
Abstract
Chromosomal translocations involving the MLL1 gene characterize a poor prognosis subset of acute myeloid leukemia (AML), referred to as 11q23-AML. Transcription of the HOXA9 and HOXA10 genes is enhanced in hematopoietic stem and progenitor cells in these leukemias. We previously found the ARIH2 gene was repressed by HoxA9 in myeloid progenitors, but activated by HoxA10 during granulopoiesis. ARIH2 encodes the Triad1 protein, an anti-proliferative E3 ubiquitin ligase. In the current study, we investigate the role of Triad1 in leukemogenesis induced by an MLL1 fusion protein (Mll-Ell). We found Mll-Ell increased expression of HoxA9, HoxA10, and Triad1 because HoxA9 represses only one of two ARIH2 cis elements that are activated by HoxA10. Although Triad1 antagonized the generally pro-proliferative effects of the Mll-Ell oncoprotein, we found blocking HoxA9 and HoxA10 phosphorylation shifted the balance to ARIH2 repression in Mll-Ell+ cells. We investigated the significance of these in vitro results in a murine bone marrow transplant model. We found Triad1 knockdown significantly shortened the latency to development of AML in mice transplanted with Mll-Ell-transduced bone marrow. And, Triad1 expression fell during the prolonged AML latency period in mice transplanted with bone marrow expressing Mll-Ell alone. Our studies identify Triad1 as a leukemia suppressor in 11q23-AML. This suggests defining relevant Triad1 substrates may indicate novel therapeutic targets in this disease.
Similar content being viewed by others


Introduction
Aberrant expression of the homeodomain (HD) transcription factors HoxB3, HoxB4, HoxA7-11, and Meis1 was identified a subset of AML with poor prognosis [1,2,3,4,5]. AML with these characteristics had translocation or partial duplication involving the MLL1 gene (11q23-AML), MYST3, and CREBBP translocations, or were cytogenetically normal with poor outcomes [1,2,3,4,5]. Hox expression was increased in CD34+ hematopoietic stem cells (HSC) in these leukemias and did not decrease during differentiation [1,2,3]. Expression of MLL1 fusion proteins described in AML in murine bone marrow increased transcription of these HOX genes and induced myeloproliferation progressing to AML in vivo [6]. The time lag indicates additional mutations are required for leukemogenesis in 11q23-AML [6].
There are four groups of HOX genes (A–D) that are highly conserved in humans and mice (HoxA-D) [7]. HSC are characterized by transcription of HOX1-4, committed progenitors by HOX7-13, and these genes become inactive in mature cells [8]. Transduction of murine bone marrow with HoxA9 or HoxA10 vectors increases granulocyte/monocyte progenitors and induces a myeloproliferative neoplasm that progresses to AML in vivo [9,10,11,12,13,14]. Therefore, leukemogenesis requires mutations in addition to HoxA9 or HoxA10. HoxA9 and HoxA10 have conserved DNA-binding domains resulting in regulation of a set of common target genes, including genes encoding growth factors and receptors such as FGF2 and TGFB2 [15,16,17,18,19].
Other HoxA9 and HoxA10 domains are not conserved and some common target genes are differentially regulated, including genes encoding phagocyte effectors, gp91PHOX and p67PHOX [15, 20,21,22,23]. Although HoxA9 and HoxA10 regulate the same cis element, HoxA10 represses and HoxA9 activates these genes [21,22,23]. Differential binding of HoxA9 versus HoxA10 to these genes is regulated by phosphorylation of HD tyrosine residues [21,22,23]. In murine bone marrow transplantation experiments, we found overexpressed HoxA10 with a HD tyrosine mutation prevented gp91PHOX and p67PHOX expression and resulted in differentiation block more rapidly than overexpressing wild-type HoxA10 [14, 24].
ARIH2 encodes the E3 ubiquitin ligase Triad1; another differentially regulated HoxA9/HoxA10 target gene [25,26,27,28]. Triad1 is required to terminate emergency (stress) granulopoiesis and its expression increases during myelopoiesis. We defined a cis element in the ARIH2 promoter activated by HoxA10, but repressed by HoxA9 [26]. HD tyrosine phosphorylation decreased HoxA9 binding of HoxA9, but increased HoxA10 binding to ARIH2 [25, 26]. In contrast, HoxA9 and HoxA10 activate an FGF2 (fibroblast growth factor 2) promoter cis element in a non-tyrosine phosphorylation dependent manner [17]. And, this resulted in increased Fgf2 production by Mll-Ell expressing myeloid progenitors, with autocrine stimulation of proliferation and hypersensitivity to cytokines with overlapping signaling pathways [29].
Triad1 ubiquitination of growth factor receptors facilitates lysosomal degradation, resulting in signal termination rather than receptor recycling [26]. We identified Triad1-dependent ubiquitination/degradation of the fibroblast growth factor receptor 1 (Fgf-R1) during termination of emergency granulopoiesis [26]. This implicated Triad1 effects on Fgf-R1 in reversing the expansion of myeloid progenitors that occurs during emergency granulopoiesis. Consistent with this, knockdown of Triad1 increased cytokine hypersensitivity of HoxA10-overexpressing cells, but not HoxA9-overexpressing cells [25, 26].
A set of twins were described with concordant MLL1 translocations, but a chromosomal deletion involving ARIH2 in only one [30]. The latter twin developed AML more rapidly, suggesting a leukemia suppressor function for Triad1 in 11q23-AML. Impaired tyrosine phosphorylation of HoxA9 and HoxA10 might also decrease Triad1 expression in MLL1-related leukemia. HoxA9 and HoxA10 are substrates for Shp2 protein tyrosine phosphatase [14]. Constitutively active Shp2 mutants are found in 11q23-AML which efficiently dephosphorylate HoxA9 and HoxA10 [14, 22, 24]. Consistent with this, we found accelerated progression to AML in mice transplanted with bone marrow transduced with vectors to express HoxA10 plus activated Shp2 versus HoxA10 alone [14, 24]. Shp2 is constitutively activated by internal tandem duplications of FLT3, and the incidence of such mutation is increased Hox-overexpressing AML [31, 32].
In the current study, we find Mll-Ell induces ARIH2 transcription and Triad1 expression in myeloid progenitor cells, but this is reversed by Shp2 activity. Although MLL1 fusion proteins increase both HoxA9 and HoxA10, the initial effect of Mll-Ell on Triad1 expression was similar to HoxA10 alone. In murine bone marrow transplant studies, we determine that Triad1 knockdown enhances Mll-Ell-induced myeloproliferation and accelerates progression to AML. Consistent with this, endogenous Triad1 expression decreases during AML progression in mice transplanted with Mll-Ell transduced bone marrow. This is associated with decreased HoxA10-binding to the ARIH2 promoter, and increased binding of HoxA9 (favoring repression). Expression of Shp2 also increases during disease progression in these mice. Therefore, Triad1 substrates, such as Fgf-R1, may identify targetable pathways in Hox-overexpressing AML.
Results
Mll-Ell increases Triad1 expression
MLL1 fusion proteins increase transcription of HOXA9 and HOXA10. Before investigating effects of MLL1 fusion proteins on Triad1, we investigated the impact of co-overexpressing these Hox proteins. For these studies, we transduced murine bone marrow cells with retroviral vectors to express HoxA9, HoxA10, or both (or control vector). Lin−CD34+ cells were used in these studies (referred to as “myeloid progenitor” cells; >70% Sca1−ckit+CD34+CD38−Gr1−) [25, 26]. Some cells were differentiated with G-CSF (>85% ckit−CD34−CD38+Gr1+).
HoxA9 significantly decreased Triad1 mRNA i with or without G-CSF-induced differentiation (p < 0.001, n = 6) and HoxA10 had the opposite effect (p < 0.001, n = 6) (Fig. 1a). Co-overexpressing HoxA9 and HoxA10 increased Triad1 mRNA (p < 0.01, n = 6), with a greater effect in differentiating cells (p = 0.001, n = 6) (Fig. 1a). HoxA9 or HoxA10 were equivalently overexpressed (Fig. 1b). We found overexpression of HoxA10 or co-overexpressing HoxA9 + HoxA10 increased total protein ubiquitination with or without G-CSF; but this was decreased by overexpressing HoxA9 alone (Fig. 1c). Triad1 protein correlated with mRNA.

HoxA9 + HoxA10 increases Triad1 expression and total protein ubiquitination in myeloid progenitor cells and differentiating granulocytes. Murine myeloid progenitor cells were transduced with HoxA9, HoxA10, HoxA9 + HoxA10, or control vector and analyzed with or without granulocyte differentiation with G-CSF. a Triad1 mRNA was increased by HoxA9 + HoxA10, but HoxA9 alone decreased Triad1. Significant differences in expression indicated by *, **, ***, #, ##, ###, &, or &&. b HoxA9 and HoxA10 were equivalently expressed in transduced cells. Significant differences indicated by *, **, ***, or #. Not significantly different expression is indicated by ‘a’, ‘b’, ‘c’, ‘d’, ‘e’, or ‘f’. Bars in these graphs are means and error bars ± SEM. c Total protein ubiquitination was increased by HoxA9 + HoxA10. Western blots of lysate proteins were sequentially probed with antibodies to total ubiquitinated (Ub) protein, HoxA9, HoxA10, Triad1, or Gapdh (loading control)
We hypothesized increased Triad1 expression and activity in cells expressing MLL1 fusion proteins. Approximately 20 MLL1 fusion proteins have been described and they have comparable effects on expression of HD transcription factors. However, some translocations are more common in AML (e.g., Mll-Ell, Mll-Af9) and others in acute lymphoblastic leukemia (e.g., Mll-Af4) [6]. We transduced primary murine bone marrow cells with a vector to express Mll-Ell or control vector, and analyzed myeloid progenitor cells with or without G-CSF-differentiation. Since tyrosine phosphorylation regulates HoxA9 and HoxA10 activity and these proteins are Shp2 substrates, we co-transduced some cells with a vector to express a constitutively active Shp2 (E76K) [25, 26].
We found significantly increased Triad1 mRNA in Mll-Ell transduced myeloid progenitor cells compared to control, with or without G-CSF (p < 0.001, n = 6) (Fig. 2a). There was significantly less Triad1 in myeloid progenitor cells co-transduced with Mll-Ell + E76K-Shp2 vectors compared to Mll-Ell alone, with or without G-CSF (p < 0.001, n = 6) (Fig. 2a). Mll-Ell increased HoxA9 and HoxA10 relative to control cells (p < 0.0001, n = 6) (Fig. 2b). This increase was equivalent for HoxA9 and HoxA10 (p = 0.4, n = 6) and not influenced by E76K-Shp2 (p > 0.2, n = 6) (Fig. 2b).

Mll-Ell increased Triad1 expression in myeloid progenitor cells with or without G-CSF. Murine bone marrow myeloid progenitor cells were transduced with vectors to express Mll-Ell, constitutively active Shp2 (E76K), both or control vector. a Triad1 mRNA was increased in Mll-Ell transduced murine bone marrow myeloid progenitors with and without differentiation, but this was reversed by constitutively active Shp2. Significant differences indicated by *, **, ***, #, ##, or ###. Not significantly different expression indicated by ‘a’ or ‘b’. b HoxA9 and HoxA10 were equivalent in cells expressing Mll-Ell. Significant differences indicated by *, **, ***, or #. Not significantly different expression indicated by ‘a’, ‘b’, ‘c’, ‘d’, ‘e’, or ‘f’. Bars in these graphs are means and error bars ± SEM. c Mll-Ell increased Triad1 protein and total protein ubiquitination in myeloid progenitors with or without granulocyte differentiation. Western blots were serially probed with antibodies to total ubiquitinated (Ub) protein, Triad1, HoxA9, HoxA10, Ell, or Gapdh (loading control). d Shp2 activity decreased Mll-Ell-induced Triad1 expression and protein ubiquitination. Western blots were probed with total Ub protein, Triad1, Shp2, and Gapdh (loading control) antibodies
We found increased total ubiquitinated protein in Mll-Ell-transduced cells compared to control, with a greater effect after G-CSF-differentiation (Fig. 2c). Co-expression of E76K-Shp2 decreased total ubiquitinated protein relative to Mll-Ell alone (Fig. 2d). Modulation of Triad1 protein by Mll-Ell, with or without E75K-Shp2, was consistent with Triad1 mRNA.
Mll-Ell activates the ARIH2 promoter in a HoxA9/HoxA10-dependent manner
To analyze the effect of Mll-Ell on the ARIH2 promoter, we transfected U937 cells with ARIH2 promoter-reporter constructs, described in our prior studies. HoxA10 activates cis elements at −182 to −191 bp and −30 to −41 bp in this promoter, but HoxA9 represses only the distal cis element (Fig. 3a) [25, 26]. Some transfectants were differentiated with retinoic acid/dimethyl formamide (RA/DMF) [33].

Mll-Ell activated the ARIH2 promoter. a Mll-Ell influenced the proximal and distal ARIH2 cis elements. ARIH2 promoter/reporter constructs containing 198 bp (distal HoxA9/HoxA10 binding and proximal HoxA10-binding cis elements), 167 bp (proximal only), or 29 bp (neither) were transfected into U937 cells were co-transfected with vectors to express and. The effect of Mll-Ell, HoxA9, HoxA10, HoxA9 + HoxA10, or control vector on reporter activity was determined ± retinoic acid (RA/DMF). Significant differences indicated by *, ** ***, #, ##, or ###. Not significant differences indicated by ‘a’, ‘b’, ‘c’, ‘d’, ‘e’, or ‘f’. b Mll-Ell increased proximal ARIH2 cis element activity. A minimal promoter/reporter construct with three copies of the proximal ARIH2 cis element or control was assayed in transfectants with or without Mll-Ell, HoxA9, HoxA10, or control vectors. Reporter activity was assayed ± RA/DMF. Significant differences indicated by * or **. Not significant differences by ‘a’, ‘b’, ‘c’, or ‘d’. c Mll-Ell regulated the distal ARIH2 cis element in a HoxA9/HoxA10-dependent manner. A minimal promoter/reporter construct with three copies of the distal ARIH2 cis element or vector control was assayed for activity in transfectants with or without Mll-Ell vector, shRNA vectors to knockdown HoxA9 or HoxA10, or an E76K-Shp2 vector (or control). Reporter activity was analyzed ± RA/DMF. Significant differences indicated by *, **, ***, #, ##, or ###. d Mll-Ell increased binding of HoxA9 or HoxA10 to the ARIH2 promoter in a differentiation stage-specific manner. Binding was determined by chromatin immunoprecipitation. Significant differences indicated by *, **, ***, or #. Bars in these graphs are means and error bars ± SEM
Mll-Ell increased activity of the 198 and 167 bp ARIH2 constructs (p < 0.001, n = 6) (Fig. 3a). This increase was significantly greater in differentiated versus untreated transfectants (73.1% ± 3.2% versus 37.6% ± 4.4% p < 0.01, n = 6), but less than with HoxA10 overexpression (p < 0.02, n = 6) (Fig. 3a). The effect of co-overexpressing HoxA9 + HoxA10 was not significantly different than Mll-Ell (p = 0.1, n = 6) (Fig. 3a). In contrast, the effect of Mll-Ell or HoxA9 + HoxA10 on the 167 bp construct was not different than HoxA10 alone (p = 0.8, n = 6) (Fig. 3a). None of the proteins influenced activity of the control reporter, which was subtracted as background.
These studies suggest Mll-Ell influences both ARIH2 cis elements. To test this directly, U937 cells were co-transfected with a reporter construct with three copies of the proximal or distal ARIH2 cis element linked to a minimal promoter. We found activity of the proximal cis element was equivalently increased by Mll-Ell and HoxA10 (p = 0.6, n = 3), but not influenced by HoxA9 (Fig. 3b) [26]. Mll-Ell decreased activity of the distal cis element in undifferentiated transfectants, but increased activity with differentiation (p < 0.001, n = 6), similar to HoxA9 + HoxA10 (Fig. 3c). In Mll-Ell-expressing cells, activity of the distal cis element was augmented by HoxA9 knockdown (p < 0.001, n = 6) and attenuated by HoxA10 knockdown (p < 0.01, n = 6). We hypothesized Shp2 decreased the effect of Mll-Ell on Triad1 expression by enhancing repression of ARIH2 by HoxA9 and blocking activation by HoxA10. Consistent with this, constitutive Shp2 activity decreased activation by Mll-Ell in transfectants with the distal ARIH2 cis element construct (p < 0.001, n = 6) (Fig. 3c). In chromatin immunoprecipitation studies, Mll-Ell increased binding of endogenous HoxA9 and HoxA10 to the distal cis element (Fig. 3D).
We also investigated the impact of HoxA9, HoxA10, Mll-Ell, or Shp2 on the endogenous ARIH2 gene in these transfectants. We found HoxA9 + HoxA10 (Fig. 4a) or Mll-Ell (Fig. 4b) significantly increased Triad1 mRNA (p < 0.001, n = 9). Activated Shp2 prevented induction of Triad1 mRNA by Mll-Ell (Fig. 4b). Mll-Ell increased HoxA9 and HoxA10 equivalently (p > 0.4, n = 6) (Fig. 4c).

Effects of HoxA9, HoxA10, and Mll-Ell on endogenous Triad1 mRNA and the 198 bp ARIH2 promoter construct are similar. a Triad1 expression increased in U937 cells expressing HoxA10 + HoxA9. Cells were transfected with vectors to express HoxA9, HoxA10, HoxA9 + HoxA10 or control. Triad1 mRNA expression was determined ± RA/DMF-differentiation. Significant differences indicated by *, **, ***, #, ##, ###, or &. b Triad1 expression increased in U937 cells expressing Mll-Ell, but this effect was decreased by constitutively active Shp2 (E76K). Cells were transfected with vectors to express Mll-Ell + E76K-Shp2 or control vector. Transfectants were analyzed ± RA/DMF. Significant differences indicated by *, **, ***, #, ##, or ###. c Expression of HoxA9 and HoxA10 is equivalent in Mll-Ell transfected U937 cells. Significant differences indicated by * or **. Not statistically significant differences indicated by ‘a’, ‘b’, or ‘c’. Bars in these graphs are means and error bars ± SEM
Triad1 antagonizes Mll-Ell-induced protein ubiquitination
To determine if Mll-Ell increased total protein ubiquitination in a Triad1-dependent manner, we transduced murine bone marrow cells with retroviral vectors to express Mll-Ell, Triad1-specific shRNAs, or both (or control vector). We found Triad1 knockdown inhibited the Mll-Ell-induced increase in total protein ubiquitination (Fig. 5a). Expression of Triad1 protein correlated with mRNA (Fig. 5b).

Triad1 knockdown or Shp2 activation increased ubiquitination-induced and cytokine-induced proliferation in Mll-Ell-transduced murine bone marrow cells. Murine myeloid progenitor cells were transduced with vectors to express Mll-Ell, Mll-Af9, Triad1, Triad1-specific shRNAs, E76K-Shp2, C463S-Shp2, or relevant vector controls. Cells were analyzed with or without G-CSF-differentiation. a Triad1 knockdown decreased the Mll-Ell-induced increase in total protein ubiquitination. Western blots of cell lysates were probed with ubiquitinated proteins (Ub), Triad1, Ell, or Gapdh (loading control) antibodies. b Mll-Ell increased Triad1 expression in myeloid progenitor cells throughout granulocyte differentiation. Significant differences indicated by *, **, ***, #, ##, ###, or &. Bars in these graphs are means and error bars ± SEM. c Triad1 knockdown increased cytokine hypersensitivity of Mll-Ell or Mll-Af9 expressing murine myeloid progenitor cells. Proliferation was determined by 3H-thymidine incorporation during GM-CSF-induced proliferation. Significant differences in incorporation at common GM-CSF doses are indicated by * or **. d Constitutive Shp2 activity increased cytokine hypersensitivity in Mll-Ell-expressing myeloid progenitor cells. Significant differences in 3H-thymidine incorporation at common GM-CSF doses indicated by * or **. e Dominant negative Shp2 decreased cytokine hypersensitivity in Mll-Ell-expressing myeloid progenitor cells. Significant differences in 3H-thymidine incorporation at common GM-CSF doses indicated by *. f Triad1 impairs Fgf2-induced proliferation of myeloid progenitor cells. Cells were analyzed for proliferation in response to various amounts of Fgf2. Significant differences in 3H-thymidine incorporation at common Fgf2 doses indicated by * or **. Circles or crosses represent means and error bars ± SEM
Since Triad1 is anti-proliferative, we investigated the impact of knocking down Triad1 on cytokine-induced proliferation in Mll-Ell transduced murine myeloid progenitor cells. Since Mll-Ell-induced Triad1 expression was blocked by constitutive Shp2 activity, we also determined the effect of activated (E76K) or dominant negative (C463S) Shp2. Transduced murine bone marrow myeloid progenitor cells were assayed for proliferation in response to a dose titration of GM-CSF. We found GM-CSF-induced proliferation of Mll-Ell-transduced cells was significantly greater than control (p < 0.001, n = 3) (Fig. 5c). Mll-Ell-induced cytokine hypersensitivity was augmented by Triad1 knockdown (p < 0.01, n = 3) (Fig. 5c), or E76K-Shp2 (p < 0.01, n = 3) (Fig. 5d), but decreased by C463S-Shp2 (Fig. 5e). We found similar results with Mll-Af9 ± Triad1 shRNA (Fig. 5c).
Fgf2 production by Mll-Ell-expressing cells facilitates GM-CSF-induced proliferation by stimulating common signaling pathways (e.g., PI3-kinase activation) [29]. We previously found an Fgf-R1 blocking antibody decreased cytokine hypersensitivity of Mll-Ell+ cells [29]. To determine if Triad1 influences proliferation via Fgf2/Fgf-R1, we transduce murine bone marrow cells with vectors to express Triad1 or Triad1-specific shRNAs (or control vectors). Consistent with our hypothesis, we found Fgf2-induced proliferation was significantly increased by Triad1 knockdown, but impaired by Triad1 overexpression (Fig. 5f).
Triad1 antagonizes Mll-Ell-induced progression to AML in vivo
We used a murine bone marrow transplantation model to investigate the influence of Triad1 on leukemogenesis. For these studies, bone marrow mononuclear cells were transduced with vectors to express Mll-Ell, Triad1-specific shRNAs, Mll-Ell + Triad1-specific shRNAs, or control vectors and transplanted into irradiated mice. Rates of development of myeloproliferation (indicated by PMN > 50% of total white blood cells with blasts <5%) or AML (circulating myeloid blasts >10%) were determined.
Mice transplanted with Mll-Ell-transduced bone marrow developed peripheral granulocytosis (Fig. 6a) with gradually increasing myeloid blasts (Fig. 6b), accompanied by a modest hemoglobin decrease (Fig. 6c). In comparison, mice receiving bone marrow transduced with Mll-Ell + Triad1 shRNAs exhibited a more rapid rise in granulocytes (p < 0.01, n = 10 by week 10) (Fig. 6a) and myeloid blasts (p < 0.01, n = 10 by week 16) (Fig. 6b) with significant anemia (Fig. 6c). Development of AML was significantly more rapid in these mice with 50% exhibiting >10% circulating blasts at 16 weeks versus 32 weeks for mice transplanted with bone marrow expressing Mll-Ell alone (p < 0.001, n = 10) (Fig. 6d). Recipients of bone marrow transduced with Triad1 shRNA vector or control vector did not develop granulocytosis, AML, or anemia.

Triad1 delayed development of myeloproliferation and acute myeloid leukemia in mice transplanted with Mll-Ell-transduced bone marrow. Murine bone marrow transduced with Mll-Ell, Triad1-specific shRNAs, both, or control vectors was transplanted into recipient mice. Mice were followed by peripheral blood counts as indicated and sacrificed at various time points. a Peripheral blood granulocytes rose significantly more rapidly in recipients of Mll-Ell with Triad1-specific shRNA transduced bone marrow in comparison to Mll-Ell alone. Significant differences in circulating granulocytes indicated by * or ** for various time points. b Peripheral blood myeloid blasts also rose significantly more rapidly in these mice. Significant differences in circulating myeloid blasts indicated by * for various time points. c Hemoglobin fell in recipients of bone marrow expressing Mll-Ell with Triad1-specific shRNA in comparison to Mll-Ell alone. Significant differences in hemoglobin indicated by * or ** for various time points. d Acute myeloid leukemia developed significantly more rapidly in recipients of Mll-Ell-transduced bone marrow with Triad1 knockdown in comparison to Mll-Ell expression alone. Development of leukemia was based on >10% circulating myeloid blasts. Circles or triangles represent means and error bars ± SEM. e Myeloproliferation transitions to acute myeloid leukemia earlier in recipients of Mll-Ell transduced bone marrow in the presence of Triad1 knockdown. Sternal bone marrow was obtained from at 16 or 24 weeks post-transplant and histology of Wright–Giesma stained sections analyzed (at ×40 magnification)
At 16 weeks, differentiating granulocytes were abundant in the bone marrow of mice transplanted with Mll-Ell transduced cells, but myeloid blasts had accumulated in recipients of Mll-Ell + Triad1 shRNA-expressing bone marrow (Fig. 6e). By 24 weeks, myeloid blasts had effaced the bone marrow of the latter, but not the former. We also investigated bone marrow populations from cohorts of mice sacrificed at 16 or 36 weeks. We found Mll-Ell significantly expanded the Sca1+ and CD34+ populations relative to control vector at 16 weeks (p < 0.01, n = 6), with further expansion at 36 weeks (p < 0.01, n = 6) (Fig. 7a). However, Triad1 knockdown in Mll-Ell transduced bone marrow expanded these populations significantly more at both points (p < 0.001, n = 6) (Fig. 7a). Triad1 knockdown alone had minimal effect on these populations.

In mice transplanted with Mll-Ell transduced bone marrow, Triad1 expression decreased and immature progenitor expansion increased with time. Mice were transplanted with bone marrow transduced with Mll-Ell, Triad1-specific shRNAs, both, or control vectors. Cohorts of mice were sacrificed at various time points for bone marrow analysis. a Progressive expansion of Sca1+ or CD34+ bone marrow progenitors in recipients of Mll-Ell transduced bone marrow was increased by Triad1 knockdown. Murine bone marrow was analyzed by flow cytometry at 16 and 24 weeks post-transplant. Significant differences indicated by *, **, ***, #, ##, ###, &, or &&. Boxes are 95% confidence interval, solid line the mean and error bars ± SEM. b Triad1 expression decreased with time post-transplant with Mll-Ell-expressing bone marrow. Total murine bone marrow mononuclear cells were analyzed at various time points post-transplant. Significant differences indicated by *, **, ***, #, ##, ###, &, &&, or &&&. c In recipients of Mll-Ell transduced bone marrow, increased Mll-Ell, HoxA9, and HoxA10 expression with time was accelerated by Triad1 knockdown. Lin−Sca1+/−CD34+ murine bone marrow cells were analyzed at various time points post-transplant by real time PCR. Significant differences indicated by *, **, ***, #, ##, ###, &, &&, or &&&. d Binding to the ARIH2 promoter of HoxA9 increased and HoxA10 decreased over time. Lin−Sca1+/−CD34+ murine bone marrow cells were analyzed by chromatin immunoprecipitation at 10 or 32 weeks post-transplant. Significant differences are indicated by * or **. e Shp2 expression increased over time. Lin−Sca1+/−CD34+ murine bone marrow cells were analyzed for Shp1 and Shp2 mRNA expression at 10 or 32 weeks post transplantation by real time PCR. Significant differences are indicated by * or **. Bars are means and error bars ± SEM
We investigated expression of Triad1, HoxA9, and HoxA10 mRNA in bone marrow cells from these mice (Lin−Sca1+/−CD34+). In recipients of Mll-Ell transduced bone marrow, Triad1 decreased significantly at each time point after 10 weeks (p < 0.01, n = 6); inversely correlating Triad1 with leukemia progression (Fig. 7b). At 10 weeks, Triad1 was reduced to approximately 50% of control in bone marrow transduced with Triad1-specific shRNAs (p = 0.07, n = 6) (Fig. 7b). Triad1 mRNA decreased at each time point in recipients of Mll-Ell + Triad1 shRNA-expressing bone marrow (p < 0.01, n = 6), but not recipients of bone marrow with Triad1 knockdown alone (Fig. 7b).
HoxA9 or HoxA10 mRNA expression was not significantly different in recipients of Mll-Ell transduced bone marrow versus control at 10 weeks (p > 0.1, n = 6), but increased significantly at each subsequent time point (p < 0.01, n = 6) (Fig. 7c). HoxA9 and HoxA10 were relatively increased in recipients of bone marrow transduced with Mll-Ell + Triad1-specific shRNAs vectors compared to Mll-Ell alone at each time point (p < 0.001, n = 6) (Fig. 7c). We previously found that PI3-kinase activation by Fgf2/Fgf-R stabilized β-catenin in Mll-Ell+ cells. CDX4 is a β-catenin target gene, and Cdx4 activates the HOXA9 and HOXA10 promoters [34, 35]. Decreased Triad1 expression, due to Triad1 shRNA or disease progression, would stabilize Fgf-R1 and consequently increase HoxA9 and HoxA10.
We investigated a mechanism for progressively decreased Triad1 in recipients of Mll-Ell transduced bone marrow by chromatin immunoprecipitation (with Lin−Sca1+/−CD34+ cells). Over time, co-precipitation of the ARIH2 promoter with HoxA9 increased, but co-precipitation with HoxA10 decreased (Fig. 7d). Since tyrosine phosphorylation regulates affinity of HoxA9 versus HoxA10 for this promoter, we investigated expression of HoxA9 and HoxA10 phosphatases; Shp2 and Shp1 [14, 22]. We found increasing expression over time of both PTPs in Lin−Sca1+/−CD34+ cells from recipients of Mll-Ell-transduced bone marrow (Fig. 7e).
Decreased Triad1 and increased HoxA9 and HoxA10 correlate with cytokine hypersensitivity in human CD34+ AML cells
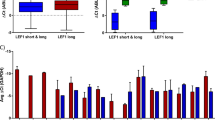
Increased Hox expression in AML is not limited to subjects with MLL1 gene rearrangements [4, 5, 36]. We investigated HoxA9 and HoxA10 expression in CD34+ bone marrow cells from AML subjects without 11q23-AML. We found significantly greater HoxA9 and HoxA10 expression in leukemia samples compared to control CD34+ bone marrow cells (p < 0.01, n = 12) (Fig. 8a). We separated these samples into low Hox versus high Hox expressing groups (p < 0.01, n = 6) (Fig. 8a). Triad1 expression was significantly increased in CD34+ bone marrow cells in the low Hox AML group versus control (p < 0.001, n = 6), but was significantly less in the high Hox AML group (p < 0.01, n = 5) (Fig. 8a). Similar to the murine experiments, we found inverse correlation between Triad1 and Shp2 or Shp1 expression in the high Hox group (Fig. 8a).

Triad1 expression and cytokine hypersensitivity correlated with HoxA9 and HoxA10 expression in human acute myeloid leukemia. Bone marrow CD34+ cells from human subjects with acute myeloid leukemia or control subjects were analyzed. a HoxA9 and HoxA10 expression distinguished different groups of human AML samples. mRNA expression was determined by real time PCR. Significant differences indicated by *, **, ***, #, ##, ###, &, or &&. Boxes are 95% confidence interval, solid line mean and dotted line median. b GM-CSF-induced proliferation was increased in human AML samples with greatest HoxA9 and HoxA10 expression. Proliferation assays were performed with various GM-CSF doses. Significant differences in 3H-thymidine incorporation at common GM-CSF dose indicated by * or **. Circles are means and error bars + SEM. c Graphic representation of effects of Mll-Ell on Triad1 and Fgf2-induced proliferation
To determine if differences in HoxA9, HoxA10, and Triad1 expression correlated with cytokine hypersensitivity, CD34+ bone marrow cells were studied for proliferation in response to a dose titration of GM-CSF. We found a greater proliferative response at each cytokine dose in the high Hox AML samples versus low Hox AML or control cells (p < 0.01, n = 3 for each cytokine dosage) (Fig. 8b). Differences in cytokine-induced proliferation between low Hox AML and control cells were modest.
Discussion
In murine bone marrow transplant studies, expression of MLL1 fusion proteins, or overexpression of HoxA9 or HoxA10, induces granulocytosis, with progression to AML after a substantial time lag [6, 9, 11]. This suggests accumulation of additional mutations is required for differentiation block. The gene encoding Triad1 is activated by HoxA10, but repressed by HoxA9 [25, 26]. And, HoxA10-induced cytokine hypersensitivity of myeloid progenitor cells is enhanced by Triad1 knockdown, suggesting Triad1 antagonizes HoxA10 pro-proliferative effects [25]. This may be relevant to the role of HoxA10-induced Triad1 expression in terminating emergency granulopoiesis; a process characterized by rapid myeloid progenitor expansion [26]. Conversely, we found repression of Triad1 expression by HoxA9 enhanced cytokine hypersensitivity of progenitor cells overexpressing this Hox protein [26].
In the current studies, we determined that an MLL1 fusion protein (Mll-Ell) increased Triad1 expression in myeloid progenitor cells, due to the initially greater impact of HoxA10 versus HoxA9 on the ARIH2 promoter. Consistent with this, Triad1 knockdown increased cytokine hypersensitivity in Mll-Ell or Mll-Af9-transduced myeloid progenitor cells. And, a progressive decrease in Triad1 expression correlated with development of AML in mice transplanted with Mll-Ell-transduced bone marrow. We demonstrated the antagonistic effect of Triad1 for Mll-Ell-induced leukemia in additional transplantation studies. In these experiments, AML developed significantly more rapidly in recipients of bone marrow transduced with vectors to express Mll-Ell plus shRNAs specific to Triad1 compared to Mll-Ell alone.
Based on these results, we hypothesize Triad1 is a suppressor of Hox-overexpressing leukemias. This is consistent with clinical data from a set of twins with discordant 11q23-AML with the same MLL1 translocation, but deletion of ARIH2 on chromosome 3p in only the one who developed AML [30]. Deletion is only one possible mechanism for decreased Triad1 expression and disease progression. In the current studies, we found impaired cytokine-induced HoxA9 or HoxA10 tyrosine phosphorylation increased ARIH2 repression and blocked transcriptional activation, respectively.
We previously identified HoxA9 and HoxA10 as substrates for both Shp2 and Shp1 [14, 22]. In prior murine transplant studies with HoxA10-overexpressing bone marrow, AML latency was decreased by co-expressing an activated, leukemia-associated form of Shp2, associated with accelerated phenotypic differentiation block and impaired progenitor apoptosis [19,20,21,22,23,24]. Our studies indicate decreased Triad1 would be an additional consequence of increased Shp2 activity in HoxA10-overexpressing bone marrow. Therefore, mutations which activate Shp2 or Flt3, as are found in human 11q23-AML, would be anticipated to cooperate with MLL1 fusion proteins [31,32,33,34,35,36]. Also, we found increased expression of Shp2 and Shp1 with disease progression in bone marrow progenitor cells from mice transplanted with Mll-Ell. This increase was associated with increased HoxA9 binding, but decreased HoxA10 binding, to the ARIH2 promoter in vivo.
We also found increased HoxA9/HoxA10, decreased Triad1 and increased Shp2 expression in human samples from AML subjects. Hox expression correlated with relative cytokine hypersensitivity of these samples in comparison to control CD34+ bone marrow cells. In our studies, the high Hox expressing subset was greater than 30% of our total patient population, consistent with the referral nature of our practice.
Other investigators found homozygous ARIH2 knockout was embryonic lethal in mice and ARIH2+/− mice developed overwhelming inflammation [37]. We found HOXA10−/− mice exhibited an overwhelming emergency granulopoiesis response, corrected by re-expressing Triad1, but not steady state inflammation [26]. These studies suggest the leukemia suppressor function of Triad1 is related to its normal role in terminating the innate immune response. Among the Triad1 substrates is Fgf-R1; a protein participating in myeloid progenitor expansion [26]. In this study, we found Triad1 influenced Fgf2-induced myeloid progenitor proliferation. This is of interest since FGF2 is a HoxA9 and HoxA10 target gene involved in cytokine hypersensitivity of Mll-Ell-expressing cells (Fig. 8c) [29].
We found Mll-Ell increases Triad1 in a Hox-dependent manner. This paradoxically antagonizes effects of Mll-Ell on cytokine hypersensitivity and leukemogenesis. However, this leukemia-suppressive effect was overcome by Shp2 activity. In mice transplanted with Mll-Ell-expressing bone marrow, Triad1 expression fell with disease progression, accompanied by increased Shp2. These studies identify initial increase in Triad1, with decline over time, as a mechanism for delayed AML in MLL1 fusion protein expressing murine models. This suggests activation of Triad1, or targeting pathways regulated by Triad1 substrates, as a potential therapeutic approach to Hox-overexpressing AML.
Materials and methods
Source of plasmids
A plasmid vector to express HoxA10 was a kind gift of C. Largman (previously University of California, San Francisco, now retired). We generated a HoxA9 expression vector by rt-PCR. A vector to express the E3 ligase Triad1 was provided by B. van der Reijden (Radboud University, Netherlands). Vectors to express Mll-Ell or Mll-Af9 were generous gifts of DE Zhang (University of California, San Diego) and J. Crispino (Northwestern University, Chicago), respectively. Constructs for Shp2 were generated based on a cDNA for Shp2 cDNA provided by S. Frank (University of Alabama, Birmingham) [14]. Vectors to express shRNAs specific to Triad1, HoxA10, and HoxA9 shRNAs and relevant controls were generated based on a tool on the Promega website (Promega, Madison, WI) using the pLKO.1puro vector; a gift from MK Rundell (previously at Northwestern University, Chicago, now retired). Knockdown experiments were performed with three combined optimal vectors.
We amplified the human ARIH2 5’ promoter region by PCR, as previously described, for assay in the pGL3-basic vector (Promega) [25]. Also as described, we generated constructs in a minimal promoter/reporter vector (pGL3-promoter) using three copies of the −22 to −48 bp or −174 to −198 bp ARIH2 promoter sequence [25].
Oligonucleotides
We obtained custom synthesized oligonucleotides from MWG Biotech (Piedmont, NC).
Myeloid cell lines and validation
The original source of U937 cells used in these experiments was A. Kraft (University of Arkansas Cancer Center, Tucson AR). Cells were maintained as described and STR profiled annually [33].
Murine bone marrow analysis
Bone marrow was obtained from C57/BL6 mice and Sca1+ cells were by affinity chromatography using the Miltenyi magnetic bead system (Miltenyi Biotechnology, Auburn, CA). Granulocyte/monocyte progenitor cells (GMP) were obtained by CD34+ isolation of bone marrow cells with the Miltenyi magnetic bead system after a 48 h culture period in DME supplemented by 10% fetal calf serum, 1% pen-strep, and murine recombinant GM-CSF (20 ng/ml), SCF (100 ng/ml), and IL-3 (10 ng/ml) (R & D Systems Inc., Minneapolis, MN). Granulocyte differentiation was performed by treatment of these cells with G-CSF (20 ng/ml).
For murine bone marrow transduction, we generated retrovirus with the MSCV vector and Phoenix packaging cell line (as per; Stratagene, La Jolla, CA). Retroviral supernatant was incubated with isolated Sca1+ cells and polybrene (6 μg/ml) [14]. Transduction studies were repeated three times with two different retrovirus batches (six independent samples per condition). All biological replicates met the standard of a normal distribution.
Murine bone marrow transplantation
C57/Black 6 mice were obtained from Jackson Labs. Lin−Sca1+ cells were transduced as described above and viable cells purified by negative selection for Annexin V using affinity chromatography (Miltenyi Biotech). Lethally irradiated syngeneic mice were injected with Lin−Sca1+ cells (2 × 105). Ten mice were studied per group for an 80% chance of detecting a 20% difference between groups. No mice were excluded from evaluation. No randomization or investigator “blinding” was required in the study design.
Peripheral blood counts were determined using a Hemavet counter (Erba Diagnostics, Miami FL). Standard errors were comparable between groups.
Quantitative PCR studies
Studies were performed by the SYBR green “standard curve” method of quantitative PCR and data analyzed using Applied Biosystems software. Each independent sample was assayed in triplicate and results normalized to 18S or input chromatin, as appropriate. All biological replicates exhibited a normal distribution.
Chromatin co-immunoprecipitation
Protein−DNA crosslinks were generated by incubating cells in formaldehyde and chromatin sized by sonication of cell lysates (to 100 bp average size) [38]. Co-immuno-precipitating chromatin was identified by PCR [18, 38]. Two independent experiments were performed for each condition and each was assayed in triplicate.
Promoter analysis
ARIH2 promoter constructs (30 μg) were assayed for Luciferase activity in U937 transfection experiments [25, 26]. Cells were also transfected with HoxA9, HoxA10, HoxA9 + HoxA10, or Mll-Ell (or control) vectors (50 μg). ARIH2 cis elements were assayed using a minimal promoter/Luciferase reporter vector with −22 to −48 bp or −174 to −198 bp of the promoter (or control). Cis elements were analyzed in U937 transfectants with Mll-Ell vectors, or vectors to overexpress or knockdown HoxA9 and/or HoxA10. Differentiation of some transfectants was induced with retinoic acid (48 h) and dimethyl formamide (24 h). Transduction efficiency was determined with a β-galactosidase reporter. Six independent samples were assayed in duplicate for each condition. All biological replicates exhibited normal distribution.
Protein analysis
Western blots were performed using cell lysates in 2 × SDS sample buffer. Western blots probed with antibodies to ubiquitin, HoxA9, HoxA10 (Santa Cruz Biotechnology, CA: SC-9133, SC-17156, SC-17158), Triad1, Ell or Gapdh (Proteintech Biotechnology, IL: catalog # 15006, 51044, 60004). Some cells were pre-treated 5 h with MG132 (5 μM). Experiments were repeated three times with different lysates and representative blots are shown.
Statistical analysis
SigmaPlot and SigmaStat software was used for statistical analysis and generation of graphical representation of data. Two sided Student’s t test or ANOVA were used and standard error represented by error bars. Statistical significance was a p value < 0.03.
Approvals
Animal studies were approved by the Animal Care and Use Committees of Northwestern University and Jesse Brown VA. Human subject research was approved by the institutional regulatory boards of Northwestern University and Jesse Brown VA and patients provided informed consent.
References
Kawagoe H, Humphries RK, Blair A, Sutherland HJ, Hogge DE. Expression of HOX genes, HOX cofactors, and MLL in phenotypically and functionally defined subpopulations of leukemic and normal human hematopoietic cells. Leukemia. 1999;13:687–98.
Drabkin HA, Parsy C, Ferguson K, Guilhot F, Lacotte L, Roy L, et al. Quantitative HOX expression in chromosomally defined subsets of acute myelogenous leukemia. Leukemia. 2002;16:186–95.
Armstrong SA, Staunton JE, Silverman LB, Pieters R, den Boer ML, Minden MD, et al. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genet. 2002;30:41–7.
Camos M, Esteve J, Jares P, Colomer D, Rozman M, Villamor N, et al. Gene expression profiling of acute myeloid leukemia with translocation t(8;16)(p11; p13) and MYST3-CREBBP rearrangement reveals a distinctive signature with a specific pattern of HOX gene expression. Cancer Res. 2006;66:6947–54.
Riche J, Zeng C, Baron A, Gadgil S, Gemmill RM, Tigaud I, et al. Hox expression in AML identified a distinct subset of patients with intermediate cytogenetics. Leukemia. 2004;18:1059–63.
Li Z, Luo RT, Mi S, Sun M, Chen P, Bao J, et al. Consistent deregulation of gene expression between human and murine MLL rearrangement leukemias. Cancer Res. 2009;69:1109–16.
Acampora D, D’Esposito M, Faiella A, Pannese M, Migliaccio E, Morelli F, et al. The human HOX gene family. Nucleic Acids Res. 1989;17:10385–10400.
Sauvageau G, Lansdorp PM, Eaves CJ, Hogge DE, Dragowska WH, Reid DS, et al. Differential expression of homeobox genes in functionally distinct CD34+ subpopulations of human bone marrow cells. Proc Natl Acad Sci USA. 1994;91:12223–7.
Calvo KR, Sykes DB, Pasillas M, Kamps MP. Hoxa9 immortalizes a granulocyte-macrophage colony-stimulating factor-dependent promyelocyte capable of biphenotypic differentiation to neutrophils or macrophages, independent of enforced meis expression. Mol Cell Biol. 2000;20:3274–85.
Lawrence HJ, Helgason CD, Sauvageau G, Fong S, Izon DJ, Humphries RK, et al. Mice bearing a targeted interruption of the homeobox gene HOXA9 have defects in myeloid, erythroid, and lymphoid hematopoiesis. Blood. 1997;89:1922–30.
Buske C, Feuring-Buske M, Antonchuk J, Rosten P, Hogge DE, Eaves CJ, et al. Overexpression of HOXA10 perturbs human lymphomyelopoiesis in vitro and in vivo. Blood. 2001;97:2286–92.
Bjornsson JM, Andersson E, Lundstrom P, Larsson N, Xu X, Repetowska E, et al. Proliferation of primitive myeloid progenitors can be reversibly induced by HOXA10. Blood. 2001;98:3301–8.
Thorsteinsdottir U, Mamo A, Kroon E, Jerome L, Bijl J, Lawrence HJ, et al. Overexpression of the myeloid leukemia-associated Hoxa9 gene in bone marrow cells induces stem cell expansion. Blood. 2002;99:121–9.
Wang H, Lindsey S, Konieczna I, Bei L, Horvath E, Eklund EA. Constitutively active SHP2 cooperates with HoxA10 overexpression to induce acute myeloid leukemia. J Biol Chem. 2009;284:2549–67.
Eklund EA. The role of Hox proteins in leukemogenesis: insights into key regulatory events in hematopoiesis. Crit Rev Oncog. 2011;16:65–76.
Shah CA, Wang H, Bei L, Platanias LC, Eklund EA. HoxA10 regulates transcription of the gene encoding transforming growth factor beta 2 (TGFB2) in myeloid cells. J Biol Chem. 2011;286:3161–76.
Shah CA, Bei L, Wang H, Platanias LC, Eklund EA. HoxA10 protein regulates transcription of gene encoding fibroblast growth factor 2 (FGF2) in myeloid cells. J Biol Chem. 2012;287:18230–48.
Bei L, Lu YF, Bellis SL, Zhou W, Horvath E, Eklund EA. Identification of a HoxA10 activation domain necessary for transcription of the gene encoding beta3 integrin during myeloid differentiation. J Biol Chem. 2007;282:16846–59.
Wang H, Lu YF, Huang W, Papoutsakis ET, Fuhrken P, Eklund EA. HoxA10 activates transcription of the gene encoding mitogen−activated protein kinase phosphatase 2 (Mkp2) in myeloid cells. J Biol Chem. 2007;282:16164–76.
Eklund EA, Jalava A, Kakar R. Tyrosine phosphorylation of HoxA10 decreases DNA binding and transcriptional repression during interferon gamma-induced differentiation of myeloid leukemia cell lines. J Biol Chem. 2000;275:20117–26.
Lindsey S, Zhu CL, Lu YF, Eklund EA. HoxA10 represses transcription of the gene encoding p67phox in phagocytic cells. J Immunol. 2005;175:5269–79.
Eklund EA, Goldenberg I, Lu Y, Andrejic J, Kakar R. SHP1 protein-tyrosine phosphatase regulates HoxA10 DNA binding and transcriptional repression activity in undifferentiated myeloid cells. J Biol Chem. 2000;39:36878–88.
Bei L, Lu YF, Eklund EA. HoxA9 activates transcription of the gene encoding gp91PHOX during myeloid differentiation. J Biol Chem. 2005;280:12359–70.
Lindsay S, Huang W, Wang H, Horvath E, Zhu C, Eklund EA. Activation of SHP2 protein-tyrosine phosphatase increases HoxA10-induced repression of the genes encoding gp91PHOX and p67PHOX. J Biol Chem. 2007;282:2237–49.
Wang H, Bei L, Shah CA, Horvath E, Eklund EA. HoxA10 influences protein ubiquitination by activating transcription of ARIH2; the gene encoding Triad1. J Biol Chem. 2011;286:16832–45.
Wang H, Bei L, Shah CA, Hu L, Eklund EA. HoxA10 terminates emergency granulopoiesis by increasing expression of Triad1. J Immunol. 2015;194:5375–87.
Van Der Reijden BA, Erpelinck-Verschueren CAJ, Lowenberg B, Jansen JH. TRIADs: a new class of proteins with a novel cysteine-rich signature. Protein Sci. 1999;8:1557–61.
Marteijn JAF, van Emst L, Erpelinck-Verschueren CAJ, Nikoloski G, Menke A, de Witte T, et al. The E3 ubiquitin-protein ligase Triad1 inhibits clonogenic growth of primary myeloid progenitor cells. Blood. 2005;106:4114–23.
Shah CA, Bei L, Wang H, Platanias LC, Eklund EA. The leukemia-associated Mll-Ell oncoprotein induces Fibroblast growth factor 2 (Fgf2)-dependent cytokine hypersensitivity in myeloid progenitor cells. J Biol Chem. 2013;288:32490–505.
Zhu X, He F, Zeng H, Ling S, Chen A, Wang Y, et al. Identification of functional cooperative mutations of SETD2 in human acute leukemia. Nat Genet. 2014;46:287–93.
Tartaglia M, Niemeyer CM, Fragale A, Song X, Beuchner J, Jung A, et al. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet. 2003;34:148–50.
Nabinger SC, Li XJ, Ramdas B, He Y, Zhang X, Zeng L, et al. The protein tyrosine phosphatase, Shp2, positively contributes to FLT3-ITD-induced hematopoietic progenitor hyper-proliferation and malignant disease in vivo. Leukemia. 2013;27:398–408.
Larrick JW, Fischer DG, Anderson SJ, Koren HA. Characterization of a human macrophage-like cell line stimulated in vitro: a model of macrophage functions. J Immunol. 1980;125:6–12.
Bei L, Huang W, Wang H, Shah C, Horvath E, Eklund E. HoxA10 activates CDX4 transcription and Cdx4 activates HOXA10 transcription in myeloid cells. J Biol Chem. 2011;286:19047–64.
Bei L, Shah C, Wang H, Huang W, Platanias LC, Eklund EA. Regulation of CDX4 gene transcription by HoxA9, HoxA10, the Mll-Ell oncogene and Shp2 during leukemogenesis. Oncogenesis. 2014;3:e135.
Oncomine 4.4, Research Edition Ann Arbor, MI: Compendia Biosciences Inc. Accessed 10 Mar 2017.
Lin AE, Ebert G, Ow Y, Preston SP, Toe JG, Cooney JP, et al. ARIH2 is essential for embryogenesis, and its hematopoietic deficiency causes lethal activation of the immune system. Nat Immunol. 2013;14:27–33.
Oberley MJ, Farnham PJ. Probing chromatin immunoprecipitates with CpG-island microarrays to identify genomic sites occupied by DNA-binding proteins. Methods Enzymol. 2003;371:577–96.
Acknowledgements
NIH grant HL87717, a VA Merit Review, the Mander Foundation, and the Director’s Research Fund of the Robert H. Lurie Comprehensive Cancer Center (to EAE).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, H., Bei, L., Shah, C.A. et al. The E3 ubiquitin ligase Triad1 influences development of Mll-Ell-induced acute myeloid leukemia. Oncogene 37, 2532–2544 (2018). https://doi.org/10.1038/s41388-018-0131-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-018-0131-5
This article is cited by
-
The role of E3 ubiquitin ligase WWP2 and the regulation of PARP1 by ubiquitinated degradation in acute lymphoblastic leukemia
Cell Death Discovery (2022)
-
ARIH2 regulates the proliferation, DNA damage and chemosensitivity of gastric cancer cells by reducing the stability of p21 via ubiquitination
Cell Death & Disease (2022)
