
Abstract
Evasion of the host immune responses is critical for both persistent human papillomavirus (HPV) infection and associated cancer progression. We have previously shown that expression of the homeostatic chemokine CXCL14 is significantly downregulated by the HPV oncoprotein E7 during cancer progression. Restoration of CXCL14 expression in HPV-positive head and neck cancer (HNC) cells dramatically suppresses tumor growth and increases survival through an immune-dependent mechanism in mice. Although CXCL14 recruits natural killer (NK) and T cells to the tumor microenvironment, the mechanism by which CXCL14 mediates tumor suppression through NK and/or T cells remained undefined. Here we report that CD8+ T cells are required for CXCL14-mediated tumor suppression. Using a CD8+ T-cell receptor transgenic model, we show that the CXCL14-mediated antitumor CD8+ T-cell responses require antigen specificity. Interestingly, CXCL14 expression restores major histocompatibility complex class I (MHC-I) expression on HPV-positive HNC cells downregulated by HPV, and knockdown of MHC-I expression in HNC cells results in loss of tumor suppression even with CXCL14 expression. These results suggest that CXCL14 enacts antitumor immunity through restoration of MHC-I expression on tumor cells and promoting antigen-specific CD8+ T-cell responses to suppress HPV-positive HNC.
Similar content being viewed by others


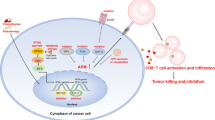
Introduction
Human papillomaviruses (HPVs) cause over 5% of all human cancer incidences, resulting in about half a million deaths every year [1]. Persistent infection with high-risk HPV is necessary for the development of HPV-associated malignancies [2], including nearly all cervical cancers (CxCa) and an increasing number of head and neck cancers (HNCs) [3, 4]. To establish persistence, HPV must evade antiviral host defenses including the innate and adaptive immune responses. The HPV oncoproteins E5, E6, and E7 dysregulate a multitude of immune response mechanisms to avoid host immune surveillance (reviewed in ref. [5]). As antiviral and antitumor immune responses share similar mechanisms [6, 7], it is likely that HPV-induced host immune evasion contributes to evasion of antitumor immune responses for cancer cell survival.
CD8+ T cells play a critical role in effective antiviral and antitumor immune responses [8]. Recent anticancer immunotherapies have had particular focus on enhancing the activity of cytotoxic CD8+ T cells to promote targeting and clearance of cancer cells [9]. These immunotherapies have shown great efficacy in a spectrum of different cancers including HNC [10, 11]. An effective CD8+ T-cell response is also of critical importance in host defense against HPV persistence and suppression of HPV-associated disease progression [12, 13]. The levels of CD8+ T-cell infiltration positively correlate with regression of HPV-positive cervical lesions [14] and disease-free survival of HPV-associated HNC cancer patients [9]. However, the mechanism of how CD8+ T cells are recruited to the tumor microenvironment (TME) to eliminate cancer cells remains elusive.
Chemokine (C-X-C motif) ligand 14 (CXCL14) is a homeostatic chemokine that promotes immune surveillance of skin epithelia through recruitment of several immune cell types, including dendritic cells (DCs), natural killer (NK) cells, and T cells [15,16,17]. Downregulation of CXCL14 expression is frequently observed in several cancers including HPV-positive cancers [18, 19]. Previously, we have shown that the HPV oncoprotein E7 is responsible for the epigenetic repression of CXCL14 transcription in HPV-positive keratinocytes and cancer cells [17]. We further revealed that restoration of CXCL14 expression in HPV-positive HNC cells dramatically suppresses tumor growth in vivo.
Here we report that the CXCL14-mediated tumor suppression is reliant on an antigen-specific CD8+ T-cell response. Our results suggest that CXCL14 enacts effective tumor suppression through recruitment of CD8+ T cells and restoration of major histocompatibility complex class I (MHC-I) antigen presentation.
Results
CXCL14-mediated tumor suppression requires CD8+ T cells
We have previously shown that CXCL14 expression significantly enhances NK and CD8+ T-cell infiltration into the tumor-draining lymph nodes (TDLNs) [17]. To examine the roles of NK and CD8+ T cells in the CXCL14-mediated antitumor immune response, NK and CD8+ T cells were depleted in wild-type C57BL/6J (B6) mice using anti-NK1.1 and anti-CD8α neutralizing antibodies, respectively (Fig. 1a). To validate specific cell depletion, we evaluated NK and CD8+ T cell numbers in mice by flow cytometry using antibodies that recognize different epitopes than the depleting antibodies. The results showed that NK and CD8+ T-cell populations were significantly depleted as expected (Fig. 1b). Next, tumor growth was monitored in the NK and CD8+ T-cell-depleted mice injected with mouse HPV-positive HNC cells transduced with vector only (MOE/E6E7Vector) or mouse HPV-positive HNC cells re-expressing CXCL14 (MOE/E6E7CXCL14). All mice injected with MOE/E6E7Vector cells exhibited robust tumor growth in isotype control and in both NK- and CD8+ T-cell-depleted mice (Fig. 1c, f–h), and succumbed to tumor burden (Fig. 1e). In mice injected with MOE/E6E7CXCL14 cells, four out of ten isotype-treated control mice completely suppress tumor growth, whereas eight out of ten NK-cell-depleted mice injected with MOE/E6E7CXCL14 cells eventually grow tumor with the lack of a statistical significance (Fig. 1d, i, j). This suggests that NK cells may contribute to the antitumor effect but are not required for CXCL14-mediated tumor suppression. Interestingly, all CD8+ T-cell-depleted mice injected with MOE/E6E7CXCL14 cells exhibited robust tumor growth (Fig. 1d, k) and succumbed by tumor burden within 60 days (Fig. 1e). These results suggest that CD8+ T cells are required for CXCL14-mediated tumor suppression and play an important role in the antitumor immune responses against HPV-positive HNC.

CD8+ T-cell depletion abrogates CXCL14-mediated tumor suppression. Wild-type B6 mice (n = 10 per group) were depleted of NK and CD8+ T cells using anti-NK1.1 and anti-CD8α antibodies, respectively, with biweekly intraperitoneal (i.p.) injections. Corresponding isotype antibodies were used as controls. Two days after the first antibody injection, MOE/E6E7Vector or MOE/E6E7CXCL14 cells (5 × 105 cells/mouse) were subcutaneously (s.c.) injected into the left flank of each mouse (a). NK- and CD8+ T-cell depletion was validated using peripheral blood by flow cytometry (b). Tumor volume was measured twice per week in mice injected with either MOE/E6E7Vector (c) or MOE/E6E7CXCL14 (d) cells. Survival rates of mice injected with MOE/E6E7CXCL14 cells were analyzed using a Kaplan–Meier estimator (e). The time to event was determined for each group (isotype, NK-, and CD8+ T-cell depletion) with the event defined as a tumor burden >2500 mm3. Deaths not associated with tumor were censored. P-values were determined by the log-rank test (e). Values that were not significantly different (n.s.) are also shown. Shown are individual tumor growth curves injected with MOE/E6E7Vector (f–h) or MOE/E6E7CXCL14 (i–k) cells. P-values of NK- or CD8+ T-cell-depleted mice compared with isotype-injected mice were determined for tumor growth (c, d) and survival (e) by two-way ANOVA analysis. *p < 0.001; n.s., not significant. Shown are representative of two independent experiments
Although all CD8+ T-cell-depleted mice injected with MOE/E6E7CXCL14 cells grew tumors, the tumor growth was delayed as compared with the control mice injected with MOE/E6E7Vector cells (Fig. 1c, d). This observation indicates two possibilities as follows: (1) in addition to CD8+ T cells, another cell type such as NK cells are necessary for the full response of CXCL14-mediated tumor suppression; and/or (2) the CD8+ T-cell depletion by the anti-CD8α antibody is incomplete and a significant number of CD8+ T cells still remain in the TME where anti-CD8α antibody does not efficiently permeate [20]. To further determine whether CD8+ T cells are required for CXCL14-mediated tumor suppression, we tested tumor growth in wild-type and Cd8α-knockout (Cd8α−/−) mice injected with MOE/E6E7Vector or MOE/E6E7CXCL14 cells. The absence of CD8+ T cells was confirmed by flow cytometry (Supplementary Fig. S1A and S1B). We found that all wild-type and Cd8α−/− mice injected with MOE/E6E7Vector cells robustly grew tumors and succumbed to tumor burden within 35 days post injection (Fig. 2a–c). Conversely, although the majority of the wild-type mice injected with MOE/E6E7CXCL14 cells did not grow tumor, all Cd8α−/− mice showed robust tumor growth (Fig. 2a, d, e). As a result, all Cd8α−/− mice succumbed to tumor burden within 35 days post injection, showing similar tumor growth kinetics as mice injected with MOE/E6E7Vector cells (Fig. 2f, g). When interpreted in the context of the delayed tumor growth observed with antibody-based CD8+ T-cell depletion (Fig. 1d, f), these results indicate that even a small population of CD8+ T cells responding to CXCL14 can slow tumor growth. Taken together, our results suggest that CD8+ T cells are the predominant driver of CXCL14-mediated tumor suppression in HPV-positive HNC.

CXCL14-mediated tumor suppression disappears in Cd8a-knockout mice. Wild-type (WT) or Cd8α−/− mice (n = 10 per group) were s.c. injected with MOE/E6E7Vector or MOE/E6E7CXCL14 cells (5 × 105 cells/mouse). Tumor volume was measured twice per week (a–e). Overall (a) and individual (b–e) tumor growth curves are shown for mice injected with MOE/E6E7Vector (a–c) or MOE/E6E7CXCL14 (a, d, e) cells. Survival rates were analyzed as was performed in Fig. 1f, g. P-values of wild-type (WT) compared with CD8α−/− mice were determined for tumor growth (a) and survival (f, g) by two-way ANOVA analysis and the log-rank test, respectively. *p < 0.05, **p < 0.0001; n.s., not significant. Shown are representative of two independent experiments
CXCL14 expression increases intratumoral infiltration of CD8+ T cells
As CD8+ T cells are necessary to enact CXCL14-mediated tumor suppression and CXCL14 expression significantly enhances T-cell migration and infiltration into the TDLN [17], we next determined the presence and abundance of CD8+ T cells in tumors with and without CXCL14 expression. To determine whether CXCL14 affects T-cell infiltration into the TME, we analyzed CD8+ T cells in frozen tumor tissue from wild-type B6 mice injected with MOE/E6E7Vector or MOE/E6E7CXCL14 cells. Using immunofluorescence, CD8+ T cells were detected by anti-CD8α antibody, along with cytokeratin counterstaining for epithelial cells (Fig. 3a, b). The percentage of CD8+ T cells per total cells in the HPV-positive tumors expressing CXCL14 was determined. The results showed that infiltration of CD8+ T cells significantly increased in tumors from mice injected with MOE/E6E7CXCL14 cells as compared with tumors from mice injected with MOE/E6E7Vector cells (Fig. 3c). These findings suggest that CXCL14 expression increases the CD8+ T-cell population in the TME, similar to what was observed in the TDLN. Furthermore, this increase in CD8+ T cells may be critical for CXCL14-mediated tumor suppression.

CXCL14 mediates CD8+ T-cell infiltration into the tumor microenvironment. Representative image of MOE/E6E7Vector (a) and MOE/E6E7CXCL14 (b) tumor sections immunostained with anti-CD8α antibody (red), anti-cytokeratin antibody (green), and DAPI (blue) to identify CD8+ T cells and tumor tissues. CD8+ T cells were quantified as a number of CD8α-positive cells per total number of DAPI-positive cells across five representative images (c). P-values were calculated using Student’s t-test. *p < 0.05. Scale bars are 50 µm
CXCL14-mediated tumor suppression requires antigen-specific CD8+ T cells
The activation of CD8+ T cells require interaction of the T-cell receptor (TCR) with its cognate peptide presented by MHC-I proteins. To evaluate whether antigen specificity of CD8+ T cells is required for CXCL14-mediated tumor suppression, we utilized the MHC-I restricted, chicken ovalbumin TCR transgenic (OT-I) mouse model [21]. The typical T-cell repertoire in wild-type mice is estimated to be responsive to over two million different peptides. In contrast, OT-I mice are genetically modified to have their CD8+ T-cell responsive repertoire highly restricted to the chicken ovalbumin peptide sequence, SIINFEKL. Although the CD8+ to CD4+ T-cell ratio is skewed in favor of CD8+ T cells as compared with wild type, all immune cell populations are present (Supplementary Fig. S1A and S1C). We tested tumor growth and mouse survival in a cohort of wild-type and OT-I mice injected with MOE/E6E7Vector or MOE/E6E7CXCL14 cells. As with the Cd8α−/− mice, both wild-type and OT-I mice injected with MOE/E6E7Vector cells robustly grew tumors (Fig. 4a, c, d) and all mice succumbed to tumor burden within 35 days post injection (Fig. 4g). Interestingly, although the majority of the wild-type mice injected with MOE/E6E7CXCL14 cells showed no or delayed tumor growth, all OT-I mice robustly grew tumors regardless of CXCL14 expression (Fig. 4b, e, f). As was observed in the Cd8α−/− mice, all OT-I mice injected with MOE/E6E7CXCL14 cells succumbed to tumor burden within 35 days (Fig. 4h). These results suggest that even in the presence of the CD8+ T-cell population, antigen specificity of the CD8+ T cells is critical for CXCL14-mediated tumor suppression.

CXCL14-mediated tumor suppression requires antigen-specific CD8+ T cells. Wild-type (WT) or OT-I mice (n = 5 per group) were s.c. injected with MOE/E6E7Vector or MOE/E6E7CXCL14 cells (5 × 105 cells/mouse). Tumor volume was measured twice per week (a–f). Tumor growth curves of each mouse are shown for WT or OT-I mice injected with MOE/E6E7Vector cells (a, c, d) or MOE/E6E7CXCL14 cells (b, e, f). Survival rates of WT or OT-I mice injected with MOE/E6E7Vector or MOE/E6E7CXCL14 (g, h) cells were analyzed as was performed in Fig. 1. P-values were determined for tumor growth (a, b) and survival (g, h) by two-way ANOVA analysis. ★ represents mice that exhibited tumor growth but had to be excluded due to self-inflicted wounds. **p < 0.01. Shown are representative of two independent experiments
CXCL14 expression restores MHC-I expression on tumor cell surface
We have shown here that the CXCL14-mediated CD8+ T-cell tumor suppression is reliant on antigen-specific CD8+ T cells. Counterintuitively, it has been well-established that the HPV oncoproteins interfere with MHC-I trafficking and expression on the surface of HPV-infected cells [22, 23]. These findings were recapitulated in our mouse model system, as MHC-I (H-2Db) expression was greatly decreased in MOE/E6E7 cells compared with HPV-negative normal immortalized MOE cells, the parental cell line of MOE/E6E7 cells (Fig. 5a). Given that CD8+ T-cell activation depends on interaction with MHC-I, we evaluated whether CXCL14 expression increases MHC-I expression in MOE/E6E7 cells by detection of the two MHC-I alloantigens in B6 mice, H-2Db and H-2Kb, using flow cytometry. Strikingly, H-2Db expression was almost completely restored in two different clones of MOE/E6E7CXCL14 cells (CL8 and CL16) comparable to the level of HPV-negative parental MOE cells (Fig. 5b, c). However, little change was observed in the H-2Kb levels regardless of CXCL14 expression (Fig. 5d). To ensure proper detection of H-2Kb, we demonstrated a significant increase of H-2Kb expression by interferon-γ (IFNγ) treatment (Fig. 5e). It is currently unknown how CXCL14 upregulates only one of the two mouse alloantigens. Our results suggest that CXCL14, in addition to promoting the migration of CD8+ T cells, may also augment antitumor CD8+ T-cell responses by restoring MHC-I expression in HPV-positive tumor cells.

CXCL14 expression restores surface expression of MHC-I proteins on HPV-positive HNC cells. Surface expression of MHC-I haplotype proteins, H-2Db (a–c) and H-2Kb (d, e), was determined in normal immortalized MOE (NI-MOE) cells (purple) (a), parental MOE/E6E7 cells (gray) (a), MOE/E6E7Vector cells (gray) (b), and two clones of MOE/E6E7CXCL14 cells (CL8-red and CL16-blue) (b) by flow cytometry. Isotype controls were evaluated on NI-MOE cells (purple outline with light purple fill) and parental MOE/E6E7 cells (black outline). Relative mean fluorescent intensity (MFI) values of MOE/E6E7Vector and MOE/E6E7CXCL14-CL16 cells were calculated (c). P-value was determined by the Student’s t-test. *p < 0.05. IFNγ treatment was used as a positive control to validate H-2Kb expression in MOE/E6E7CXCL14 cells (e). Shown are representative of three independent experiments
CXCL14-mediated tumor suppression is abrogated by knockdown of MHC-I expression in tumor cells
To determine whether restoration of MHC-I expression by CXCL14 is important for CXCL14-mediated tumor suppression, we knocked down β2-microglobulin (B2m) in MOE/E6E7CXCL14 cells using short hairpin RNAs (shRNAs). B2M is a core component of the MHC-I complex, which is necessary for the proper formation of the MHC-I complex to be trafficked and presented on the cell surface. Accordingly, disruption of B2M blocks MHC-I surface expression and antigen presentation to CD8+ T cells. We transduced the MOE/E6E7CXCL14 cells with a pool of three shRNA clones against B2M (see Methods). To enrich cells with B2M knockdown, we sorted out the transduced MOE/E6E7CXCL14 cells by selecting cells with the lowest 10% of MHC-I expression levels using fluorescence-activated cell sorting. The sorted cells (MOE/E6E7CXCL14/shB2M) showed decreased H-2Db expression similar to MOE/E6E7Vector cells, despite their CXCL14 expression (Fig. 6a). Interestingly, H-2Kb, which had previously shown nominal changes due to CXCL14 expression, was also slightly decreased in MOE/E6E7CXCL14/shB2M cells (Fig. 6b). MOE/E6E7CXCL14/shB2M cells exhibited no significant difference in doubling time, HPV oncoprotein expression, or CXCL14 expression, as compared with the parental MOE/E6E7CXCL14 cells (Supplementary Fig. S2). To determine whether B2M knockdown abrogates CXCL14-mediated tumor suppression in vivo, we monitored tumor growth in wild-type B6 mice injected with MOE/E6E7CXCL14/shB2M cells. Interestingly, the majority of the mice injected with MOE/E6E7CXCL14/shB2M cells exhibited robust tumor growth, whereas only two out of ten mice injected with MOE/E6E7CXCL14 cells experienced tumor growth (Fig. 6c, e, f). Further, nine out of ten mice injected with MOE/E6E7CXCL14/shB2M cells succumbed to tumor burden within 50 days post injection, whereas eight out of ten mice injected with MOE/E6E7CXCL14 cells survived without tumor development up to 80 days (Fig. 6d). CXCL14 appears to promote an antitumor immune response by both on recruiting peripherally immune cells to the TME as well as acting on the tumor cell itself by increasing MHC-I expression (Fig. 6g). These results suggest that upregulation of MHC-I expression by CXCL14 expression is critical for CXCL14-mediated tumor suppression through the CD8+ T-cell response.

CXCL14-mediated tumor suppression is abrogated by MHC-I knockdown in HNC cells. MOE/E6E7CXCL14 cells were transduced with shRNAs directed to B2M and enriched for knockdown by FACS sorting for the bottom 10% of MHC-I-expressing cells (MOE/E6E7CXCL14/shB2M). Flow cytometric analysis of H-2Db (a) and H-2Kb (b) MHC-I molecules in MOE/E6E7Vector (gray), MOE/E6E7CXCL14 (blue), and MOE/E6E7CXCL14/shB2M (red) cells by flow cytometry. Wild-type B6 mice (n = 10 per group) were s.c. injected with MOE/E6E7Vector, MOE/E6E7CXCL14, or MOE/E6E7CXCL14/shB2M cells (5 × 105 cells/mouse). Tumor volume was measured twice per week (c). Individual growth curves are shown from mice injected with MOE/E6E7CXCL14 (e) and MOE/E6E7CXCL14/shB2M (f) cells. Survival rates of mice injected with MOE/E6E7Vector, MOE/E6E7CXCL14, and MOE/E6E7CXCL14/shB2M cells were analyzed as was performed in Fig. 1 (d). Summary model of findings of CXCL14 on peripheral immune cells and on immune gene expression (g). P-value of MOE/E6E7CXCL14/shB2M cells compared with MOE/E6E7Vector or MOE/E6E7CXCL14 cells was determined for tumor growth (c) and survival (d) by two-way ANOVA analysis. ★ represents mice that exhibited tumor growth but had to be excluded due to self-inflicted wounds. *p < 0.01, **p < 0.001. Shown are representative of two independent experiments
CXCL14 expression in tumor cells induces CD8+ T-cell activation
Various immune checkpoint proteins such as PD-1 and CTLA-4 can interfere with the antitumor effector functions of CD8+ T cells, despite abundant CD8+ T-cell infiltration into the TME (reviewed in ref. [8]). To determine whether CXCL14 expression induces a polyfunctional CD8+ T-cell response, we assessed the production of IFNγ, tumor necrosis factor-α (TNFα), and interleukin 2 (IL-2) from CD8+ T cells co-cultured with MOE/E6E7vector or MOE/E6E7CXCL14 cells by enzyme-linked immunosorbent assay (ELISA) [24]. CD8+ T cells were isolated from mice injected with MOE/E6E7Vector cells (denoted as “null-CD8” as no tumor suppression was maintained) or MOE/E6E7CXCL14 cells (denoted as “primed-CD8” as tumor suppression was maintained). The isolated null- or primed-CD8+ T cells were co-cultured with the mitomycin-C-treated target cells (MOE/E6E7Vector or MOE/E6E7CXCL14). CD8+ T cells isolated from OT-I mice co-cultured with MOE/E6E7CXCL14 cells expressing the chicken ovalbumin (MOE/E6E7CXCL14/Ova) were used as a positive control for CD8+ T-cell activation. We also included CD8+ T cells stimulated with phorbol-12-myristate-13-acetate and ionomycin (PMA/Iono) as another positive control. As expected, IFNγ and TNFα production was significantly increased in both positive controls (Fig. 7a, b). Interestingly, IFNγ and TNFα production was highly induced when primed-CD8+ T cells were co-cultured with MOE/E6E7CXCL14 cells, but not with MOE/E6E7Vector cells. IFNγ, but not TNFα, was produced when null-CD8+ T cells were co-cultured with MOE/E6E7CXCL14 cells but not with MOE/E6E7Vector cells (Fig. 7a, b). IL-2 production was only detected in control PMA/Iono-treated CD8+ T cells but not in null- or primed-CD8+ T cells co-cultured with target cells nor CD8+ T cells from OT-I mice (Fig. 7c). These results suggest that CXCL14 expression in tumor cells induces IFNγ and TNFα production in CD8+ T cells.

CXCL14 expression in tumor cells activates CD8+ T cells for IFNγ and TNFα production and tumor cell killing. CD8+ T cells isolated from mice injected with MOE/E6E7Vector (Null-CD8+ T cells) or MOE/E6E7CXCL14 (Primed-CD8+ T cells) were co-cultured with mitomycin-treated MOE/E6E7Vector or MOE/E6E7CXCL14 cells in the presence of anti-CD28 antibody and IL-2 for 5 days. Naive CD8+ T cells alone were used as a negative control (Naive). Naive CD8+ T cells cultured with anti-CD3/CD28 antibody beads and IL-2 were stimulated with PMA and Ionomycin (PMA/Iono) as a positive control. CD8+ T cells isolated from an OT-I mouse co-cultured with MOE/E6E7CXCL14 cells expressing OVA (MOE/E6E7CXCL14-OVA) served as an additional positive control. Supernatants were collected and measured for IFNγ (a), TNFα (b), and IL-2 (c) production using ELISA. Shown are means ± SD of quadruplicates. The cytotoxic activity of null- and primed-CD8+ T cells was determined by the LDH release assay in co-culture with MOE/E6E7Vector and MOE/E6E7CXCL14 cells as target cells (d, e) at effector: target ratios 100:1, 10:1, 1:1, and 0 (no effector cells). Mean percent specific lysis values ± SD of triplicates were normalized to the signal from cell lysis by 1% SDS. P-value was determined by the Student’s t-test comparing target MOE/E6E7CXCL14 cells with target MOE/E6E7Vector cells (a–c). *p < 0.0001. Shown are representative of three (a–c) and two (d, e) independent experiments
To assess how the cytolytic capacity of CD8+ T cells is affected by CXCL14 expression in target tumor cells, we performed a lactate dehydrogenase (LDH) release assay that measures cytoplasmic LDH in cell culture supernatant released by cell lysis. Briefly, null-CD8+ T cells or primed-CD8+ T cells were added to prepared target cells, MOE/E6E7Vector, MOE/E6E7CXCL14, or MOE/E6E7CXCL14/shB2M cells. After 24 h of incubation, the cell culture supernatant was collected and LDH levels were measured. Interestingly, primed-CD8+ T cells efficiently killed both MOE/E6E7Vector and MOE/E6E7CXCL14 cells, but not MOE/E6E7CXCL14/shB2M cells, at 10:1 effector–target ratio (Fig. 7d). These results suggest that the primed-CD8+ T cells are sufficient to kill tumor cells but require MHC-I expression. Surprisingly, null-CD8+ T cells still showed significant cell-killing activity against MOE/E6E7CXCL14 cells at 10:1 effector–target ratio (Fig. 7e). In contrast, the null-CD8+ T cells could not kill MOE/E6E7Vector and MOE/E6E7CXCL14/shB2M cells. A curious finding is that the primed-CD8+ T cells were able to kill the MOE/E6E7Vector cells, albeit at a lesser efficiency. A possible scenario is that in which the primed CD8+ T cells maintain their ability to kill the MOE/E6E7Vector cells due to the fact that MHC-I is not entirely eliminated from the cells, but massively reduced. Thus, an enrichment of a small number of tumor antigen-specific CD8+ T cells may enact tumor cell killing, despite the low MHC-I expression level. These data further reinforce our findings that CXCL14 expression substantially enhances CD8+ T-cell activity through priming of CD8+ T cells and by increasing MHC-I expression in tumor cells. Taken together, our results strongly suggest that CXCL14 expression in tumor cells induces tumor suppression by driving CD8+ T-cell activation and facilitating tumor cell killing.
Discussion
CD8+ T-cell infiltration into tumors positively correlates with patient survival in several cancers, including HPV-positive HNC. HPV-associated cancers often take decades to develop and HPV must persist within the infected cell by evading immune clearance [25]. Although the majority of HPV infections are ultimately cleared, a small percentage of persistent high-risk HPV infections progress to cancer [26].
It has been well established that T-cell responses are required to eliminate HPV-infected cells (reviewed in ref. [27]). An indicator of immune-mediated regression of HPV infections is high infiltration of both CD4+ and CD8+ T cells in infected tissues [28]. Furthermore, activated intraepithelial CD8+ T cells are highly correlative to the regression rate of cervical precancerous lesions [29]. It has been shown in the mouse papillomavirus (MmuPV1) model that T cells are necessary for clearance of MmuPV1 infections [30]. Interestingly, only when both CD4+ and CD8+ T-cell compartments were eliminated in B6 mice, MmuPV1 infection persists and tumor develops, demonstrating the potent capacity of T cells to eliminate the infection. In persistent infections, immune tolerance driven by HPV limits the function of these immune cells to enact an effective immune response [31]. Recently, it has been shown that Langerhans cells, DCs residents of the epithelium, isolated from women with persistent HPV infection cannot activate CD8+ T cells by presenting HPV antigens alone [32]. Additional immune-stimulatory factors are necessary for CD8+ T-cell activation. In HPV-associated oropharyngeal squamous cell carcinoma, antigen-specific CD8+ T-cell infiltration into tumor is highly correlative with positive patient outcomes [33]. CD8+ T-cell functions are limited by either E7-mediated expression of suppressors such as PD-L1 or by low levels of antigen presentation [34, 35]. To overcome this T-cell tolerance, several immunotherapies have been tested and approved by the FDA [36]. Nevertheless, a majority of cancer patients do not respond to the current immunotherapies due to other suppressor mechanisms including the low T-cell infiltration into the TME. Thus, it is of great importance to find factors that can recruit and facilitate the activation of CD8+ T cells. We have previously shown that restoration of CXCL14 expression in HPV-positive HNC cells is sufficient for NK- and T-cell recruitment and tumor suppression [17]. Here we report that antigen-specific CD8+ T cells are required for CXCL14-mediated tumor suppression. In addition, we show that CXCL14 expression restores MHC-I expression on HPV-positive HNC cells and activates CD8+ T cells to kill autologous tumor cells.
Surprisingly, we found that CXCL14 restores the surface expression of the H-2Db but not the H-2Kb MHC-I haplotype. One possibility for the expression differences could be attributed to protein stability of the different alloantigens on the cell surface. It has previously been shown that H-2Db shows higher protein stability on cell surface as compared with H-2Kb [37]. Further evidence that would support the protein stability theory is that peptide-binding affinity to the MHC-I regulates MHC-I protein stability on cell surface. A separate study has shown that the E7 peptide (RAHYNIVTF) presented by H-2Db is critical to enact an antitumor CD8+ T-cell response [38]. Furthermore, the E7 peptide RAHYNIVTF binds to the H-2Db complex with high affinity [39]. Thus, it is possible that the increase of H-2Db, but not H-2Kb, by CXCL14 may be the consequence of the stable peptide-H-2Db complex remaining on the cell surface in sufficient time to be recognized by CD8+ T cells. In contrast, the H-2Kb with weak binding to a peptide may result in fast turnover and remain undetected.
Interestingly, overexpression of the hepatitis C virus (HCV) core protein has been shown to increase MHC-I levels on hepatocytes [40]. The results imply that expression of a single viral protein alone can increase MHC-I expression in the absence of the rest of the genome. This is important, as other studies have shown that HCV virion infection downregulates MHC-I on hepatocytes [41]. In our system, CXCL14 restoration of MHC-I expression must overcome the activity of both HPV oncoproteins that have been shown to suppress MHC-I surface expression through multiple mechanisms. Finally, in support of modest changes of MHC-I enacting protection against CD8+ T-cell killing, a recent publication has reported that Zika virus upregulates MHC-I expression to protect the cell from NK-cell killing [42]. Although the shifts in MHC-I were modest, it was sufficient to enact protection from NK-cell killings. Unfortunately, CD8+ T-cell killing was not evaluated in this study and thus a comparison with our results is not possible [42].
CXCL14 is a highly conserved chemokine constitutively expressed in normal skin keratinocytes [43]. CXCL14 expression in skin layers promote routine host immune surveillance by recruiting various immune cells [15]. Previous studies have shown that CXCL14 recruits NK cells, DCs, monocytes, and neutrophils [43,44,45]. Shurin et al. [44] revealed that CXCL14 mediated activation and maturation of DCs in vivo and in vitro. However, as this study used SCID mice that lack adaptive immunity, the roles of T cells in CXCL14-mediated tumor suppression had not been evaluated.
Our finding of the CXCL14-mediated antitumor immune response suggests that CXCL14 would be a useful tool to develop novel immunotherapies for HPV-positive CxCa and HNC patients. Importantly, our CD8+ T-cell depletion data strongly suggest that CXCL14 may assist to bolster the immune-mediated tumor suppression even when a few numbers of CD8+ T cells are present. Clinically, this is of significant importance, because pre-existing CD8+ T-cell infiltration is a key factor for efficacious anticancer immunotherapies including the treatment of immune checkpoint inhibitors [46, 47]. Therefore, adding CXCL14 to current immunotherapy regimens could be highly effective for nonresponders.
CXCL14 may also induce tumor suppression in other cancers beyond HPV-positive cancers, as CXCL14 expression is downregulated in several cancers including renal and gastric cancers [18, 19]. Indeed, restoration of CXCL14 expression triggers tumor suppression in lung and colorectal cancers, as well as HNC [44, 48, 49]. In addition, CXCL14 could be used to treat HPV-positive premalignant lesions for cancer prevention. Although current HPV vaccines are effective to prevent new HPV infection of only a handful of HPV genotypes, there is no means to treat individuals persistently infected with HPV. Given that CXCL14 expression restores MHC-I expression in HPV-infected keratinocytes and mediates CD8+ T-cell response, the use of CXCL14 as a therapeutic could extend beyond HPV-associated cancers and may have a protective role in premalignant lesions.
On the other hand, several studies have shown that CXCL14 expression levels are correlated to poor clinical outcome in pancreatic cancer, osteosarcomas, and a subtype of prostate cancers [50,51,52]. It has been shown that in these cancers, NK-cell activity is of key importance in controlling tumor growth: pancreatic [53], osteosarcoma [54], and prostate [55] cancers, and an intriguing possibility is that the CXCL14 expression may suppress the NK-cell cytotoxic capacity through its upregulation of MHC-I, an inhibitor of NK-cell activity. Interestingly, CXCL14 expression from stromal fibroblasts results in more severe disease pathologies in mouse tumor models [50]. As HPV induces peripheral tolerance in the microenvironment through actions of the HPV oncoproteins, the immune cells recruited by peripheral CXCL14 may, at best, be ineffective and, at worst, immunosuppressive inflammation by stromal NK and T cells may promote cancer progression [56]. Thus, further studies are necessary to better understand the mechanisms of CXCL14-mediated immune regulations in the TME of different cancers.
Many cancers have been shown to suppress MHC-I expression to evade antitumor immunity [57]. Particularly, a significant decrease of MHC-I repression is commonly observed in HPV-positive cells and cancers [58]. The HPV oncoproteins utilize various mechanisms to downregulate MHC-I expression in infected cells. HPV E5 binds to MHC-I, trapping it in the Golgi apparatus, preventing trafficking to the cell surface [22]. Previous work from our laboratory has shown that all but one (HLA-F) MHC-Iα subunit is repressed in HPV-positive keratinocytes [23]. Furthermore, the MHC-Iα-subunit HLA-E expression is suppressed by E7-mediated promoter methylation. Other studies have suggested that E7 inhibits transporter associated with antigen processing (TAP1), which transports cytosolic peptides into the endoplasmic reticulum, where the peptides bind to MHC-I for surface presentation. Here we have shown that CXCL14 expression restores downregulated MHC-I expression on HPV-positive HNC cells. Although the mechanism by which CXCL14 increases MHC-I remains to be determined, our results suggest that CXCL14 plays a key role in recruiting and activating CD8+ T cells in the TME. These findings would provide useful insight into the mechanism that increases CD8+ T-cell recognition of tumor cells and boost the efficacy of current immunotherapies.
Methods
Cell lines and reagents
The HPV-positive mouse oropharyngeal epithelial (MOE/E6E7) cell line and its derivative cell lines were cultured in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum (FBS) as previously described [17]. MOE/E6E7 (transformed with Ras and HPV16 E6/E7) cells were generated by Dr. John Lee, and validated by assessing cytokeratin expression, the presence of the E6 and E7 expression vectors, which confer resistance to puromycin, and activation of the mitogen-activated protein kinase pathway, a hallmark of E6 expression. All cell lines were confirmed mycoplasma-free before use.
Plasmids, lentivirus production, and transduction
The shRNA clones against B2M (TRCN0000066424, TRCN0000066425, and TRCN0000288438) were obtained through the Functional Genomics Facility at the University of Colorado School of Medicine. Lentiviruses were produced using 293FT cells transfected with the packaging constructs pMDG.2 (Addgene, 12259), psPAX2 (Addgene, 12260), and the transfer vector as previously described [17].
Reverse transcriptase-quantitative PCR
Total RNA was extracted using a RNeasy Mini kit (Qiagen) with on-column DNase digestion using the RNase-free DNase (Qiagen) according to the suppliers’ instructions. First-strand cDNA was synthesized using Transcriptor First Strand cDNA Synthesis Kit (Roche) from 1 μg of total RNA. Real-time PCR was performed in a 20 μL reaction mixture containing 10 μL of FastStart Universal SYBR green master (Rox, Roche Applied Science), 0.5 μM of each primer, 5 μL of cDNA template, and nuclease-free water using the Bio-Rad CFT Connect real-time system. Primers used in this study are as follow: Gapdh-Fwd: 5′-AATGACCCCTTCATTGACCTC-3′, Gapdh-Rev: 5′-ATGGGATTTCCATTGATGACA-3′, Cxcl14-Fwd: 5′-GGAAATGAAGCCAAAGTACCC-3′, Cxcl14-Rev: 5′-AGGCGTTGTACCACTTGATGA-3′, HPV16-E7-Fwd: 5′-AAATGACAGCTCAGAGGAGGAG-3′, HPV16-E7-Rev: 5′-GAGTCACACTTGCAACAAAAGG-3′. Data were normalized by the level of Gapdh mRNA.
Mice and tumor growth
Breeding pairs of C57BL/6J, OT-I mice (C57BL/6-Tg(TcraTcrb)1100Mjb/J), and Cd8α-knockout mice (B6.129S2-Cd8atm1Mak/J) were obtained from Jackson Laboratory and were bred in-house. Mice were genotyped according to recommended protocols (https://www.jax.org/strain/002665). All mice were maintained in accordance with the USDA guidelines. Tumors were initiated in 6- to 8-week-old mice by injection of engineered MOE/E6E7 cells (5–10 × 105 cells/mouse) subcutaneously into the rear right flank of mice (n = 10 per group unless otherwise indicated). Animals within a single cage were injected with different tumor cells to account for cage-to-cage variation. Investigators were not blinded to the different experimental groups. Tumor growth was assessed weekly using previously established techniques [17]. Tumor volume was calculated using the equation: volume = (width2 × length)/2. Animals were euthanized when tumor size was >1.5 cm in any dimension. Conversely, mice were considered tumor-free when no measurable tumor was detected for a period of 11 weeks. Survival graphs were calculated by standardizing for a tumor volume of 2500 mm3. The University of Colorado Denver and Sanford Health Institutional Animal Care and Use Committees-approved experiments, in accordance with the National Institutes of Health guidelines for the use of live animals.
Antibodies and flow cytometry
The following anti-mouse antibodies were purchased from ThermoFisher and were used according to the manufacturer’s specifications: CD8α (clone 53-6.7, Cat# 47-0081-30 or clone Tonbo 2.43, Cat# 50-1886-U100), CD4 (clone RM4-5, Cat# 48-0042-82), H-2Kb (clone AF6-88.5.5.3, Cat# 12-5958-82), H-2Db (clone 28-14-8, Cat# 11-5999-82), and anti-mouse isotype IgG2a (clone eBM2a, Cat# 114724-81). Anti-mouse CD45 (clone 30-F11, Cat# 103129) and NKp46 (clone 29A1.4, Cat# 137617) were purchased from Biolegend. Mouse spleens were passed through a 100 μm cell strainer (Corning Life Sciences) and incubated in Red Blood Cell Lysing Buffer Hybri-Max (Sigma-Aldrich) for 3 min at room temperature. For detection of MHC-I, MOE/E6E7 cells were lifted with 1 × citric saline to prevent cleavage of MHC-I on cell surface. The prepared cells were incubated with the corresponding panel of antibodies conjugated with unique fluorophores for 30 min to 1 h at room temperature and washed with phosphate-buffered saline (PBS). Samples were passed through a 35 μm cell strainer (Corning Life Sciences) immediately before analysis on an LSRII flow cytometer (Becton Dickinson) using FACSDiva collection software.
T-cell isolation and cultures
Mouse spleens were mechanically disrupted over a 100-μm filter and subjected to magnetic bead-negative enrichment for CD8+ T cells, using a CD8+ T-cell isolation kit (Stemcell Technologies). Isolated CD8+ T cells were cultured in RPMI (Gibco) containing 10% FBS, l-glutamine, penicillin/streptomycin, and β-mercaptoethanol (50 μM). All cultures were maintained at a concentration of 1 × 106 cells/mL. T cells were stimulated with 1 μg/mL anti-CD28 (clone 37.51, ThermoFisher, Cat# 14-0281-82) or anti-CD3/anti-CD28 microbeads according to the manufacturer’s recommendation (Dynabeads Mouse T-Activator CD3/CD28; Invitrogen). Stimulated cells were incubated in media supplemented with 10 ng/mL of recombinant mouse IL-2 (ThermoFisher).
ELISA and in vitro cytotoxicity LDH assay
CD8+ T cells were isolated from mice injected with MOE/E6E7 cells. CD8+ T cells were co-cultured with mitomycin-C-treated MOE/E6E7Vector or MOE/E6E7CXCL14 cells in media containing IL-2 and anti-CD28 for 5 days. Cell culture supernatants were used to quantify TNFα (R&D Systems, Cat# MTA00B), IFNγ (Biolegend, Cat# 430801), and IL-2 (R&D Systems, Cat# M2000) production by ELISA according to the manufacturer’s recommendations. Collected CD8+ T cells were incubated with MOE/E6E7Vector or MOE/E6E7CXCL14 cells (10,000 cells per reaction) for 8 h. Reactions were performed in quadruplicates at the indicated effector/target ratios according to the manufacturer’s instructions. Spontaneous lysis was assessed in the absence of effector CD8+ T cells, and maximum lysis was detected by treating target cells with 1% SDS.
Immune cell depletion in vivo
Mice received intraperitoneal administration of 100 μg/treatment of anti-NK1.1 antibody (BioXcell, clone PK136, Cat# BE0036) or anti-CD8α antibody (BioXcell, clone GK 2.43, Cat# BE0061), or control isotype IgG antibody (mIgG2a, clone C1.18.4, Cat# BE0085 or rat IgG2b, clone LTF-2, Cat# BE0090) twice a week for 30 days. On day 5, flow cytometry was performed with peripheral blood to verify specific cell depletion. CD8+ T cell was detected with anti-CD8α antibody (clone 53-6.7). Depletion (anti-NK1.1) and detection (anti-NKp46) of NK epitopes do not overlap. The anti-CD8α antibody for CD8+ T-cell depletion (clone GK 2.43) does not compete for the epitope with the anti-CD8α antibody for CD8+ T-cell detection (clone 2.43) [59].
Immunofluorescence
Tumor tissues were collected from mice injected with MOE/E6E7Vector and MOE/E6E7CXCL14 cells, and fixed overnight in 4% paraformaldehyde. All tissues were cryoprotected using 20% sucrose in PBS and subsequently frozen in optimal cutting temperature compound. Tissue was cryosectioned at 12 μm increments. Each tissue section was blocked using lamb serum and permeabilized using Triton X. Immunofluorescence was performed on the tissue sections using the following antibodies: mouse anti-pan cytokeratin (1:100, Novus, clone PCK-26, Cat# NB120-6401) and rat anti-CD8α (1:200, Novus, clone 53-6.7, Cat# NBP1-49045) antibodies. After incubation with primary antibodies, sections were incubated with appropriate Alexa Fluor-conjugated secondary antibodies: anti-mouse IgG Alexa Fluor 488 (Invitrogen, Cat# A-11029), anti-rat IgG Alexa Fluor 594 (Invitrogen, Cat# A-21209), and DAPI (Invitrogen). Immunofluorescence images were captured using a Zeiss 780 LSM confocal microscope.
Statistical analysis
Student’s t-test and two-way analysis of variance were used to calculate significance for comparison of two matched groups and three or more unmatched groups, respectively. Results were considered statistically significant at a p-value < 0.05. Distributions of time to event outcomes (e.g., survival time) was summarized with Kaplan–Meier curves, compared across groups using the log-rank test with α = 0.01.
References
Parkin DM. The global health burden of infection-associated cancers in the year 2002. Int J Cancer. 2006;118:3030–44.
Cogliano V, Baan R, Straif K, Grosse Y, Secretan B, El Ghissassi F, et al. Carcinogenicity of human papillomaviruses. Lancet Oncol. 2005;6:204.
Bosch FX, Lorincz A, Munoz N, Meijer CJ, Shah KV. The causal relation between human papillomavirus and cervical cancer. J Clin Pathol. 2002;55:244–65.
Chaturvedi AK, Engels EA, Pfeiffer RM, Hernandez BY, Xiao W, Kim E, et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J Clin Oncol. 2011;29:4294–301.
Westrich JA, Warren CJ, Pyeon D. Evasion of host immune defenses by human papillomavirus. Virus Res 2017;231:21–33.
Kuss-Duerkop SK, Westrich JA, Pyeon D. DNA tumor virus regulation of host DNA methylation and its implications for immune evasion and oncogenesis. Viruses. 2018;10:pii: E82.
Johnston RJ, Comps-Agrar L, Hackney J, Yu X, Huseni M, Yang Y, et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8( + ) T cell effector function. Cancer Cell 2014;26:923–37.
Klebanoff CA, Gattinoni L, Restifo NP. CD8 + T-cell memory in tumor immunology and immunotherapy. Immunol Rev 2006;211:214–24.
Kansy BA, Concha-Benavente F, Srivastava RM, Jie HB, Shayan G, Lei Y, et al. PD-1 status in CD8( + ) T cells associates with survival and anti-PD-1 therapeutic outcomes in head and neck cancer. Cancer Res 2017;77:6353–64.
Farkona S, Diamandis EP, Blasutig IM. Cancer immunotherapy: the beginning of the end of cancer? BMC Med 2016;14:73.
Ferris RL, Blumenschein G Jr., Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med. 2016;375:1856–67.
Maldonado L, Teague JE, Morrow MP, Jotova I, Wu TC, Wang C, et al. Intramuscular therapeutic vaccination targeting HPV16 induces T cell responses that localize in mucosal lesions. Sci Transl Med. 2014;6:221ra13.
Cuburu N, Graham BS, Buck CB, Kines RC, Pang YY, Day PM, et al. Intravaginal immunization with HPV vectors induces tissue-resident CD8 + T cell responses. J Clin Invest. 2012;122:4606–20.
Trimble CL, Clark RA, Thoburn C, Hanson NC, Tassello J, Frosina D, et al. Human papillomavirus 16-associated cervical intraepithelial neoplasia in humans excludes CD8 T cells from dysplastic epithelium. J Immunol 2010;185:7107–14.
Lu J, Chatterjee M, Schmid H, Beck S, Gawaz M. CXCL14 as an emerging immune and inflammatory modulator. J Inflamm (Lond). 2016;13:1.
Lee HT, Liu SP, Lin CH, Lee SW, Hsu CY, Sytwu HK, et al. A crucial role of CXCL14 for promoting regulatory T cells activation in stroke. Theranostics 2017;7:855–75.
Cicchini L, Westrich JA, Xu T, Vermeer DW, Berger JN, Clambey ET, et al. Suppression of antitumor immune responses by human papillomavirus through epigenetic downregulation of CXCL14. MBio 2016;7:3.
Lyu XJ, Li HZ, Ma X, Li XT, Gao Y, Ni D, et al. Elevated S100A6 (Calcyclin) enhances tumorigenesis and suppresses CXCL14-induced apoptosis in clear cell renal cell carcinoma. Oncotarget 2015;6:6656–69.
Hu C, Lin F, Zhu G, Xue X, Ding Y, Zhao Z, et al. Abnormal hypermethylation of promoter region downregulates chemokine CXC ligand 14 expression in gastric cancer. Int J Oncol. 2013;43:1487–94.
Moses K, Klein JC, Mann L, Klingberg A, Gunzer M, Brandau S. Survival of residual neutrophils and accelerated myelopoiesis limit the efficacy of antibody-mediated depletion of Ly-6G + cells in tumor-bearing mice. J Leukoc Biol. 2016;99:811–23.
Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, Carbone FR. T cell receptor antagonist peptides induce positive selection. Cell 1994;76:17–27.
DiMaio D, Petti LM. The E5 proteins. Virology 2013;445:99–114.
Cicchini L, Blumhagen RZ, Westrich JA, Myers ME, Warren CJ, Siska C, et al. High-risk human papillomavirus E7 alters host DNA methylome and represses HLA-E expression in human keratinocytes. Sci Rep. 2017;7:3633.
Kim DH, Kim EM, Lee EH, Ji KY, Yi J, Park M, et al. Human papillomavirus 16E6 suppresses major histocompatibility complex class I by upregulating lymphotoxin expression in human cervical cancer cells. Biochem Biophys Res Commun. 2011;409:792–8.
Tindle RW. Immune evasion in human papillomavirus-associated cervical cancer. Nat Rev Cancer. 2002;2:59–65.
de Martel C, Ferlay J, Franceschi S, Vignat J, Bray F, Forman D, et al. Global burden of cancers attributable to infections in 2008: a review and synthetic analysis. Lancet Oncol. 2012;13:607–15.
van der Burg SH, de Jong A, Welters MJ, Offringa R, Melief CJ. The status of HPV16-specific T-cell reactivity in health and disease as a guide to HPV vaccine development. Virus Res 2002;89:275–84.
Hibma MH. The immune response to papillomavirus during infection persistence and regression. Open Virol J. 2012;6:241–8.
Woo YL, Sterling J, Damay I, Coleman N, Crawford R, van der Burg SH, et al. Characterising the local immune responses in cervical intraepithelial neoplasia: a cross-sectional and longitudinal analysis. BJOG 2008;115:1616–21. discussion 21-2
Handisurya A, Day PM, Thompson CD, Bonelli M, Lowy DR, Schiller JT. Strain-specific properties and T cells regulate the susceptibility to papilloma induction by Mus musculus papillomavirus 1. PLoS Pathog. 2014;10:e1004314.
Smola S, Trimble C, Stern PL. Human papillomavirus-driven immune deviation: challenge and novel opportunity for immunotherapy. Ther Adv Vaccin. 2017;5:69–82.
Da Silva DM, Woodham AW, Rijkee LK, Skeate JG, Taylor JR, Koopman ME, et al. Human papillomavirus-exposed Langerhans cells are activated by stabilized Poly-I:C. Papillomavirus Res. 2015;1:12–21.
Welters MJP, Ma W, Santegoets S, Goedemans R, Ehsan I, Jordanova ES, et al. Intratumoral HPV16-specific T cells constitute a type I-oriented tumor microenvironment to improve survival in HPV16-driven oropharyngeal cancer. Clin Cancer Res. 2018;24:634–47.
Bal V, McIndoe A, Denton G, Hudson D, Lombardi G, Lamb J, et al. Antigen presentation by keratinocytes induces tolerance in human T cells. Eur J Immunol. 1990;20:1893–7.
Liu C, Lu J, Tian H, Du W, Zhao L, Feng J, et al. Increased expression of PDL1 by the human papillomavirus 16 E7 oncoprotein inhibits anticancer immunity. Mol Med Rep. 2017;15:1063–70.
Ran X, Yang K. Inhibitors of the PD-1/PD-L1 axis for the treatment of head and neck cancer: current status and future perspectives. Drug Des Devel Ther. 2017;11:2007–14.
Su RC, Miller RG. Stability of surface H-2K(b), H-2D(b), and peptide-receptive H-2K(b) on splenocytes. J Immunol 2001;167:4869–77.
de Oliveira LM, Morale MG, Chaves AA, Cavalher AM, Lopes AS, Diniz Mde O, et al. Design, immune responses and anti-tumor potential of an HPV16 E6E7 multi-epitope vaccine. PLoS ONE. 2015;10:e0138686.
Feltkamp MC, Smits HL, Vierboom MP, Minnaar RP, de Jongh BM, Drijfhout JW, et al. Vaccination with cytotoxic T lymphocyte epitope-containing peptide protects against a tumor induced by human papillomavirus type 16-transformed cells. Eur J Immunol. 1993;23:2242–9.
Herzer K, Falk CS, Encke J, Eichhorst ST, Ulsenheimer A, Seliger B, et al. Upregulation of major histocompatibility complex class I on liver cells by hepatitis C virus core protein via p53 and TAP1 impairs natural killer cell cytotoxicity. J Virol 2003;77:8299–309.
Tardif KD, Siddiqui A. Cell surface expression of major histocompatibility complex class I molecules is reduced in hepatitis C virus subgenomic replicon-expressing cells. J Virol 2003;77:11644–50.
Glasner A, Oiknine-Djian E, Weisblum Y, Diab M, Panet A, Wolf DG, et al. Zika virus escapes NK cell detection by upregulating major histocompatibility complex class I molecules. J Virol 2017;91:22.
Kurth I, Willimann K, Schaerli P, Hunziker T, Clark-Lewis I, Moser B. Monocyte selectivity and tissue localization suggests a role for breast and kidney-expressed chemokine (BRAK) in macrophage development. J Exp Med. 2001;194:855–61.
Shurin GV, Ferris RL, Tourkova IL, Perez L, Lokshin A, Balkir L, et al. Loss of new chemokine CXCL14 in tumor tissue is associated with low infiltration by dendritic cells (DC), while restoration of human CXCL14 expression in tumor cells causes attraction of DC both in vitro and in vivo. J Immunol 2005;174:5490–8.
Cao X, Zhang W, Wan T, He L, Chen T, Yuan Z, et al. Molecular cloning and characterization of a novel CXC chemokine macrophage inflammatory protein-2 gamma chemoattractant for human neutrophils and dendritic cells. J Immunol 2000;165:2588–95.
Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–71.
Ribas A, Dummer R, Puzanov I, VanderWalde A, Andtbacka RHI, Michielin O, et al. Oncolytic virotherapy promotes intratumoral T cell infiltration and improves anti-PD-1 immunotherapy. Cell 2017;170:1109–19 e10.
Hata R, Izukuri K, Kato Y, Sasaki S, Mukaida N, Maehata Y, et al. Suppressed rate of carcinogenesis and decreases in tumour volume and lung metastasis in CXCL14/BRAK transgenic mice. Sci Rep. 2015;5:9083.
Miyamoto C, Maehata Y, Motohashi K, Ozawa S, Ikoma T, Hidaka K, et al. Fasudil, a Rho kinase inhibitor, suppresses tumor growth by inducing CXCL14/BRAK in head and neck squamous cell carcinoma. Biomed Res 2014;35:381–8.
Augsten M, Hagglof C, Olsson E, Stolz C, Tsagozis P, Levchenko T, et al. CXCL14 is an autocrine growth factor for fibroblasts and acts as a multi-modal stimulator of prostate tumor growth. Proc Natl Acad Sci USA. 2009;106:3414–9.
Lu J, Song G, Tang Q, Zou C, Han F, Zhao Z, et al. IRX1 hypomethylation promotes osteosarcoma metastasis via induction of CXCL14/NF-kappaB signaling. J Clin Invest. 2015;125:1839–56.
Wente MN, Mayer C, Gaida MM, Michalski CW, Giese T, Bergmann F, et al. CXCL14 expression and potential function in pancreatic cancer. Cancer Lett 2008;259:209–17.
Van Audenaerde JRM, Roeyen G, Darcy PK, Kershaw MH, Peeters M, Smits ELJ. Natural killer cells and their therapeutic role in pancreatic cancer: A systematic review. Pharm Ther. 2018;189:31–44.
Tarek N, Lee DA. Natural killer cells for osteosarcoma. Adv Exp Med Biol. 2014;804:341–53.
Pasero C, Gravis G, Granjeaud S, Guerin M, Thomassin-Piana J, Rocchi P, et al. Highly effective NK cells are associated with good prognosis in patients with metastatic prostate cancer. Oncotarget 2015;6:14360–73.
Chen L, Guo L, Tian J, He H, Marinova E, Zhang P, et al. Overexpression of CXC chemokine ligand 14 exacerbates collagen-induced arthritis. J Immunol 2010;184:4455–9.
Garcia-Lora A, Algarra I, Garrido F. MHC class I antigens, immune surveillance, and tumor immune escape. J Cell Physiol. 2003;195:346–55.
Nasman A, Andersson E, Nordfors C, Grun N, Johansson H, Munck-Wikland E, et al. MHC class I expression in HPV positive and negative tonsillar squamous cell carcinoma in correlation to clinical outcome. Int J Cancer. 2013;132:72–81.
Podolin PL, Foley JP, Carpenter DC, Bolognese BJ, Logan GA, Long E 3rd, et al. T cell depletion protects against alveolar destruction due to chronic cigarette smoke exposure in mice. Am J Physiol Lung Cell Mol Physiol. 2013;304:L312–23.
Acknowledgements
We thank Ross Kedl, Linda van Dyk, Eric Clambey, Paul Ahlquist, Paul Lambert, Xiao-Jing Wang, and members of the Pyeon laboratory for useful comments and suggestions. This work was supported in part by NIH R01 DE026125 (DP and WCS), NIH T32 AI052066 (JAW), Immunology and Microbiology Pilot Award (DP and JES), grants from the University of Colorado Cancer Center (DP), Colorado Clinical and Translational Sciences Institute (DP, JIS, and ROG), and a generous gift from the Marsico Fund (DR).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
DR is a consultant for Astra Zeneca and Suvica, and an advisory board member for Astra Zeneca, Merck, EMD Serono, Genentech, and Nanobiotix. A patent has been applied for by University of Colorado and Sanford Health with DP and JHL as named inventors. The patent application number is PCT/US16/58550. All other authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Westrich, J.A., Vermeer, D.W., Silva, A. et al. CXCL14 suppresses human papillomavirus-associated head and neck cancer through antigen-specific CD8+ T-cell responses by upregulating MHC-I expression. Oncogene 38, 7166–7180 (2019). https://doi.org/10.1038/s41388-019-0911-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-019-0911-6
This article is cited by
-
Targeting MHC-I molecules for cancer: function, mechanism, and therapeutic prospects
Molecular Cancer (2023)
-
Comprehensive analysis of CXCL14 uncovers its role during liver metastasis in colon cancer
BMC Gastroenterology (2023)
-
CDX-2 expression correlates with clinical outcomes in MSI-H metastatic colorectal cancer patients receiving immune checkpoint inhibitors
Scientific Reports (2023)
-
CXCL14 facilitates the growth and metastasis of ovarian carcinoma cells via activation of the Wnt/β-catenin signaling pathway
Journal of Ovarian Research (2021)
