague, I. W., Susser, E. S. & Brown, A. S. Maternal iron deficiency and the risk of schizophrenia in offspring. Arch. Gen. Psychiatry 65, 1136–1144 (2008).
Schmidt, R. J., Tancredi, D. J., Krakowiak, P., Hansen, R. L. & Ozonoff, S. Maternal intake of supplemental iron and risk of autism spectrum disorder. Am. J. Epidemiol. 180, 890–900 (2014).
Sørensen, H. J., Nielsen, P. R., Pedersen, C. B. & Mortensen, P. B. Association between prepartum maternal iron deficiency and offspring risk of schizophrenia: population-based cohort study with linkage of danish national registers. Schizophr. Bull. 37, 982–987 (2011).
Barker, D. J. P. Fetal nutrition and cardiovascular disease in later life. Br. Med. Bull. 53, 96–108 (1997).
Gluckman, P. D. & Hanson, M. A. Living with the past: evolution, development, and patterns of disease. Science 305, 1733–1736 (2004).
Painter, R. C., Roseboom, T. J. & Bleker, O. P. Prenatal exposure to the Dutch famine and disease in later life: an overview. Reprod. Toxicol. 20, 345–352 (2005).
Lopuhaa, C. E. Atopy, lung function, and obstructive airways disease after prenatal exposure to famine. Thorax 55, 555–561 (2000).
Ravelli, A. et al. Glucose tolerance in adults after prenatal exposure to famine. Lancet 351, 173–177 (1998).
Lumey, L., Stein, A., Kahn, H. & Romijn, J. Lipid profiles in middle-aged men and women after famine exposure during gestation: the Dutch Hunger Winter Families Study. Am. J. Clin. Nutr. 89, 1737–1743 (2009).
Roseboom, T. J. et al. Coronary heart disease after prenatal exposure to the Dutch famine, 1944-45. Heart 84, 595–598 (2000).
Hensch, T. K. Critical period regulation. Annu. Rev. Neurosci. 27, 549–579 (2004).
Clancy, B., Darlington, R. B. & Finlay, B. L. The course of human events: predicting the timing of primate neural development. Dev. Sci. 3, 57–66 (2000).
Holliday, M. in Human Growth: A Comprehensive Treatise (eds Falkner, F. & Tanner, J. M.) 101–117 (Plenium Press, Boston, MA, 1986).
Kretchmer, N., Beard, J. L. & Carlson, S. The role of nutrition in the development of normal cognition. Am. J. Clin. Nutr. 63, 997S–1001S (1996).
McLean, E. et al. Worldwide prevalence of anaemia, WHO Vitamin and Mineral Nutrition Information System, 1993–2005. Public Health Nutr. 12, 444 (2009).
Yip, R. Iron deficiency: contemporary scientific issues and international programmatic approaches. J. Nutr. 124, 1479S–1490S (1994).
Mei, Z. et al. Assessment of iron status in US pregnant women from the National Health and Nutrition Examination Survey (NHANES), 1999–2006. Am. J. Clin. Nutr. 93, 1312–1320 (2011).
Cogswell, M. E. et al. Assessment of iron deficiency in US preschool children and nonpregnant females of childbearing age: National Health and Nutrition Examination Survey 2003-2006. Am. J. Clin. Nutr. 89, 1334–1342 (2009).
Lozoff, B., Jimenez, E. & Wolf, A. W. Long-term developmental outcome of infants with iron deficiency. N. Engl. J. Med. 325, 687–694 (1991).
Tamura, T. et al. Cord serum ferritin concentrations and mental and psychomotor development of children at five years of age. J. Pediatr. 140, 165–170 (2002).
Lozoff, B., Jimenez, E., Hagen, J., Mollen, E. & Wolf, A. W. Poorer behavioral and developmental outcome more than 10 years after treatment for iron deficiency in infancy. Pediatrics 105, E51 (2000).
Lukowski, A. F. et al. Iron deficiency in infancy and neurocognitive functioning at 19 years: evidence of long-term deficits in executive function and recognition memory. Nutr. Neurosci. 13, 54–70 (2010).
Lozoff, B. et al. Functional significance of early-life iron deficiency: outcomes at 25 years. J. Pediatr. 163, 1260–1266 (2013).
Corapci, F., Radan, A. E. & Lozoff, B. Iron deficiency in infancy and mother-child interaction at 5 years. J. Dev. Behav. Pediatr. 27, 371–378 (2006).
Chang, S. et al. Iron-deficiency anemia in infancy and social emotional development in preschool-aged Chinese children. Pediatrics 127, e927–e933 (2011).
Lozoff, B. Iron deficiency and child development. Food Nutr. Bull. 28, S560–S571 (2007).
Jorgenson, L. A., Wobken, J. D. & Georgieff, M. K. Perinatal iron deficiency alters apical dendritic growth in hippocampal CA1 pyramidal neurons. Dev. Neurosci. 25, 412–420 (2003).
Fretham, S. J. B. et al. Temporal manipulation of transferrin-receptor-1-dependent iron uptake identifies a sensitive period in mouse hippocampal neurodevelopment. Hippocampus 22, 1691–1702 (2012).
Tyagi, E., Zhuang, Y., Agrawal, R., Ying, Z. & Gomez-Pinilla, F. Interactive actions of Bdnf methylation and cell metabolism for building neural resilience under the influence of diet. Neurobiol. Dis. 73, 307–318 (2015).
Zeisel, S. Choline, other methyl-donors and epigenetics. Nutrients 9, 445 (2017).
Ly, A. et al. Maternal folic acid supplementation modulates DNA methylation and gene expression in the rat offspring in a gestation period-dependent and organ-specific manner. J. Nutr. Biochem. 33, 103–110 (2016).
Tran, P. V. et al. Prenatal choline supplementation diminishes early-life iron deficiency-induced reprogramming of molecular networks associated with behavioral abnormalities in the adult rat hippocampus. J. Nutr. 146, 484–493 (2016).
Barks, A., Fretham, S. J., Georgieff, M. K. & Tran, P. V. Early-life neuronal-specific iron deficiency alters the adult mouse hippocampal transcriptome. J. Nutr. 148, 1521–1528 (2018).
Mudd, A. T., Fil, J. E., Knight, L. C. & Dilger, R. N. Dietary iron repletion following early-life dietary iron deficiency does not correct regional volumetric or diffusion tensor changes in the developing pig brain. Front. Neurol. 8, 735 (2018).
Leyshon, B. J., Radlowski, E. C., Mudd, A. T., Steelman, A. J. & Johnson, R. W. Postnatal iron deficiency alters brain development in piglets. J. Nutr. 146, 1420–1427 (2016).
Ranade, S. C. et al. Different types of nutritional deficiencies affect different domains of spatial memory function checked in a radial arm maze. Neuroscience 152, 859–866 (2008).
Rao, R., Tkac, I., Schmidt, A. T. & Georgieff, M. K. Fetal and neonatal iron deficiency causes volume loss and alters the neurochemical profile of the adult rat hippocampus. Nutr. Neurosci. 14, 59–65 (2011).
Connor, J. R. & Menzies, S. L. Relationship of iron to oligodendrocytes and myelination. Glia 17, 83–93 (1996).
Brunette, K. E., Tran, P. V., Wobken, J. D., Carlson, E. S. & Georgieff, M. K. Gestational and neonatal iron deficiency alters apical dendrite structure of CA1 pyramidal neurons in adult rat hippocampus. Dev. Neurosci. 32, 238–248 (2010).
Carlson, E. S. et al. Iron is essential for neuron development and memory function in mouse hippocampus. J. Nutr. 139, 672–679 (2009).
Bastian, T. W., von Hohenberg, W. C., Mickelson, D. J., Lanier, L. M. & Georgieff, M. K. Iron deficiency impairs developing hippocampal neuron gene expression, energy metabolism, and dendrite complexity. Dev. Neurosci. 38, 264–276 (2016).
Greminger, A. R., Lee, D. L., Shrager, P. & Mayer-Proschel, M. Gestational iron deficiency differentially alters the structure and function of white and gray matter brain regions of developing rats. J. Nutr. 144, 1058–1066 (2014).
Monk, C. et al. Maternal prenatal iron status and tissue organization in the neonatal brain. Pediatr. Res. 79, 482–488 (2016).
Ball, G. et al. Development of cortical microstructure in the preterm human brain. Proc. Natl Acad. Sci. USA 110, 9541–9546 (2013).
Kroenke, C. D. et al. Microstructural changes of the baboon cerebral cortex during gestational development reflected in magnetic resonance imaging diffusion anisotropy. J. Neurosci. 27, 12506–12515 (2007).
Fukumitsu, K. et al. Synergistic action of dendritic mitochondria and creatine kinase maintains ATP homeostasis and actin dynamics in growing neuronal dendrites. J. Neurosci. 35, 5707–5723 (2015).
Oruganty-Das, A., Ng, T., Udagawa, T., Goh, E. L. K. & Richter, J. D. Translational control of mitochondrial energy production mediates neuron morphogenesis. Cell. Metab. 16, 789–800 (2012).
Dallman, P. R. Biochemical basis for the manifestations of iron deficiency. Annu. Rev. Nutr. 6, 13–40 (1986).
Connor, J. R., Pavlick, G., Karli, D., Menzies, S. L. & Palmer, C. A histochemical study of iron-positive cells in the developing rat brain. J. Comp. Neurol. 355, 111–123 (1995).
Taylor, E. M. & Morgan, E. H. Developmental changes in transferrin and iron uptake by the brain in the rat. Dev. Brain Res. 55, 35–42 (1990).
Morath, D. J. & Mayer-Pröschel, M. Iron modulates the differentiation of a distinct population of glial precursor cells into oligodendrocytes. Dev. Biol. 237, 232–243 (2001).
Morath, D. J. & Mayer-Pröschel, M. Iron deficiency during embryogenesis and consequences for oligodendrocyte generation in vivo. Dev. Neurosci. 24, 197–207 (2002).
Greminger, A. R. & Mayer-Pröschel, M. Identifying the threshold of iron deficiency in the central nervous system of the rat by the auditory brainstem response. ASN Neuro 7, 1–10 (2015).
Lisman, J. E. et al. Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci. 31, 234–242 (2008).
Beard, J., Erikson, K. M. & Jones, B. C. Neonatal iron deficiency results in irreversible changes in dopamine function in rats. J. Nutr. 133, 1174–1179 (2003).
Pisansky, M. T. et al. Iron deficiency with or without anemia impairs prepulse inhibition of the startle reflex. Hippocampus 23, 952–962 (2013).
Schmidt, A. T., Waldow, K. J., Grove, W. M., Salinas, J. A. & Georgieff, M. K. Dissociating the long-term effects of fetal/neonatal iron deficiency on three types of learning in the rat. Behav. Neurosci. 121, 475–482 (2007).
Algarin, C. et al. Differences on brain connectivity in adulthood are present in subjects with iron deficiency anemia in infancy. Front. Aging Neurosci. 9, 54 (2017).
Buckner, R. L., Andrews-Hanna, J. R. & Schacter, D. L. The brain’s default network: anatomy, function, and relevance to disease. Ann. N. Y. Acad. Sci. 1124, 1–38 (2008).
Carlson, E. S., Stead, J. D. H., Neal, C. R., Petryk, A. & Georgieff, M. K. Perinatal iron deficiency results in altered developmental expression of genes mediating energy metabolism and neuronal morphogenesis in hippocampus. Hippocampus 17, 679–691 (2007).
Schachtschneider, K. M. et al. Impact of neonatal iron deficiency on hippocampal DNA methylation and gene transcription in a porcine biomedical model of cognitive development. BMC Genom. 17, 856 (2016).
Lubin, F. D., Roth, T. L. & Sweatt, J. D. Epigenetic regulation of BDNF gene transcription in the consolidation of fear memory. J. Neurosci. 28, 10576–10586 (2008).
McGowan, P. O. et al. Broad epigenetic signature of maternal care in the brain of adult rats. PLoS ONE 6, e14739 (2011).
Anderson, O. S. et al. Epigenetic responses following maternal dietary exposure to physiologically relevant levels of bisphenol A. Environ. Mol. Mutagen. 53, 334–342 (2012).
McBirney, M. et al. Atrazine induced epigenetic transgenerational inheritance of disease, lean phenotype and sperm epimutation pathology biomarkers. PLoS ONE 12, e0184306 (2017).
Perkins, A., Lehmann, C., Lawrence, R. C. & Kelly, S. J. Alcohol exposure during development: Impact on the epigenome. Int. J. Dev. Neurosci. 31, 391–397 (2013).
Ke, X. et al. IUGR disrupts the PPARγ-Setd8-H4K20me1and Wnt signaling pathways in the juvenile rat hippocampus. Int. J. Dev. Neurosci. 38, 59–67 (2014).
Tran, P. V., Kennedy, B. C., Lien, Y.-C., Simmons, R. A. & Georgieff, M. K. Fetal iron deficiency induces chromatin remodeling at the Bdnf locus in adult rat hippocampus. Am. J. Physiol. Regul. Integr. Comp. Physiol. 308, R276–R282 (2015).
Tran, P. V., Carlson, E. S., Fretham, S. J. B. & Georgieff, M. K. Early-life iron deficiency anemia alters neurotrophic factor expression and hippocampal neuron differentiation in male rats. J. Nutr. 138, 2495–2501 (2008).
Tran, P. V., Fretham, S. J. B., Carlson, E. S. & Georgieff, M. K. Long-term reduction of hippocampal brain-derived neurotrophic factor activity after fetal-neonatal iron deficiency in adult rats. Pediatr. Res. 65, 493–498 (2009).
Moore, L. D., Le, T. & Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 38, 23–38 (2013).
Tahiliani, M. et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935 (2009).
Bachman, M. et al. 5-Hydroxymethylcytosine is a predominantly stable DNA modification. Nat. Chem. 6, 1049–1055 (2014).
Wen, L. & Tang, F. Genomic distribution and possible functions of DNA hydroxymethylation in the brain. Genomics 104, 341–346 (2014).
Chen, Z. et al. Structural insights into histone demethylation by JMJD2 family members. Cell 125, 691–702 (2006).
Tsukada, Y. et al. Histone demethylation by a family of JmjC domain-containing proteins. Nature 439, 811–816 (2006).
Shimamoto, C. et al. Functional characterization of FABP3, 5 and 7 gene variants identified in schizophrenia and autism spectrum disorder and mouse behavioral studies. Hum. Mol. Genet. 23, 6495–6511 (2014).
Picker, J. D. & Coyle, J. T. Do maternal folate and homocysteine levels play a role in neurodevelopmental processes that increase risk for schizophrenia? Harv. Rev. Psychiatry 13, 197–205 (2005).
Schmidt, R. J. et al. Combined prenatal pesticide exposure and folic acid intake in relation to autism spectrum disorder. Environ. Health Perspect. 125, 097007 (2017).
McKee, S. E. & Reyes, T. M. Effect of supplementation with methyl-donor nutrients on neurodevelopment and cognition: considerations for future research. Nutr. Rev. 76, 497–511 (2018).
Wozniak, J. R. et al. Choline supplementation in children with fetal alcohol spectrum disorders: a randomized, double-blind, placebo-controlled trial. Am. J. Clin. Nutr. 102, 1113–1125 (2015).
Xiong, X. et al. Impact of pregnancy-induced hypertension on fetal growth. Am. J. Obstet. Gynecol. 180, 207–213 (1999).
Chockalingam, U. M., Murphy, E., Ophoven, J. C., Weisdorf, S. A. & Georgieff, M. K. Cord transferrin and ferritin values in newborn infants at risk for prenatal uteroplacental insufficiency and chronic hypoxia. J. Pediatr. 111, 283–286 (1987).
Bosley, A. R., Sibert, J. R. & Newcombe, R. G. Effects of maternal smoking on fetal growth and nutrition. Arch. Dis. Child. 56, 727–729 (1981).
Sullivan, E. L., Nousen, E. K. & Chamlou, K. A. Maternal high fat diet consumption during the perinatal period programs offspring behavior. Physiol. Behav. 123, 236–242 (2014).
Shen, Y. et al. Associations among maternal pre-pregnancy body mass index, gestational weight gain and risk of autism in the Han Chinese population. BMC Psychiatry 18, 11 (2018).
Monk, C., Georgieff, M. K. & Osterholm, E. A. Maternal prenatal distress and poor nutrition - mutually influencing risk factors affecting infant neurocognitive development. J. Child Psychol. Psychiatry 54, 115–130 (2013).
Shorter, K. R., Felder, M. R. & Vrana, P. B. Consequences of dietary methyl donor supplements: Is more always better? Prog. Biophys. Mol. Biol. 118, 14–20 (2015).
Pylipow, M. et al. Early postnatal weight gain, intellectual performance, and body mass index at 7 years of age in term infants with intrauterine growth restriction. J. Pediatr. 154, 201–206 (2009).
Laus, M. F., Vales, L. D., Costa, T. M. & Almeida, S. S. Early postnatal protein-calorie malnutrition and cognition: a review of human and animal studies. Int. J. Environ. Res. Public. Health 8, 590–612 (2011).
Galler, J. R. et al. Early childhood malnutrition predicts depressive symptoms at ages 11-17. J. Child Psychol. Psychiatry 51, 789–798 (2010).
Morse, N. L. Benefits of docosahexaenoic acid, folic acid, vitamin D and iodine on foetal and infant brain development and function following maternal supplementation during pregnancy and lactation. Nutrients 4, 799–840 (2012).
Brenna, J. T. Long-chain polyunsaturated fatty acids and the preterm infant: a case study in developmentally sensitive nutrient needs in the United States. Am. J. Clin. Nutr. 103, 606s–615s (2016).
Dey, A. C. et al. Maternal and neonatal serum zinc level and its relationship with neural tube defects. J. Health Popul. Nutr. 28, 343–350 (2010).
Fuglestad, A. J., Kroupina, M. G., Johnson, D. E. & Georgieff, M. K. Micronutrient status and neurodevelopment in internationally adopted children. Acta Pediatr. 105, e67–e76 (2016).
Acknowledgements
This work was supported by grants from the National Institutes of Health (R01HD29421 to M.K.G.; R01NS099178 to P.V.T.; F30HD093285 to A.K.B.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Barks, A., Hall, A.M., Tran, P.V. et al. Iron as a model nutrient for understanding the nutritional origins of neuropsychiatric disease. Pediatr Res 85, 176–182 (2019). https://doi.org/10.1038/s41390-018-0204-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41390-018-0204-8
This article is cited by
-
Chromatin accessibility and H3K9me3 landscapes reveal long-term epigenetic effects of fetal-neonatal iron deficiency in rat hippocampus
BMC Genomics (2024)
-
Micronutrients and Brain Development
Current Nutrition Reports (2019)
