
Abstract
Alterations in ERBB family members have been associated with many tumor malignancies. EGFR and ERBB2 have been extensively explored in clinical oncology and several drugs currently target them therapeutically. However, the significance of ERBB4 as a potential therapeutic target remains mostly unexplored, even though ERBB4 is overexpressed or mutated in many solid tumors. Using a unique functional protein microarray platform, we found that ibrutinib inhibits ERBB4 activity in the same nM range as its canonical target, BTK. Cell-based assays revealed that ibrutinib treatment inhibited cell growth and decreased phosphorylation of ERBB4 and downstream targets MEK and ERK in cancer cell lines with high levels of endogenous ERBB4. In vivo, ibrutinib-responsive mouse xenograft tumors showed decreased tumor volumes with ibrutinib treatment. Interestingly, global gene expression comparisons between responsive and non-responsive cells identified a signature featuring the WNT pathway that predicts growth responsiveness to ibrutinib. Non-responsive ERBB4-expressing cell lines featured elevated activity of the WNT pathway, through the overexpression of WNT5A. Moreover, inhibition of WNT5A expression led to an ibrutinib response in non-responsive cell lines. Our data show that inhibiting ERBB4 reduces cell growth in cells that have low WNT5A expression and reveal a link between the ERBB4 and WNT pathways.
Similar content being viewed by others


Introduction
Kinase inhibitors are widely used critical drugs in the treatment of cancer. Initially designed to be selective, the discovery of unexpected targets can offer serendipitous benefits by broadening the potential indications for use. For example, imatinib, originally designed as a BCR–Abl inhibitor for the treatment of chronic myeloid leukemia, was later determined to inhibit Kit and platelet-derived growth factor receptor enabling its use for the treatment of gastrointestinal stromal tumors [1, 2]. However, given the heterogeneity of cancer, even the successful inhibition of the targets may not guarantee a response to the drug.
Ibrutinib, is a covalent inhibitor of Bruton’s tyrosine kinase (BTK), used in the treatment of several blood-related cancers including chronic lymphocytic leukemia (CLL), mantle cell lymphoma and Waldenström’s macroglobulinemia [3, 4]. Although ibrutinib was initially developed for the treatment of B-cell malignancies, emerging data from various mouse models of cancer implies that ibrutinib could be repurposed to treat other solid tumors [5, 6]. Several clinical trials are evaluating ibrutinib for efficacy in metastatic pancreatic adenocarcinoma (NCT02436668), cutaneous melanoma (NCT02581930), and non-small cell lung cancers with epidermal growth factor receptor (EGFR) mutation (NCT02321540). Ibrutinib is also reported to have inhibitory activity on several other kinases such as ITK, TEC, JAK3, HCK, BLK, EGFR, and ERBB2 [7,8,9]. This lack of selectivity could be exploited to treat tumors beyond BTK dependency. Strong efficacy and low toxicity of ibrutinib suggest that other tumor types might benefit from ibrutinib treatment if the appropriate targets could be identified.
Results
Screening of ibrutinib on NAPPA microarrays revealed ERBB4 as an attractive target
We screened kinase inhibitors in high throughput using our protein microarray platform nucleic acid programmable protein array (NAPPA) displaying 108 protein kinases and 30 non-kinases (Table S1). In NAPPA, complementary DNAs encoding the genes of interest are expressed in situ using human ribosomes and chaperone proteins, and the freshly expressed proteins are immobilized onto the array surface by a tag-specific antibody (Fig. 1a). To ensure array quality and batch reproducibility, sample arrays were tested for DNA levels and protein levels (Fig. 1b) revealing batch-to-batch reproducibility of r2 ≥ 0.91 (Fig. S1 and Table S2). Incubating these phosphatase-treated kinase arrays with or without added ATP during the kinase reaction followed by anti-pTyr antibody allowed the identification of a number of autophosphorylated tyrosine kinases (TKs), confirming that the proteins on the array were active and could be used for functional screenings (Fig. 1b). Proteins without TK activity (e.g., p27 and CDK2) showed no increase in phosphorylation after the reaction (Table S5).

Screening of kinase inhibitor on NAPPA. a Cartoon representation of NAPPA platform and the steps performed to screen kinase inhibitors. b From left to right, DNA levels (measured with pico green), protein display (anti-Flag), and phosphorylation levels after autophosphorylation reaction (anti-pTyr) performed in the presence or absence of ATP. Each protein is displayed in quadruplicate on the arrays. c Representative images of the phosphorylation signal on arrays after 1-h treatment with imatinib (100 nM–10 μM) or vehicle (DMSO). d Kinase activity measured as percentage of the control (DMSO) array
To test whether NAPPA was suitable for screening kinase inhibitors, we measured the kinase activity in arrays treated with staurosporine (0.1–10 μM). As expected, the overall kinase activity decreased, in a dose-dependent manner, for nearly all autophosphorylated TK present on the array (Fig S2, Table S3). Next, imatinib was used to test selective kinase inhibition. A significant reduction in Abl1 and BCR–Abl1 activity was observed in imatinib-treated arrays; whereas, the other kinases remained mostly unaffected (Figs. 1c, d, Table S4). Taken together, our data suggest that this protein microarray platform could be used for the unbiased screening of TK inhibitors.
Next, we focused on ibrutinib and screened NAPPA arrays to identify potential new targets. Among the TKs, the two with the strongest inhibition by ibrutinib were its cognate target, BTK and V-Erb-B2 avian erythroblastic leukemia viral oncogene homolog 4 (ERBB4), a member of the EGFR family (Figs. 2a, b, Fig. S3, Table S5). A radionucleotide kinase assay showed that 1 µM of ibrutinib inhibited the activity of both BTK and ERBB4 by >80%, whereas Abl1 (negative control) was 100% active at the same drug concentration (Fig. S4). Next, active site-dependent competition binding assays measured the binding affinities (Kd) between ibrutinib and BTK, ERBB4, ITK, and Abl1 as 0.64, 2, 43. and 1100 nM, respectively (Fig. 2c). As the Kd values for the inhibition of ERBB4 and BTK were observed in the same nM range, we regarded ERBB4 as a potential target of ibrutinib in vivo.

Ibrutinib inhibits ERBB4. a Arrays were screened with ibrutinib (100 nM–10 μM) or vehicle (DMSO) and representative images are shown. b Kinase activity of selected kinases is shown. c Active site-dependent competition binding assay between ibrutinib and Abl1, BTK, ITK, and ERBB4. For each curve, the experimental Kd was obtained
Ibrutinib impedes growth of some ERBB4-dependent cells
To assess whether the inhibition of ERBB4 by ibrutinib affected cell growth, we used the microarray-based gene expression profile of 1035 cancer cell lines from the Cancer Cell Line Encyclopedia (CCLE) database to select a panel of eleven cell lines expressing high levels of ERBB4 and undetectable levels of BTK, ERBB4High/BTKLow, and two cell lines with undetectable levels of both ERBB4 and BTK, ERBB4Low/BTKLow (Fig. S5). The predicted ERBB4 and BTK protein levels of each cell line was confirmed by western blot (Fig. S6). Cells were treated with 1 µM ibrutinib for 6 days and the number of viable cells was measured by Trypan blue assay (Fig. 3a). As expected, no response was observed for ERBB4Low/BTKLow cell lines. However, interestingly, ERBB4High/BTKLow cells presented a wide range of ibrutinib response from no effect (H146) to a marked reduction (H661 and H716) in viable cell counts after the drug treatment. A small increase in cell death was observed in some cell lines after the treatment with ibrutinib; however, the percentage of cell death was too small, <20%, to be the main effect of ibrutinib (Fig. S7). This result was reminiscent of the findings for ibrutinib-treated CLL and multiple myeloma patients’ cells driven by BTK, where in drug doses up to 1 µM, accentuated cell death was not prominent [10, 11].

Ibrutinib inhibits cell growth in ERBB4High/BTKLow cell lines. a Cell proliferation assays were performed in ERBB4High/BTKLow and ERBB4Low/BTKLow cell lines after ibrutinib treatment (6 days, 1 μM). The number of viable cells is reported as a percentage of the respective control (DMSO treated). b Dose-response curves after ibrutinib treatment (6 days, 1 nM–10 μM). c Protein levels of ERBB4 and downstream proteins (MEK, ERK, and AKT) in ibrutinib-treated H661 cells (0.1–1 μM). d Western blots for ERBB4 and GAPDH in cell lines infected with GFP or shRNA against ERBB4 or scramble. e Protein levels of EGFR and ERBB2 after shRNA infection. f Cell proliferation assay for cells infected with shERBB4, scramble or GFP. g Tumor growth of H661 cell-derived mouse xenografts (n = 10) after daily treatment of ibrutinib (5 mg/kg) and buffer only control for 60 days. The P-value calculated by the two-tailed T-test is shown, and the two-tailed Mann–Whitney U-test P-value was 0.01. (**P < 0.002; ***P < 0.001; ****P < 0.0001)
Ibrutinib-responsive cell lines (H522, H661, and KLE) and control cell lines (A549 and MDA-MB-231) were further tested with different concentrations of ibrutinib (Fig. 3b, Fig. S8). Control cells showed inhibition only at 10 µM, possibly due to nonspecific toxicity and IC50s of 6.7 μM (A549) and 8.4 μM (MDA-MD-231). In contrast, all responsive cell lines showed a significant dose-dependent inhibition of cell proliferation at concentrations as low as 100 nM, with IC50s of 58 nM (H661), 84 nM (KLE) and 580 nM (H522). The responses in these cells occurred within the same concentration range as that previously observed for a BTK-driven response [10, 11]. At the protein level, ibrutinib induced a decrease in the phosphorylation levels of ERBB4, without changes in total protein levels (Fig. 3c). Notably, reduced phosphorylated forms were also observed for mitogen-activated protein kinase kinase (MEK), extracellular signal-regulated kinase (ERK), and AKT serine/threonine kinase (AKT) three key downstream proteins in the ERBB4 pathway, suggesting a decrease in the activity of ERBB4 and ERBB4-induced pathways. Furthermore, to assess whether EGFR or ERBB2 inhibition contributes to the therapeutic effect of ibrutinib in sensitive cell lines, we used short hairpin RNA (shRNA) to knockdown EGFR or ERBB2. Even without EGFR or ERRB2, these cells showed an ibrutinib response similar to the parental cells and clearly inhibited the activity of ERBB4 and its downstream partners MEK, ERK, and AKT (Fig. S9, Fig. S10, Fig. S11).
To determine if ERBB4 inhibition could reduce cell growth, we infected ibrutinib-responsive cell lines with shRNAs targeting ERBB4. Three distinct hairpins were used and each promoted a strong decrease in ERBB4 protein levels (Fig. 3d). The shRNAs were designed to be specific to ERBB4 and do not cross react with other members of the EGFR family, as shown by western blot (Fig. 3e). Cell proliferation assays showed that cells infected with shERBB4 had a decreased growth ratio similar to what was observed in parental cells treated with ibrutinib (Fig. 3f). Together, these experiments suggest that ibrutinib inhibits ERBB4 and can reduce growth rates in some cells with high levels of ERBB4. Moreover, as the cell lines chosen for this study derive from distinct tissues, the inhibition observed was not tissue specific. To determine whether differential response observed in vitro would remain true in vivo, we assessed the ability of ibrutinib to reduce tumor growth of H661 mouse xenografts. When a daily dose of (5 mg/kg) was administered H661 xenografts showed a decrease in tumor volume as compared with untreated tumors (Fig. 3g). We observed the same trend when we administered H661 xenografts with a low dose of ibrutinib (Fig. S12). Moreover, non-responsive 1321N1 mouse xenografts did not show any reduction in tumor growth with ibrutinib treatment (Fig. S12).
Next, we focused on the differential responsiveness to ibrutinib among cells with elevated levels of ERBB4. No correlations between ibrutinib response and relative ERBB4 abundance, phosphorylation levels or levels of ERBB4 cleavable intracellular domain, a molecule with transcription activity associated with increased cell proliferation [12], were observed (Fig. S13). Recently, ibrutinib was associated with inhibition of EGFR and ERBB2 [13], so we measured the total and phosphorylated levels of endogenous EGFR, ERBB2, and ERBB3, but found no correlations with drug response (Fig. S14). These results raised the possibility that other interacting pathways might modulate ibrutinib responsiveness.
Elevated WNT pathway activity in ERBB4-positive cell lines is associated with ibrutinib resistance
As the levels of ERBB4 and other EGFR family members could not explain the differential responsiveness to ibrutinib, we examined the gene expression profiles of ibrutinib-responsive and non-responsive cells and searched for gene signatures that might associate with ibrutinib sensitivity. The mRNA expression levels of untreated cells under normal growth conditions were measured by a 2 × 100 bp paired-end RNA-Seq on Illumina HiSeq-2000 sequencer. After sequence alignment and expression estimation, we performed a two-group comparison using the non-parametric Mann–Whitney U-test and selected a pool of 401 differentially expressed genes with P-values < 0.05 (Table S6). These genes effectively clustered ibrutinib-responsive and non-responsive cells into two separate groups (Fig. S15), and thus were potentially associated with the responsiveness to ibrutinib in ERBB4High/BTKLow cancer cell lines.
To identify biological processes that may govern the ibrutinib responsiveness, pathway analysis was performed on the differentially expressed genes, based on information in public pathway databases. After removing redundant pathway terms, 23 signaling pathways were found significantly dysregulated (Table S7), which included interferon, G protein-coupled receptors, cytokine receptors, growth factor receptors (including ERBB4), and WNT signaling pathways. As these pathways were similarly altered in cell lines from different cancer types (lung, endometrial, colon, ovarian, and astrocytoma), their contribution to ibrutinib resistance appears to be independent of tissue origin. In parallel, individual differentially expressed genes were manually annotated for their known functions based on the literature. Notably, among the top 42 differentially expressed genes, 5 genes (Dickkopf WNT signaling pathway inhibitor 1 (DKK1), GPC3, SLC40A1, RGS3, and Wingless-type MMTV integration site family member 5A (WNT5A)) were associated with the WNT pathway (Table S8).
To verify the findings from our RNA-Seq data in the ibrutinib-responsive and non-responsive cell lines, we used the CCLE microarray data set as independent gene expression data to compare gene expression in the same eight cell lines. Among the 401 differentially expressed genes we previously identified, 46 also correctly classified the cells into ibrutinib-responsive and non-responsive groups using the CCLE data (Mann–Whitney test P < 0.05, Table S9). Like our own gene expression data, WNT-related genes were highly enriched (Fisher’s exact test P = 8.9E-04, Table 1, Table S9), including WNT5A, DKK1, TBL1XR1, and TLE2. This strongly suggested that the WNT pathway plays a key role in ibrutinib resistance in ERBB4High/BTKLow cancer cell lines.
The WNT pathway regulates diverse signal transduction events triggered by the extracellular WNT protein, playing important roles in tumor growth and metastasis [14, 15]. From our RNA-Seq data, examination of gene expression of WNT pathway network members revealed that several WNT ligands (WNT5A, WNT3, and WNT7A), WNT regulators (DKK1 and GPC3), signaling molecules (GNB1, GNG11, and CSNK2A2), protein sorting/processing (VPS26A, VPS35, PSMD10, AMER1, and CUL3), and transcriptional regulators (SMARCA4, TLE2, and histones) were differentially expressed between ibrutinib-responsive and non-responsive cells (Fig. 4a). Importantly, as shown in Table S8, WNT5A was upregulated, and DKK1 (an inhibitor of WNT pathway) was downregulated in ibrutinib non-responsive cells, indicating a synergistic elevation of the WNT pathway activity in ibrutinib non-responsive cells. Confirming the differential expression levels at the protein level, western blotting showed that abundances of WNT5A and DKK1 proteins correlated with their mRNA levels (Fig. 4b).

Differential expression of WNT pathway genes in ibrutinib-responsive and non-responsive cells. a Reactome map for all genes in the WNT pathway with relative expression levels in ibrutinib-responsive and non-responsive cells and differential expression (P-value: P-values from two-tailed Mann–Whitney U-test) are shown. Edges represent functional interactions between genes. b Protein levels of DKK1 and WNT5A in ibrutinib-responsive and non-responsive cells with relative intensities for DKK1/WNT5A ratio
DKK1/WNT5A signatures predict ibrutinib response
In addition to providing biological insight on ERBB4 function in cancer, the newly identified association between the WNT pathway and ibrutinib resistance prompted us to propose a molecular signature for prediction of ibrutinib responsiveness. With 46 genes that correctly classified ibrutinib-responsive and non-responsive cells in both RNA-Seq and CCLE data, we performed a logistic regression coupled with cross-validation-based feature selection and identified the 10 most informative genes, which included DKK1 and WNT5A. Therefore, along with the logistic regression classifiers, we tested a simple gene expression signature based on the relative ratio of these two genes (i.e., DKK1 level divided by WNT5A level), which was sufficient to classify ibrutinib-responsive and non-responsive cells. In our RNA-Seq data, all responsive cell lines showed DKK1/WNT5A ratios >1.0, whereas all non-responsive cell lines had DKK1/WNT5A ratios <1.0 (Table S10). Furthermore, the ratios were strongly correlated (Pearson’s R = −0.80) with the number of viable cells after a 24-h treatment of 1 µM ibrutinib. In addition, the DKK1/WNT5A ratios in the eight cell lines compared in the CCLE data were highly comparable to our RNA-Seq data (Pearson’s R = 0.98, Table S10), and classified the cells into ibrutinib-responsive and non-responsive groups with a strong negative correlation to the number of viable cells after ibrutinib treatment (Pearson’s R = −0.90, Table S10).
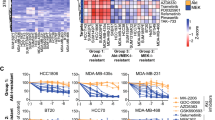
To further validate these results, we used our signature to select seven additional ERBB4High/BTKLow cell lines from the CCLE database, three cell lines with a low DKK1/WNT5A ratio and predicted to be non-responsive to ibrutinib (SK-N-MC, A673, U-118 MG) and four cell lines with high DKK1/WNT5A ratio and predicted to be responsive (Calu-3, HCC1419, HuTu80, 22Rv1), and tested them for drug response. Protein levels for the cell lines were confirmed by western blot (Fig. 5a). All three cell lines with low DKK1/WNT5A ratio showed no response to ibrutinib treatment up to 1 µM and inhibition in cell proliferation with an IC50 of 3 µM for U-118 MG, 4 µM for A673 and 7 µM for SK-N-MC (Fig. 5b and Fig. S16). Conversely, three cell lines (out of four) with high DKK1/WNT5A ratio were responsive to ibrutinib with IC50s of 10 nM for HCC1419, 50 nM for Calu-3 and 800 nM for HuTu80. The only outlier was the 22Rv1 prostate carcinoma cell that had a higher DKK1/WNT5A ratio and a responsive signature based on the logistic regression classifier, which showed resistance to ibrutinib (IC50, 26 µM). Studies have shown that 22Rv1 cells are resistant to a broad spectrum of chemotherapeutics mainly because of the overexpression of ABCG2, a protein that contributes to multi-drug resistance [16], which might explain the ibrutinib resistance observed in our experiments. Taken together, 6 out of 7 newly tested cell lines (and 14 of 15 cell lines overall) validated the responsiveness of ibrutinib based on the DKK1/WNT5A signature in ERBB4High/BTKLow cells indicating the applicability of this signature to predict response for ibrutinib inhibition of cell growth.

DKK1/WNT-based prediction on ibrutinib responsiveness. a Characterization of cell lines regarding the expression levels of BTK, ERBB4, DKK1 and WNT5A. b Viable cell counts of ERBB4High/BTKLow cells with high DKK/WNT5A ratio (Calu-3, HCC1419, HuTu80, and 22RV1) or low DKK/WNT5A ratio (SK-N-MC, A673, U-118 MG) after treatment with ibrutinib for 6 days (****P < 0.0001)
To further explore the relationship between WNT5A and ibrutinib resistance, we assessed whether reducing WNT5A levels would make resistant cell lines more responsive to ibrutinib. Stable cell lines were generated for 132N1 and A673 cells after knockdown with four different independent shRNAs against WNT5A. As shown in Figs. 6a, b, two out of the four shRNAs effectively reduced WNT5A levels. These cells were subsequently treated with different doses of ibrutinib for a period of 6 days. Compared with the control, WNT5A-reduced cells acquired sensitivity to ibrutinib around 500 nM. In addition, we also performed the reciprocal experiments on the ibrutinib-responsive cells by supplementing H522 and H661 cells with conditioned media from WNT5A-secreting cells and subsequently treating with ibrutinib (Fig. S17). The addition of WNT5A-conditioned media increased the resistance towards ibrutinib. These data further strengthen the role of WNT pathway in determining the sensitivity to ibrutinib response in ERBB4-expressing cells.

Ibrutinib responsiveness in WNT5A knockdown cells. a Effect of WNT5A knockdown in 132N1 cells in WNT5A protein levels and cell proliferation after ibrutinib treatment (6 days, 0.01–10 µM). b Effect of WNT5A knockdown in A673 cells and cell proliferation after ibrutinib treatment (6 days, 0.01–10 µM). c Patient profile of predicted ibrutinib responsiveness across 23 cancer types. Patient samples in TCGA with high levels of ERBB4 were further classified as ‘ibrutinib-sensitive-like’ (DKK1/WNT5A ≥ 2.0, blue), ‘ibrutinib-resistant-like’ (DKK1/WNT5A ≤ 0.5, red), or ‘neutral’ (0.5 < DKK1/WNT5A < 2.0, green). The graph shows the predicted classification profiles by cancer types. (*P < 0.05; ** P < 0.002; ***P < 0.001)
We were curious if any human tumors occur with high levels of ERBB4 and an ibrutinib-responsive signature. We used The Cancer Genome Atlas (TCGA) RNA-Seq data on 25 different cancer types. With a cutoff of 1.0 FPKM (fragments per kilobase million) among the total of 8281 tumor samples, 3292 samples expressed ERBB4. The DKK1/WNT5A ratio was then applied to classify the ERBB4-positive samples into ibrutinib-responsive-like (DKK1/WNT5A ≥ 2.0), ibrutinib-non-responsive-like (DKK1/WNT5A ≤ 0.5), and neutral (0.5 < DKK1/WNT5A < 2.0) groups (Fig. 6c). Interestingly, although ovarian cancer samples were mostly (90%) ERBB4 positive, as recently reported [17], the majority had ibrutinib-non-responsive signatures, suggesting an elevated level of WNT signaling as previously reported [18]. Among more prevalent cancer types, 35–45% of samples from low-grade glioma, glioblastoma multiforme, and breast carcinoma were classified to have either responsive-like or neutral signatures and thus may be consistent with ibrutinib responsiveness.
Discussion
Our previous publications showed that NAPPA is a flexible platform for the generation of protein microarrays that can be successfully used for the identification of protein biomarkers for several diseases, including cancer and diabetes [19, 20]. Here we show for the first time that NAPPA arrays can be used for the study of protein kinases and for the screening of kinase inhibitors. The main advantage of NAPPA is the flexibility that comes from the expression of the proteins from cDNAs printed on the array. Any cDNA can be printed on the arrays, and therefore any protein can be studied, including mutant versions of relevant proteins.
The ibrutinib target ERBB4 is a transmembrane TK, and a member of the EGFR family, associated with the regulation of key pathways such as phosphoinositide 3-kinase/AKT and focal adhesion kinase [21, 22]. Recent reports have shown EGFR family members as new targets for ibrutinib. For ibrutinib inhibition of TKs, a cysteine located in the kinase hinge segment is crucial [23]. Among numerous TKs encoded in the human genome, 14 TKs share this conserved cysteine residue including EGFR, ERBB2, and ERBB4 [23]. Some studies have focused on utilizing ibrutinib to target EGFR and ERBB2, the two most studied members in the EGFR family [13]. On the contrary, ERBB4 largely remains unexplored in clinical oncology as a potential target. Multiple studies have reported activating mutations in ERBB4 in melanoma and lung cancer [24]. Altered expression of ERBB4 has been observed in breast cancer, neuroblastoma, non-small cell lung cancer, and colon cancer [13, 25, 26]. Overexpression of ERBB4 is associated with increased metastatic potential in Ewing’s sarcoma [27], poor prognosis in triple-negative breast cancer [28], and drug resistance in ovarian cancers [29]. Even though many of the drugs targeting the EGFR family can potentially inhibit ERBB4, no systematic studies were performed to address this. Here, we present ibrutinib as a potent ERBB4 inhibitor whose activity inhibited the growth of a number of ERBB4-dependent cell lines in vitro and in vivo.
We could not explain the differential response to ibrutinib observed in our cell panel by the level of total or phosphorylated forms of ERBB4, EGFR, ERBB2, or ERBB3, suggesting alternative pathways or mechanisms are responsible for the observed differences in drug response. However, in our RNA-seq data, as well as CCLE microarray data set, an independent data source, we observed strong enrichment and differential expression of WNT pathway genes in responsive and non-responsive cells. Based on the newly discovered association with the WNT pathway, we proposed and validated a gene expression signature based on the relative mRNA expression levels of two genes, DKK1 and WNT5A, to predict the response for ibrutinib in ERBB4-expressing tumors. Growth suppression occurred specifically in cells with high DKK1/WNT5A ratio, both in cells and in vivo, whereas cells expressing more WNT5A than DKK1 were resistant to ibrutinib treatment. Based on the ongoing studies in our lab, we suspect bypass signaling pathways may contribute toward ibrutinib resistance. For instance, the ibrutinib non-responsive 1321N1 cell line, which does not harbor any ERBB4 mutations, shows reduction in phospho ERBB4 levels (data not shown) when treated with ibrutinib, despite no reduced cell/tumor growth in both our cell-based and in vivo models. Our studies show decreasing the WNT5A expression makes the non-responsive cells more sensitive to ibrutinib treatment, but the mechanism of resistance is yet to be elucidated.
Although the WNT pathway may modulate the effect of ERBB4 inhibition on cell growth, connections between these two pathways have not been studied. Other ERBB members are known to inactivate glycogen synthase kinase 3, a key WNT pathway component, through direct phosphorylation by AKT [30]. If this held for ERBB4, it would place WNT downstream of, and thus epistatic on, ERBB4. Alternatively, ERBB4 may signal via ERK/LRP6 [31]. ERBB receptors may employ ERK to activate WNT signaling by phosphorylating LRP6, a WNT co-receptor, which binds to numerous WNT ligands. Indirectly, ERBB4 and WNT pathways may crosstalk through the transcriptional regulation of key proteins present in each pathway. Many other mechanisms may exist for the interaction of ERBB4 with both canonical and non-canonical WNT signaling, and future studies are needed to elucidate the exact mechanism.
In conclusion, we show that ERBB4 is an attractive target for ibrutinib using a functional protein microarray platform NAPPA. We demonstrated that ERBB4-expressing cells respond to ibrutinib in a WNT pathway dependent manner. We further show that inhibiting certain WNT activating ligands assist in increasing the response for ibrutinib. Our results raise the possibility that ibrutinib can be used as a drug beyond B-cell malignancies to treat ERBB4-expressing cancers, either by itself or as a combination with WNT inhibitors.
Materials and methods
Nucleic acid programmable protein array
NAPPA kinase microarrays were generated with virtually all protein–TKs from the human genome. The coding sequence for each TK was cloned into an expression plasmid and immobilized on the microarray surface, with one protein kinase per feature. Briefly, NAPPA printing mix was prepared with plasmid DNA coding for genes of interest, anti-flag antibody, cross-linker, and poly-lysine. Samples were printed in quadruplicate in aminopropyltriethoxysilane-coated glass slides.
Before kinase expression NAPPA arrays were blocked for 1 h at room temperature (RT) using Superblock solution (Pierce), and then dried with filtered air. The slides were covered with HybriWells and protein expression was performed by injection of 160 µL human cell-free expression into the chamber. Slides were incubated for 90 min at 30 °C and 30 min at 15 °C, followed by 1 h of blocking with TBSTB (1X Tris-buffered saline supplemented with 0.2% Tween 20 and 3% bovine serum albumin). The levels of protein expression were assayed with anti-Flag antibody followed by secondary antibodies labeled with cy3. Before the kinase assay, the slides were treated with lambda phosphatase and DNASe for 1 h to remove any phosphorylation that occurred during protein expression. Next, arrays were washed with TBST to remove the phosphatase and the kinase reaction was initiated with kinase buffer (25 mM Tris-HCl (pH 7.5), 5 mM beta-glycerophosphate, 2 mM dithiothreitol (DTT), 0.1 mM Na3VO4, 10 mM MgCl2) with ATP (500 µM) in the presence or absence of kinase inhibitors.The kinase activity on the array was measured with antiphosphotyrosine antibody followed by secondary antibodies labeled with cy3. All antibody incubations were performed in a 1:1000 dilution in TBSTB at RT. NAPPA arrays were scanned with a PowerScanner and the signal intensity was quantified using the Array-ProAnalyzer 6.3, using the default settings. The median intensity across the quadruplicates was measured and the background was corrected through the subtraction of the median value of the negative controls.
NAPPA data analysis
The data were normalized using positive control features present in each array. The signal obtained in the microarray treated with kinase inhibitor was interpolated in the curve obtained from arrays treated with buffer only (lower phosphorylation level—0%) or treated with ATP and dimethylsulfoxide (DMSO; maximum phosphorylation level—100%).
Radioactive kinase assay
Purified protein kinases were incubated with radioactive ATP and increasing concentrations of kinase inhibitors. The kinase activity was measured through the levels of radioactive phosphorylation present in each kinase.
Active site-dependent competition binding assay
Soluble protein kinases tagged with a DNA tag were incubated with an affinity column in the presence of increasing concentration of kinase inhibitor. The amount of kinase bound to the column is proportional to the kinase activity and it was measured by quantitative PCR.
Mammalian cell culture
The cell lines NCI-H661, NCI-H522, NCI-146, KLE, Caov4, NCI-H716, HuTu80, HCC 1419, Calu-3, 22Rv1, U-118MG, A-673, SK-N-MC, MDA-MB-231, A549, K-562, L Wnt-5A, and L cells were obtained from the American Type Culture Collection (ATCC). The cells, Cov434 (cat. no. 07071909), COV318 (cat. no. 07071903), and 1321N1 (cat. no. 86030402) were obtained from Sigma, HEC-59 (cat. no. C0026001) from AddexBio and OVKATE and OVSAHO from the Japanese Collection of Research Bioresources (JCRB) Cell Bank. All cells were tested for mycoplasma contamination before use with the Universal Mycoplasma Detection Kit (ATCC 30-1012K).
Whole-cell extract preparation and western blotting
Cells were lysed in the cell lysis buffer (50 mmol/L of Tris-HCl, pH 7.4, 150 mmol/L NaCl, 1% Nonidet P-40, 0.1% sodium azide, 1 mmol/L NaF, 2 mmol/L Na3VO4, 1 tablet of protease inhibitor (Roche)). The total protein content was measured by the BCA protein assay kit (Pierce) and the proteins were separated on sodium dodecyl sulfate–polyacrylamide gel electrophoresis gels and transferred to polyvinylidene difluoride membranes using the Trans-Blot SD Semi-Dry Transfer Cell (Bio-Rad). The primary antibodies were used according to the product instruction. Western blots were developed by supersignal west femto maximum sensitivity substrate (Thermo Scientific) and exposed and imaged by the FluorChemFC2 imager (Alpha Innotech). The antibodies used here are as follows: Btk (D3H5) (cat. no. 5082), HER4/ERBB4 (cat. no. 4795), Phospho-HER4/ERBB4 (Tyr984 (cat. no. 3790), Phospho-HER4/ERBB4 (Tyr1284) (cat. no. 4757), GAPDH XP (cat. no. 5174), EGF receptor XP (cat. no. 4267), Phospho-EGF receptor (Tyr1045) (cat. no. 2237), HER2/ERBB2 (cat. no. 2165), Phospho-HER2/ERBB2 (Tyr1221/1222) (cat. no. 2243), HER3/ERBB3 XP (cat. no. 12708), Phospho-HER3/ERBB3 (Tyr1289), AKT (cat. no. 4691), Phospho-AKT (S473) (cat. no. 4060), Erk1/2 (cat. no. 4695), Phospho-Erk1/2 (cat. no.4370), MEK1/2 (cat. no. 9122), Phospho-MEK1/2 (cat. no. 9154) were purchased from Cell Signaling Technology. Anti-WNT5A (cat. no. ab72583), was obtained from Abcam and the Dkk-1 antibody (cat. no. sc-25516) was purchased from Santa Cruz Biotechnology.
Cell viability assay
Cells were seeded on six-well plates 24 h prior to kinase inhibitor treatment. The next day, each well was treated with different concentrations of ibrutinib (1 nM, 10 nM, 100 nM, 1 µM and 10 µM) along with DMSO as a control. The cell media were replaced every other day with fresh ibrutinib containing media and the cultures were continued for 6 days. The live cells were counted by Cell Titer Glow (Promega) or Trypan blue assay. All experiments were performed in triplicate and repeated at least three times. All data were analyzed using GraphPad 7 (GraphPad Software, La Jolla, CA, USA), expressed as mean ± S.E.M. P-value of <0.05 were considered significant in all studies.
Crystal violet stain
Cells were washed with phosphate-buffered saline and fixed for 1 h at RT in fixing solution (10% acetic acid and 10% methanol in water). The fixing solution was removed and 0.5% crystal violet was added to the cells and incubated over-night at RT. Excess stain was removed by washing with water and the plates were dried and imaged.
shRNA-mediated knockdown
shRNA control and shRNAs against ERBB4, EGFR, ERBB2, and WNT5A were obtained from DNASU (https://dnasu.org). 293LX cells were transfected with each shRNA and lentiviral particles were collected. For ERBB4 knockdown, NCI-H522, KLE, and NCI-H661 were plated in six-well plates and allowed to attach over-night. The cells were infected with respective ERBB4 shRNA-encoding lentivirus along with control shRNAs. Cells infected with each shRNA were selected with 0.5 µg/mL of puromycin for 6 days. The number of viable cells was counted by Trypan blue exclusion assay with the countess automated cell counter (Invitrogen). For EGFR and ERBB2 knockdown, NCI-H661 and KLE cells were infected with corresponding shRNAs and selected with 0.6 µg/mL puromycin to create stable cell lines. For WNT5A knockdown, 132N1 and A673 cells were infected with WNT5A shRNAs and selected with 0.6 µg/mL puromycin to create stable cell lines.
WNT5A assay
WNT5A-secreting media and control condition media were obtained from culturing L Wnt-5A (ATCC), and L cells (ATCC) according to ATCC recommendation. The presence of WNT5A in secreted media were confirmed by western blot. H522 and H661 cells supplemented with WNT5A or L condition media were seeded on six-well plates 24 h prior to ibrutinib treatment. The next day, each well was treated with different concentrations of ibrutinib (100 nM, 1 µM) along with DMSO and untreated cells as controls. The cell media were replaced every other day with fresh ibrutinib containing media conditioned with WNT5A or L and the cultures were continued for 6 days. The live cells were counted by Trypan blue assay. All experiments were performed in triplicate and repeated at least on 2 independent days.
Tumor xenograft studies
Seven weeks old NOD-SCID mice were injected with 1 × 106 H661 cells or 1321N1 cells subcutaneously in the flank with a volume of 100 µl of 1:1 culture media and Matrigel. Each group had three females and seven males with n = 10 per group, for which the sample size was determined by a power analysis with an estimated effect size of 1.0. After about 2 weeks after injection, animals were treated daily with ibrutinib (5 mg/kg, 2% DMSO in castor oil) or buffer only by oral delivery into mouth. Tumor xenografts were measured with a digital caliper, and the tumor volumes were determined by (length × width2)/2. Tumor volumes are plotted as mean ± SEM, and P-value of <0.05 based on both T-test and Mann–Whitney U-test were considered significant. At the end of the experiment, mice were euthanized with CO2. Mice were maintained in accordance with Arizona State University Institutional Animal Care Guidelines (IACUC Protocol #: 14-1370R). The mice were randomized for treatment, with investigator blinded for tumor measurements.
RNA-seq
RNA from ibrutinib-responsive and ibrutinib non-responsive cell lines was obtained with RNA easy (Qiagen) and the samples were sequenced using Illumina HiSeq2000. The sequence reads were aligned to the Ensembl human genome by the TopHat/bowtie2 software, and the aligned read counts were quantified and normalized against gene sizes and the number of reads by the cufflinks software. Genes (excluding anti-sense RNA and pseudo-genes) with FPKM higher than 1 in any of the eight samples tested (four ibrutinib-responsive and four non-responsive cell lines) were used for further analysis. Using as cutoff a fold change of 1.5 and P-value < 0.05 in two-tailed Mann–Whitney tests, a set of 401 distinct genes was obtained.
Data availability
RNA-Seq data that support the findings of this study have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo with the accession code GSE102744.
References
Blanke CD, Demetri GD, von Mehren M, Heinrich MC, Eisenberg B, Fletcher JA, et al. Long-term results from a randomized phase II trial of standard-versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol. 2008;26:620–5.
Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347:472–80.
Smith MR. Ibrutinib in B lymphoid malignancies. Exp Opin Pharmacother. 2015;16:1879–87.
Woyach JA, Johnson AJ, Byrd JC. The B-cell receptor signaling pathway as a therapeutic target in CLL. Blood. 2012;120:1175–84.
Massó-Vallés D, Jauset T, Serrano E, Sodir NM, Pedersen K, Affara NI, et al. Ibrutinib exerts potent antifibrotic and antitumor activities in mouse models of pancreatic adenocarcinoma. Cancer Res. 2015;75:1675–81.
Sagiv-Barfi I, Kohrt HEK, Czerwinski DK, Ng PP, Chang BY, Levy R. Therapeutic antitumor immunity by checkpoint blockade is enhanced by ibrutinib, an inhibitor of both BTK and ITK. Proc Natl Acad Sci USA. 2015;112:E966–E972.
Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci USA. 2010;107:13075–80.
Berglof A, Hamasy A, Meinke S, Palma M, Krstic A, Mansson R, et al. Targets for ibrutinib beyond B cell malignancies. Scand J Immunol. 2015;82:208–17.
Wang X, Wong J, Sevinsky CJ, Kokabee L, Khan F, Sun Y, et al. Bruton’s tyrosine kinase inhibitors prevent therapeutic escape in breast cancer cells. Mol Cancer Ther. 2016;15:2198–208.
Herman SEM, Gordon AL, Hertlein E, Ramanunni A, Zhang X, Jaglowski S, et al. Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765. Blood. 2011;117:6287–96.
Rushworth SA, Bowles KM, Barrera LN, Murray MY, Zaitseva L, MacEwan DJ. BTK inhibitor ibrutinib is cytotoxic to myeloma and potently enhances bortezomib and lenalidomide activities through NF-κB. Cell Signal. 2013;25:106–12.
Junttila TT, Sundvall M, Lundin M, Lundin J, Tanner M, Härkönen P, et al. Cleavable ErbB4 isoform in estrogen receptor–regulated growth of breast cancer cells. Cancer Res. 2005;65:1384–93.
Grabinski N, Ewald F. Ibrutinib (ImbruvicaTM) potently inhibits ErbB receptor phosphorylation and cell viability of ErbB2-positive breast cancer cells. Invest New Drugs. 2014;32:1096–104.
Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer. 2013;13:11–26.
Niehrs C. The complex world of WNT receptor signalling. Nat Rev Mol Cell Biol. 2012;13:767–79.
Liu T, Xu F, Du X, Lai D, Liu T, Zhao Y, et al. Establishment and characterization of multi-drug resistant, prostate carcinoma-initiating stem-like cells from human prostate cancer cell lines 22RV1. Mol Cell Biochem. 2010;340:265–73.
Davies S, Holmes A, Lomo L, Steinkamp MP, Kang H, Muller CY, et al. High incidence of ErbB3, ErbB4, and MET expression in ovarian cancer. Int J Gynecol Pathol. 2014;33:402–10.
Arend RC, Londoño-Joshi AI, Straughn JM Jr, Buchsbaum DJ. The Wnt/β-catenin pathway in ovarian cancer: a review. Gynecol Oncol. 2013;131:772–9.
Anderson KS, Sibani S, Wallstrom G, Qiu J, Mendoza EA, Raphael J, et al. Protein microarray signature of autoantibody biomarkers for the early detection of breast cancer. J Prote Res. 2011;10:85–96.
Bian X, Wasserfall C, Wallstrom G, Wang J, Wang H, Barker K, et al. Tracking the antibody immunome in type 1 diabetes using protein arrays. J Prote Res. 2016;16:195–203.
Arteaga Carlos L, Engelman Jeffrey A. ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell. 2014;25:282–303.
Carpenter G. ErbB-4: mechanism of action and biology. Exp Cell Res. 2003;284:66–77.
Wu H, Wang W, Liu F, Weisberg EL, Tian B, Chen Y, et al. Discovery of a potent, covalent BTK inhibitor for B-cell lymphoma. ACS Chem Biol. 2014;9:1086–91.
Prickett TD, Agrawal NS, Wei X, Yates KE, Lin JC, Wunderlich JR, et al. Analysis of the tyrosine kinome in melanoma reveals recurrent mutations in ERBB4. Nat Genet. 2009;41:1127–32.
Williams CS, Bernard JK, Demory Beckler M, Almohazey D, Washington MK, Smith JJ, et al. ERBB4 is over-expressed in human colon cancer and enhances cellular transformation. Carcinogenesis. 2015;36:710–8.
Kurppa KJ, Denessiouk K, Johnson MS, Elenius K. Activating ERBB4 mutations in non-small cell lung cancer. Oncogene. 2016;35:1283–91.
Mendoza-Naranjo A, El-Naggar A, Wai DH, Mistry P, Lazic N, Ayala FRR, et al. ERBB4 confers metastatic capacity in Ewing sarcoma. EMBO. Mol Med. 2013;5:1019–34.
Kim J-Y, Jung HH, Do I-G, Bae S, Lee SK, Kim SW, et al. Prognostic value of ERBB4 expression in patients with triple negative breast cancer. BMC Cancer. 2016;16:138.
Gilmour LMR, Macleod KG, McCaig A, Gullick WJ, Smyth JF, Langdon SP. Expression of erbB-4/HER-4 growth factor receptor isoforms in ovarian cancer. Cancer Res. 2001;61:2169–76.
Nakata A, Yoshida R, Yamaguchi R, Yamauchi M, Tamada Y, Fujita A, et al. Elevated β-catenin pathway as a novel target for patients with resistance to EGF receptor targeting drugs. Sci Rep. 2015;5:13076.
Krejci P, Aklian A, Kaucka M, Sevcikova E, Prochazkova J, Masek JK, et al. Receptor tyrosine kinases activate canonical WNT/-catenin signaling via MAP kinase/LRP6 pathway and direct-catenin phosphorylation. PLoS ONE. 2012;7:e35826.
Acknowledgements
We thank everyone in the LaBaer lab for their suggestions and contributions to make this work possible, in particular, Ian Shoemaker, Justin Saul, Kathy Stinchfield, Kristina Buss, Jason Steel, Jacquelyn Kilbourne, Hannah Johnson, and Mike Gaskin. We also want to thank the members of the Biodesign Center for Personalized Medicine for their critical feedback.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Femina Rauf and Fernanda Festa contributed equally to this work.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. If you remix, transform, or build upon this article or a part thereof, you must distribute your contributions under the same license as the original. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/.
About this article

Cite this article
Rauf, F., Festa, F., Park, J.G. et al. Ibrutinib inhibition of ERBB4 reduces cell growth in a WNT5A-dependent manner. Oncogene 37, 2237–2250 (2018). https://doi.org/10.1038/s41388-017-0079-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-017-0079-x
This article is cited by
-
Anticancer effect of zanubrutinib in HER2-positive breast cancer cell lines
Investigational New Drugs (2023)
-
Brown adipose tissue-derived Nrg4 alleviates endothelial inflammation and atherosclerosis in male mice
Nature Metabolism (2022)
-
Analysis of H3K4me3 and H3K27me3 bivalent promotors in HER2+ breast cancer cell lines reveals variations depending on estrogen receptor status and significantly correlates with gene expression
BMC Medical Genomics (2020)
-
Comprehensive genetic analysis of pediatric germ cell tumors identifies potential drug targets
Communications Biology (2020)
-
The role of ErbB4 in cancer
Cellular Oncology (2020)
