
Abstract
Aggression and callous, uncaring, and unemotional (CU) traits are clinically related behavioral constructs caused by genetic and environmental factors. We performed polygenic risk score (PRS) analyses to investigate shared genetic etiology between aggression and these three CU-traits. Furthermore, we studied interactions of PRS with smoking during pregnancy and childhood life events in relation to CU-traits. Summary statistics for the base phenotype were derived from the EAGLE-consortium genome-wide association study of children’s aggressive behavior and were used to calculate individual-level genome-wide and gene-set PRS in the NeuroIMAGE target-sample. Target phenotypes were ‘callousness’, ‘uncaring’, and ‘unemotional’ sumscores of the Inventory of Callous-Unemotional traits. A total of 779 subjects and 1,192,414 single-nucleotide polymorphisms were available for PRS-analyses. Gene-sets comprised serotonergic, dopaminergic, glutamatergic, and neuroendocrine signaling pathways. Genome-wide PRS showed evidence of association with uncaring scores (explaining up to 1.59% of variance; self-contained Q = 0.0306, competitive-P = 0.0015). Dopaminergic, glutamatergic, and neuroendocrine PRS showed evidence of association with unemotional scores (explaining up to 1.33, 2.00, and 1.20% of variance respectively; self-contained Q-values 0.037, 0.0115, and 0.0473 respectively, competitive-P-values 0.0029, 0.0002, and 0.0045 respectively). Smoking during pregnancy related to callousness scores while childhood life events related to both callousness and unemotionality. Moreover, dopaminergic PRS appeared to interact with childhood life events in relation to unemotional scores. Our study provides evidence suggesting shared genetic etiology between aggressive behavior and uncaring, and unemotional CU-traits in children. Gene-set PRS confirmed involvement of shared glutamatergic, dopaminergic, and neuroendocrine genetic variation in aggression and CU-traits. Replication of current findings is needed.
Similar content being viewed by others

Introduction
Aggressive behaviors in children are common, multifactorial, and continuous traits [1,2,3]. A clinically important subgroup of aggressive children displays high levels of callous-unemotional (CU) traits. CU-traits describe a lack of guilt, limited empathy, and shallow affect [1, 4]. Although high levels of CU-traits are considered a subphenotype within youth diagnosed with conduct disorder (CD), CU-traits also occur in frequently comorbid disorders such as oppositional-defiant disorder (ODD) and attention-deficit/hyperactivity disorder (ADHD) [1], and adversely affect quality of life in these children [1, 3, 5]. In addition to the aforementioned clinical diagnoses, a distinction can be made between reactive and proactive aggression. Reactive aggression occurs in reaction to frustration or perceived threat, whereas proactive aggression is considered an instrumental behavior driven by reward anticipation and this subtype of aggression is conceptually also more closely related to CU-traits [6].
From twin studies and a recent genome-wide association study (GWAS), it is known that up to approximately half of the variance in aggression can be explained by genetic factors [7, 8]. GWASs of aggression-related phenotypes have implicated some susceptibility loci, yet genome-wide significant findings are still few [8,9,10]. To detect more variants with smaller effects, larger samples are needed [11]. The effects of multiple variants can, however, be aggregated into a polygenic risk score (PRS). Based on GWAS summary statistics, PRS can be calculated in an independent target sample [12]. PRS can also be restricted to gene-sets to specifically investigate pathways of interest [13]. A monoaminergic and neuroendocrine signaling gene-set was recently linked to reactive aggression in females (and nominally significant to proactive aggression in males) [14] and a glutamatergic gene-set was implicated in hyperactivity/impulsivity [15].
Some well-studied environmental risk factors that have been linked to aggression-related phenotypes (i.e. CD, CU-traits, antisocial behavior, ODD, and ADHD) include maternal smoking during pregnancy and adverse childhood experiences [1, 16,17,18,19]. Other factors such as poor parental monitoring or poverty have also been implicated in CD and CU-traits [1]. In addition to independent contributions from genes and environment, gene-environment (GxE) interactions are thought to play an important role [2, 7, 20] by providing insight into inter-individual differences in susceptibility to environmental and/or genetic factors [2, 17, 20, 21]. Part of the early GxE-interaction studies may, however, have suffered from the use of weak candidate genes and underpowered samples [22, 23]. Similar to genetic main effects, GxE-interactions in aggression most likely represent polygenic phenomena and therefore also benefit from polygenic GxE-interaction modeling.
Currently, we calculated PRS based on the results of the largest available GWAS of children’s aggressive behavior (i.e. our ‘base’ phenotype). This GWAS was conducted by the EAGLE-consortium [8] and consisted of a meta-analysis of nine cohorts in which aggression was measured by questionnaires such as the Child Behavior Checklist and Strengths and Difficulties Questionnaire. Using the aforementioned aggression-PRS we investigated evidence for shared genetic etiology with (i.e. whether the PRS predicts) three CU ‘target’ traits (using the NeuroIMAGE-sample; [24] enriched for ADHD). When such genetic sharing exists, it indicates that children displaying CU-traits share etiology with other aggressive phenotypes. In addition to PRS as a general measure of genetic liability, we also investigated evidence for shared genetic etiology restricted to previously implicated pathways (gene-sets) mentioned above. Moreover, we were interested in potential interactions of PRS with smoking during pregnancy and/or childhood traumatic experiences in relation to CU-traits. Because CU-traits can be assessed as a three-factor construct, with three meaningful dimensions, namely ‘callousness’ (describing a callous attitude towards others), ‘uncaring’ (describing a lack of caring about performance), and ‘unemotional’ traits (describing a lack of emotional expression), we investigated these three dimensions separately [25]. All of these three CU-dimensions have been related to (sub)scales of antisocial behavior [25]. Only few genetic association studies to date have specifically investigated CU-traits; in the available studies CU-traits were assessed as a single dimension and only suggestive hits were identified [26, 27]. We used NeuroIMAGE as our target sample, because of the availability of extensive phenotypic and environmental data, in addition to individual-level genome-wide genotyping data. Moreover, the relatively high number of ADHD-cases allowed for more robust control of ADHD as a comorbid condition, which genetic association studies of aggression often lack.
Methods
NeuroIMAGE
NeuroIMAGE is the follow-up of the Dutch part of the International Multicenter ADHD Genetics (IMAGE) case–control study, including 331 families with at least one child with ADHD and at least one biological sibling and 153 control families. This resulted in a total of 412 children with ADHD and 227 unaffected siblings, 262 healthy controls, and 81 children with ‘subhthreshold’ levels of ADHD-symptoms. The diagnosis of ADHD was ascertained according to DSM-IV-TR criteria using information obtained through a semi-structured diagnostic interview and rating scales. Inclusion criteria were a European Caucasian descent, IQ ≥ 70, age < 18 years, and no diagnosis of autism, epilepsy, learning disorders, neurological diseases, or genetic syndromes. More information can be found elsewhere [24].
Target phenotypes: ‘Callousness’, ‘Uncaring’, and ‘Unemotional’ dimensions of the Inventory of Callous-Unemotional traits
Main outcomes, i.e. ‘target phenotypes’, were the three self-reported dimensions measuring ‘callousness’ (describing a callous attitude towards others), ‘uncaring’ (describing a lack of caring about performance), and ‘unemotional’ behavior (describing a lack of emotional expression) that constitute the CU-traits construct as assessed by the Inventory of Callous-Unemotional traits (ICU; [25] data collected for NeuroIMAGE between 2009 and 2012). The ICU has shown sufficient reliability (estimates for our current sample are provided in the Supplement) and construct validity regarding CU-traits and consists of a total of 24 items rated on a four-point scale (0–3; all individual items are described in the Supplement). Sum scores for the callousness (33 maximum), uncaring (24 maximum), and unemotional (15 maximum) dimensions were analyzed separately given the three-factor structure of the ICU [25].
Genotyping
Genotyping was performed at the Radboud University, using the Illumina Psych-Array 24 v1.1A. This genotyping chip assesses ~560,000 markers, and has been developed in collaboration with the Psychiatric Genomics Consortium for the (genome-wide) analyses of psychiatric phenotypes [28]. Imputation was performed using the RICOPILI-pipeline [29]. Only single-nucleotide polymorphisms (SNPs) passing quality control filters regarding Impute Information scores (0.8), minor allele frequency (0.01), Hardy–Weinberg equilibrium test (P cut-off 1E-06), and SNP-call rate (0.95) were retained. Individual genome-wide genotype data was available for 4,573,985 SNPs for 779 subjects in NeuroIMAGE.
Environmental factors
Environmental factors that were investigated and included in gene-environment (GxE)-interaction analyses were maternal smoking during pregnancy (dichotomized for any amount of smoking in any trimester) and childhood traumatic life events scores. Childhood life events scores were calculated based on a child self-reported traumatic life events questionnaire, consisting of 11 (potentially) traumatic life events that were scored (0/1) based on whether the child had ever experienced the event (the maximum possible score was 11). The following 11 themes were addressed: (1) physical violence, (2) sexual violence, (3) relationship break-up, (4) friendship break-up, (5) personal failure, (6) problems in family, (7) problems at school, (8) problems in peer group, (9) leaving religious community, (10) death of a loved one, and (11) severe illness or injury [24, 30].
Statistical analyses
To reduce excess variance and remove outliers (potentially representing error in the data) regarding our outcomes, we excluded participants scoring >3 standard deviations (SDs) on callousness (N = 6 excluded), uncaring (N = 2 excluded), or unemotional (no subjects excluded) ICU-dimensions. Furthermore, the childhood traumatic life events scores were dichotomized closest to the 67th percentile, to avoid collinearity with covariate-interaction terms in GxE-interaction analyses. Control variables were sex, age at outcome assessment, the first 10 principal components (PCs), and gene-covariate and environment-covariate interactions in GxE-interaction analyses [31]. Furthermore, we filtered 69 participants with deviant PC-scores (i.e. scoring >|±2| SDs on any of the first 10 PCs, using the 1000 Genomes European reference populations [32]). Population stratification was further investigated by studying PC-analysis plots of NeuroIMAGE merged with the 1000 Genomes phase 1 reference dataset [32].
Polygenic risk scores
Polygenic risk scores (PRS) analyses were performed using PRSice2-software [33]. GWAS summary data was available for 2,188,528 SNPs of the EAGLE-consortium aggression GWAS (available at https://www.wikigenes.org/e/art/e/348.html#GWA_meta-analyses_summary_results) [8], and a total of 1,192,414 SNPs could be included for PRS-analyses (SNP-matching across base and target samples was based on SNP rs-numbers). SNPs were clumped based on linkage disequilibrium (LD) using PRSice default settings (i.e. a bidirectional 250Kb-window and R2-threshold of 0.1), resulting in a total of 66,088 LD-clumped SNPs. PRS were calculated according to Supplementary Equation S1. According to our best knowledge, no prior studies investigating shared genetic etiology between aggression and CU-traits, which we could use for selecting an a-priori P-value threshold for the PRS, were available. Therefore, to avoid underfitting in the absence of a-priori information, PRS were calculated at multiple P-value thresholds [33, 34]. We first calculated PRS for at most 14 ‘broad’ P-value thresholds (i.e. 0.0001, 0.0002, 0.0005, 0.001, 0.002, 0.005, 0.01, 0.02, 0.05, 0.1, 0.2, 0.3, 0.4, and 0.5) using an additive and recessive model since recessive SNP-effects have been reported previously in aggression [35, 36]. If at least nominal significance was reached for one of the thresholds, the PRS was included in the final analyses and calculated for a small range of further thresholds around the best-fitting of the (at most) 14 broad thresholds. This procedure was performed for genome-wide and gene-set (see below) PRS in relation to our three target phenotypes. Multiple testing correction was applied in two stages: first, we computed ‘empirical P-values’ for the association of each best-fitting PRS. Empirical P-values were obtained by comparing the P-value of the PRS for the actual phenotype with a null-distribution of P-values of the PRS regressed on 11,000 randomly permuted phenotypes, to correct for overfitting due to testing multiple P-value thresholds across two inheritance models [33]. Second, we adjusted the empirical P-values using the procedure described by Benjamini and Hochberg [37] to control the false discovery rate (FDR) for the number of gene-sets and phenotypes investigated in the final stage of the PRS-analyses (i.e. ‘FDR Q-values’). A detailed description of all PRS-procedures is provided in the Supplement. In addition to investigating whether the PRS were associated with our traits of interest, we also computed ‘competitive P-values’ to investigate the level of enrichment of the SNP-sets representing the best-fitting PRS. The competitive P-values were obtained by comparing the P-value of the PRS with a null-distribution of P-values of 11,000 random SNP-sets of the same size, drawn from the genome-wide genetic background signal outside of the PRS SNP-set, regressed on the phenotype [33].
Gene-set PRS
In addition to genome-wide PRS, we also computed PRS from four gene-sets that were previously implicated in aggression-related phenotypes. We defined serotonergic, dopaminergic, neuroendocrine, and glutamatergic gene-sets according to Donkelaar et al. and Naaijen et al., given the careful and comprehensive selection of genes related to the pathways of interest in these studies [14, 15]. Lists of genes included in each gene-set are provided in Supplementary Table S1. Gene-set PRS may provide additional information to genome-wide PRS, as association signals from these individual sets may be more difficult to detect in a genome-wide signal. Different sets/pathways may also have different directions of association with the target phenotype. We calculated gene-set PRS also for different P-value thresholds as not all genetic variation in a pathway is necessarily linked with the target phenotype (similar to the genome-wide PRS).
PRS-Environment interactions (GxE)
Since gene-environment correlations (rGE) could be confounding GxE-interactions (i.e. the genetic factor could be related to both the environmental factor and outcome in the ‘GxE’) [38], we first investigated rGE between significant PRS (identified in the PRS-analyses) and childhood life events and smoking during pregnancy. Subsequently, environmental main effects and GxE-interactions between the best-fitting PRS and environment were analyzed in relation to callousness, uncaring, and unemotional ICU-dimensions. GxE-interactions were coded in R, using linear mixed models as implemented in ‘Lme4’ and ‘LmerTest’ packages [39, 40]. In addition to abovementioned control variables a random intercept for each family was added to adjust for sibling relatedness within our sample [41, 42]. BH-adjustment was used to correct for testing multiple GxE-interactions.
Sensitivity analyses
As PRSice currently does not support linear mixed models, we were unable to correct for sibling relatedness in our main PRS-analyses. Therefore, we coded our significant PRS-models in R [39, 40] and corrected for sibling relatedness as described [41, 42]. Second, to investigate whether comorbid ADHD may be driving our results, we also included ADHD-case/control status as a covariate. Third, because of the clustered family structure in NeuroIMAGE, estimation of PCs could be slightly artefactual [42, 43] and since it was not feasible to calculate PCs from unrelated subjects only, we investigated potential inaccuracy by removing the PC-covariates (and reincluding subjects with deviant PC-scores as well) and only keeping adjustment for sibling relatedness. Fourth, because the Major Histocompatibility Complex (MHC) locus shows extended LD-structure and many diseases have been associated with this region [44], we adjusted our significant PRS by excluding the MHC-locus, to investigate to which degree SNPs in this region might be affecting our results. These same aforementioned sensitivity analyses were performed for significant GxE-interactions. As it was not possible to perform permutation-based analyses with the mixed models in R (and hence, apply comprehensive multiple testing correction) we compared the uncorrected PRS association P-values from each sensitivity analysis with the main analyses. Furthermore, by applying P-value thresholding to gene-set PRS, only a subset of the gene-set is represented by the best-fitting PRS. To investigate whether the whole gene-set is more strongly associated with the target phenotype, we repeated the gene-set PRS-analyses keeping the P-value threshold at 1.
Results
Descriptive statistics of NeuroIMAGE
Table 1 provides descriptive statistics of the phenotypic and environmental variables in NeuroIMAGE. See Supplementary Fig. S1 for global and intra-European PCA-plots of NeuroIMAGE merged with the 1000 Genomes reference populations [32]. NeuroIMAGE appeared most proximal to CEPH and British European populations.
PRS-analyses
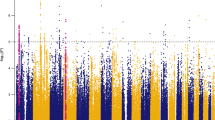
Six PRSs reached at least nominal significance at one of the at most 14 broad P-value thresholds and were included in the final analyses. These were a dopaminergic (additive) and glutamaterig (recessive) PRS in relation to callousness scores, a genome-wide (recessive) PRS in relation to uncaring scores, and a glutamatergic, dopaminergic, and neuroendocrine (recessive) PRS in relation to unemotional scores (Supplementary Table S2 provides an overview of intermediate results). For these six PRS, we calculated some additional nearby P-value thresholds. See Fig. 1a–r and Table 2 for all results including the total number of and PRS model-fit across all thresholds tested. Four PRS showed evidence for association with and enrichment for CU-traits. Genome-wide PRS predicted uncaring scores (best-fitting-R2 = 1.59%, 58 SNPs, empirical-P = 1.02E-02, FDR-Q = 3.06E-02, competitive-P = 1.55E-03; Supplementary Table S3 provides a list of the nearest genes to the SNPs in this PRS), whereas glutamatergic PRS (best-fitting-R2 = 2.00%, 179 SNPs, empirical-P = 1.91E-03, FDR-Q = 1.15E-02, competitive-P = 1.82E-04), dopaminergic PRS (best-fitting-R2 = 1.33%, 3 SNPs, empirical-P = 1.85E-02, FDR-Q = 3.70E-02, competitive-P = 2.91E-03), and neuroendocrine PRS (best-fitting-R2 = 1.20%, 41 SNPs, empirical-P = 3.15E-02, FDR-Q = 4.73E-02, competitive-P = 4.55E-03) predicted unemotional scores. In addition, glutamatergic PRS showed evidence of nominal significant association with callousness scores (best-fitting-R2 = 0.93%, 67 SNPs, empirical-P = 6.74E-02, FDR-Q = 8.09E-02) but showed significant enrichment (competitive-P = 1.39E-02). Supplementary Tables S4(A–D) provides the minor allele frequency, Hardy-Weinberg test result, and call rate for the SNPs included in the best-fitting PRS that showed evidence for association with and enrichment for CU-traits.

Add. additive model, PRS Polygenic Risk Score, Rec. recessive model, SNP single-nucleotide polymorphism. PRS were calculated first for at most 14 ‘broad’ P-value thresholds using the additive and recessive inheritance model (a, b, d, e, g, h, j, k, m, n, p, q; note that for the gene-set based PRS, some of the lower thresholds included no SNPs). Subsequently PRS were calculated for some further thresholds around the best-fitting of the at most 14 ‘broad’ thresholds (Fig. 1c, f, i, l, o, r; the additional thresholds represent the unique P-values of the SNPs in the GWAS summary statistics). The number of SNPs is shown on top of the bar plots with sufficient space available. The model-fit for the PRS across all tested P-value thresholds as well as the number of thresholds tested is shown. Table 2 provides specific details (e.g. regression coefficients, association, and enrichment test results) for the best-fitting PRS in relation to callousness, uncaring and unemotional traits.
PRS-Environment interactions (GxE)
No (rGE) between PRS and childhood life event scores and/or smoking during pregnancy were observed (See Supplementary Table S5 for all results). Regarding environmental main effects and GxE-interactions, see Table 3 for all results. Childhood life events related to callousness (FDR-Q = 3.19E-04) and unemotional scores (FDR-Q = 4.55E-02). Smoking during pregnancy related to callousness scores (FDR-Q = 3.47E-03). The best-fitting dopaminergic PRS interacted with childhood life events in relation to unemotional scores (FDR-Q = 4.55E-02; See Fig. 2 and Supplementary Table S6 for environment-stratified PRS-effects).

PRS Polygenic Risk Score. See Supplementary Table S6 for all stratified results.
Sensitivity analyses
See Supplementary Table S7 for all results. After adjustment for sibling relatedness the association strength for the best-fitting PRS remained similar (genome-wide PRS in relation to uncaring scores P = 5.30E-04; glutamatergic PRS P = 1.63E-04, dopaminergic PRS P = 2.16E-03, and neuroendocrine PRS P = 3.60E-03 in relation to unemotional scores). After further adjustment for ADHD results again remained similar (genome-wide PRS in relation to uncaring scores P = 2.74E-04; glutamatergic PRS P = 1.91E-04, dopaminergic PRS P = 1.70E-03, and neuroendocrine PRS P = 3.10E-03, and GxE-interaction P = 1.33E-02 in relation to unemotional scores). When PC-covariates were removed and subjects with deviant PC-scores were reincluded, results changed only slightly (genome-wide PRS in relation to uncaring scores P = 3.14E-04; glutamatergic PRS P = 4.50E-04, dopaminergic PRS P = 4.59E-04, and neuroendocrine PRS P = 8.79E-03; and GxE-interaction P = 1.13E-02 in relation to unemotional scores). When the MHC-locus was removed, results remained unchanged (no MHC-SNPs in the PRSs) except for the genome-wide PRS in relation to uncaring scores (P = 2.38E-03, 56 SNPs; 2 MHC-SNPs removed). Including all SNPs in the gene-set PRS (i.e. a threshold of 1) resulted in a reduced association strength of the PRS compared to the identified best-fitting threshold in the main analyses (full glutamate-set PRS [325 SNPs] R2 = 1.22%, P = 0.0037, full dopamine-set PRS [224 SNPs] R2 = (1.02E-06)%, P = 0.9979 and full neuroendocrine-set PRS [1107SNPs] R2 = 0.58%, P = 0.1827 in relation to unemotional scores).
Discussion
We performed PRS-analyses to investigate evidence for shared genetic etiology between aggressive behavior and callous, uncaring and unemotional traits in children/adolescents. In addition to genome-wide PRS, we also studied PRS based on gene-sets that have been previously implicated in aggression-related phenotypes [14, 15]. Furthermore, we studied GxE-interaction between PRS and two key environmental adversities. Our results suggest that aggression shares genetic etiology with the ICU-dimensions ‘uncaring’ and ‘unemotional’, and confirmed glutamatergic, dopaminergic, and neuroendocrine signaling as shared biological pathways of interest. Moreover, dopaminergic PRS appeared to interact with childhood life events in relation to unemotional scores.
The only genome-wide polygenic association was observed in relation to uncaring scores. This PRS included 58 SNPs at a relatively low P-value threshold, indicating that SNPs that associated more strongly with aggressive behavior in general also combine into a stronger polygenic signal in relation to uncaring traits. A number of the genes related to the SNPs in this PRS have been linked to aggression (e.g. MECOM, AVPR1A) [45], other related psychiatric disorders such as autism or schizophrenia (e.g. MACROD2, ADD2) [46, 47], or have been implicated in neurobiological functions such as synapse remodeling and interneuron maturation (e.g. RAPGEF4, DGKG) [48, 49]. The observation that gene-set PRS explained relatively large amounts of variance in CU-dimensions when compared to the genome-wide PRS could be related to opposite directions of effects of different gene-sets/pathways, that cancel each other out when added together in the genome-wide PRS. Furthermore, as the genome-wide PRS represents a more general genetic liability, the signal could also be susceptible to more noise from irrelevant SNPs.
Glutamatargic PRS related to unemotional scores and explained the most variance (up to 2% at the best-fitting threshold) in an individual trait. The glutamatergic gene-set used for set-based PRS-analysis in the present study, was linked previously to ADHD (i.e. hyperactivity scores) [15]. As the sample of that study was derived from the IMAGE-project, it partially overlaps with our current study sample. However, the degree of overlap is only limited (NeuroIMAGE is based on a distinct subset of IMAGE and these samples were also genotyped separately; furthermore the aforementioned study only included ADHD-cases) and, moreover, currently we investigated CU-traits rather than ADHD. The current results support the notion that there is glutamatergic genetic liability underlying multiple pediatric aggression-related phenotypes (i.e. hyperactivity, unemotional scores, and more general aggressive behavior). This is an interesting observation, given that glutamatergic genes have also been proposed as candidates in other neurodevelopmental disorders such as autism and schizophrenia [50, 51], which are often also present with aggressive behavior. In addition, glutamatergic PRS were also enriched for callousness scores, which may suggest some degree of shared glutamatergic liability among callousness and unemotional traits. However, the association with callousness scores was only nominally significant.
Regarding dopamine, a PRS combining three (LD-clumped) SNPs predicted unemotional scores. The direction of effect was negative, suggesting that children with a higher aggression PRS had lower unemotional scores. Reactive aggression is more common than proactive aggression in the population (e.g. [52]), and some studies have reported a negative association between reactive aggression and specifically the ICU-unemotional scale (e.g. [53]). Further, a low degree of stress-reactivity has been observed in children displaying CU-traits, whereas in children with CD but without CU-traits, heightened threat-sensitivity and reactive aggression is seen [1, 16]. Therefore, we theorize that children displaying a more general tendency toward aggression (i.e. with higher aggression-PRS), might be also more reactive aggressive and stress/threat sensitive and, hence, be less unemotional. One of the PRS-SNPs was located within the PRKAG2-gene. The gene PRKAG2 encodes the non-catalytic Gamma 2 subunit of the AMP-activated protein kinase enzyme and associations of PRKAG2-variants with temporal lobe volume [54] and cognitive impairment [55] have been reported, which points to the neurobiological role of this gene. In addition, the effect of dopaminergic PRS appeared to be moderated by childhood life events in relation to unemotional scores, such that the PRS related most strongly to unemotional scores in children with higher life event scores. Dopamine plays an important role in motivation, reward, and decision making, and antagonism of dopaminergic (D2) receptors by antipsychotic drugs has been shown to reduce aggressive behaviors, pointing to the involvement of the dopaminergic system in aggression [56]. Furthermore, in two recent functional imaging studies, reduced activation patterns in parts of the dopaminergic reward system (such as the ventral striatum, amygdala, and prefrontal cortex) were linked to CU-traits and disruptive behavior [57, 58]. Although a significant link with CU-traits was reported in only one of these studies [57] (which may be related to CU-traits being assessed as a unified construct and/or the use of an ethnically stratified sample [58]) this could suggest that the currently observed shared dopaminergic genetic liability between aggression and unemotional CU-traits is related to functional neural differences in dopaminergic brain circuits involved in reward processing.
Shared genetic factors between aggressive behavior and unemotional scores was also suggested by neuroendocrine PRS. A recent study showed that cortisol reactivity moderated the link between aggression and CU-traits in a pediatric longitudinal cohort, such that the aggression-CU-traits link was present only in the context of low-cortisol reactivity [59]. Furthermore, interaction between testosteron and cortisol in relation to aggression in adolescents has also been reported, suggesting that only in subjects with low cortisol levels testosterone levels were linked to aggression [60]. Moreover, the recent study that linked the currently investigated neuroendocrine gene-set to aggression, also reported that the sub-set of genes in the set that linked most strongly with aggression were glucocorticoid genes [14]. Therefore, our current results could point to a key role for cortisol-related genetic variation in a shared genetic liability among aggressive behavior and CU-traits in children.
Strengths and limitations
A unique feature of the current study includes the simultaneous investigation of genome-wide PRS and recently implicated gene-sets to study potential shared genetic etiology between aggression and callous, uncaring and unemotional traits. Furthermore, we investigated interactions between the PRS and two key environmental factors for CU-traits, thereby providing an approach to address GxE-polygenicity (although the number of SNPs was somewhat limited in our best-fit PRS). Nevertheless, some limitations should be discussed. First, as NeuroIMAGE consists of a partially referred sample, findings may not necessarily generalize to CU-traits distributed within the general population. Furthermore, although our base sample was large, our target sample had a relatively modest size. While this may have prevented detection of small effects in individual variants, it proved adequate for polygenic analyses (power for detecting PRS-main effects explaining between 1.20 and 2.00% of variance ranged from 81 to 96%) and mostly sufficient to perform GxE-analyses (power for detecting PRS-by-environment interactions based on PRS-main effects ranged from 80 to 95% for childhood life events and from 65 to 86% for smoking during pregnancy). Although GxE-interactions based on individual SNPs typically require large sample sizes, detection of PRS-based GxE-interactions may require less power due to aggregation of SNP-effects into the PRS. Furthermore, although subsetting genes reduced the absolute number of available SNPs, the resulting PRS may be actually more predictive and therefore power should not necessarily be adversely affected by set-based PRS-analyses. Careful selection of gene-sets is, however, important. In addition, observed effects may be (partially) explained by, e.g., mediation effects or coexistent aggression. Last, our present study has been among the first studies investigating genetic sharing between aggression and CU-traits and therefore current results need replication (e.g. to validate the predictive ability of the PRS in an independent sample).
Conclusion
Our study provides evidence suggesting shared genetic etiology between children’s aggressive behavior and ‘uncaring’ and ‘unemotional’ ICU-dimensions, thereby supporting the notion of a polygenic architecture underlying CU-traits. In addition to genome-wide PRS, gene-set based PRS pointed to shared genetic variation within glutamatergic, dopaminergic, and neuroendocrine signaling pathways as well as GxE-interaction with childhood life events. Furthermore, individual genes derived from the PRS have been linked to aggression and neurodevelopment previously. Although ‘callousness’ was not significantly related to PRS, childhood life events and smoking during pregnancy were most strongly linked to this ICU-dimension, suggesting a differential contribution of genes and environment to callousness, uncaring, and unemotional traits.
Funding and disclosure
This publication is the work of the authors and this research is supported by the European Community’s Seventh Framework Programme (FP7/2007-2013) under grant agreement no. 603016 (MATRICS) and no. 602805 (Aggressotype). The NeuroIMAGE-project was supported by NIH Grant R01MH62873 (to S.V.F.), NWO Large Investment Grant 1750102007010 and ZonMW Grant 60-60600-97-193 (to J.K.B.), and grants from Radboud University Nijmegen Medical Center, University Medical Center Groningen and Accare, and VU University Amsterdam. I.H.R., A.D., M.K., J.O. and P. J.H. reported no financial interests or potential conflicts of interest. J.K.B. has been a consultant to/advisory board member of/and/or a speaker for Janssen Cilag BV, Eli Lilly, Shire, Lundbeck, Roche, and Servier. He is not an employee of any of these companies and not a stock shareholder of any of these companies. He has no other financial or material support, including expert testimony, patents, or royalties. S.V.F. received income, potential income, travel expenses, continuing education support, and/or research support from Tris, Otsuka, Arbor, Ironshore, Shire, Akili Interactive Labs, VAYA, Ironshore, Sunovion, Supernus, and Genomind. With his institution, he has US patent US20130217707 A1 for the use of sodium-hydrogen exchange inhibitors in the treatment of ADHD.
References
Blair RJR, Leibenluft E, Pine DS. Conduct disorder and callous–unemotional traits in youth. N Engl J Med. 2014;371:2207–16.
Waltes R, Chiocchetti AG, Freitag CM. The neurobiological basis of human aggression: a review on genetic and epigenetic mechanisms. Am J Med Genet Part B Neuropsychiatr Genet. 2016;171:650–75.
Olsson M. DSM diagnosis of conduct disorder (CD)-a review. Nord J Psychiatry. 2009;63:102–12.
American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 5th ed. Washington, DC: American Psychiatric Association; 2013.
Herpers PCM, Klip H, Rommelse NNJ, Greven CU, Buitelaar JK. Associations between high callous–unemotional traits and quality of life across youths with non-conduct disorder diagnoses. Eur Child Adolesc Psychiatry. 2016;25:547–55.
Kempes M, Matthys W, De Vries H, Van Engeland H. Reactive and proactive aggression in children. A review of theory, findings and the relevance for child and adolescent psychiatry. Eur Child Adolesc Psychiatry. 2005;14:11–9.
Veroude K, Zhang-James Y, Fernàndez-Castillo N, Bakker MJ, Cormand B, Faraone SV. Genetics of aggressive behavior: an overview. Am J Med Genet B Neuropsychiatr Genet. 2016;171:3–43.
Pappa I, St Pourcain B, Benke K, Cavadino A, Hakulinen C, Nivard MG, et al. A genome-wide approach to children’s aggressive behavior: the EAGLE consortium. Am J Med Genet B Neuropsychiatr Genet. 2016;171:562–72.
Tielbeek JJ, Johansson A, Polderman TJC, Rautiainen M-R, Jansen P, Taylor M, et al. Genome-wide association studies of a broad spectrum of antisocial behavior. JAMA Psychiatry. 2017;74:1242–50.
Rautiainen M, Paunio T, Repo-Tiihonen E, Virkkunen M, Ollila H, Sulkava S, et al. Genome-wide association study of antisocial personality disorder. Transl Psychiatry 2016;6:e883.
Andlauer T, Müller-Myhsok B, Ripke S. Statistical genetics: genome-wide studies. In: Schulze T, McMahon F, editors. Psychiatric genetics: a primer for clinical and basic scientists. 1st ed. Oxford University Press; 2018. p. 57–69.
Purcell S, Wray N, Stone J, Visscher P, O’Donovan M, Sullivan P, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009;460:748–52.
Euesden J, Lewis CM, O’Reilly PF. PRSice: Polygenic Risk Score software. Bioinformatics 2015;31:1466–8.
van Donkelaar MMJ, Hoogman M, Shumskaya E, Buitelaar JK, Bralten J, Franke B Monoamine and neuroendocrine gene-sets associate with frustration-based aggression in a gender-specific manner. Eur Neuropsychopharmacol. 2020;30:75–86.
Naaijen J, Bralten J, Poelmans G, Glennon JC, Franke B, Buitelaar JK, et al. Glutamatergic and GABAergic gene sets in attention-deficit/hyperactivity disorder: association to overlapping traits in ADHD and autism. Transl Psychiatry 2017;7:e999–7.
Blair RJ. The neurobiology of psychopathic traits in youths. Nat Rev Neurosci. 2013;14:786–99.
Byrd AL, Manuck SB. MAOA, childhood maltreatment, and antisocial behavior: Meta-analysis of a gene-environment interaction. Biol Psychiatry 2014;75:9–17.
Ruisch IH, Dietrich A, Glennon JC, Buitelaar JK, Hoekstra PJ. Maternal substance use during pregnancy and offspring conduct problems: a meta-analysis. Neurosci Biobehav Rev. 2018;84:325–36.
Afifi TO, McMillan KA, Asmundson GJG, Pietrzak RH, Sareen J. An examination of the relation between conduct disorder, childhood and adulthood traumatic events, and posttraumatic stress disorder in a nationally representative sample. J Psychiatr Res 2011;45:1564–72.
Holz NE, Zohsel K, Laucht M, Banaschewski T, Hohmann S, Brandeis D. Gene x environment interactions in conduct disorder: implications for future treatments. Neurosci Biobehav Rev. 2018;91:239–58.
Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al. Finding the missing heritability of complex diseases. Nature 2009;461:747–53.
Dick DM, Agrawal A, Keller MC, Adkins A, Aliev F, Monroe S, et al. Candidate gene–environment interaction research. Perspect Psychol Sci. 2015;10:37–59.
Vassos E, Collier DA, Fazel S. Systematic meta-analyses and field synopsis of genetic association studies of violence and aggression. Mol Psychiatry 2014;19:471–7.
von Rhein D, Mennes M, van Ewijk H, Groenman AP, Zwiers MP, Oosterlaan J, et al. The NeuroIMAGE study: a prospective phenotypic, cognitive, genetic and MRI study in children with attention-deficit/hyperactivity disorder. Design and descriptives. Eur Child Adolesc Psychiatry. 2015;24:265–81.
Essau CA, Sasagawa S, Frick PJ. Callous-unemotional traits in a community sample of adolescents. Assessment 2006;13:454–69.
Viding E, Hanscombe KB, Curtis CJC, Davis OSP, Meaburn EL, Plomin R. In search of genes associated with risk for psychopathic tendencies in children: a two-stage genome-wide association study of pooled DNA. J Child Psychol Psychiatry. 2010;51:780–8.
Viding E, Price TS, Jaffee SR, Trzaskowski M, Davis OSP, Meaburn EL, et al. Genetics of callous-unemotional behavior in children. PLoS ONE 2013;8:e65789.
Logue MW, Amstadter AB, Baker DG, Duncan L, Koenen KC, Liberzon I, et al. The psychiatric genomics consortium posttraumatic stress disorder workgroup: posttraumatic stress disorder enters the age of large-scale genomic collaboration. Neuropsychopharmacology 2015;40:2287–97.
Lam M, Awasthi S, Watson HJ, Goldstein J, Panagiotaropoulou G, Trubetskoy V, et al. RICOPILI: Rapid Imputation for COnsortias PIpeLIne. Bioinformatics. 2019;(August):1–4.
Reijneveld SA, Bosch NM, Bakker MP, Verhulst FC, Ormel J, Oldehinkel AJ. Timing matters: Long term effects of adversities from prenatal period up to adolescence on adolescents’ cortisol stress response. The TRAILS study. Psychoneuroendocrinology 2012;37:1439–47.
Keller M. Gene-by-environment interaction studies have not properly controlled for potential confounders: The problem and the (simple) solution. Biol Psychiatry 2014;75:18–24.
Auton A, Abecasis GR, Altshuler DM, Durbin RM, Bentley DR, Chakravarti A, et al. A global reference for human genetic variation. Nature 2015;526:68–74.
Choi SW, O’Reilly PF. PRSice-2: Polygenic Risk Score software for biobank-scale data. Gigascience 2019;8:1–6.
Choi SW, Mak TSH, O’Reilly PF. A guide to performing Polygenic Risk Score analyses. bioRxiv 2018.
Ruisch IH, Dietrich A, Glennon JC, Buitelaar JK, Hoekstra PJ. Interplay between genome-wide implicated genetic variants and environmental factors related to childhood antisocial behavior in the UK ALSPAC cohort. Eur Arch Psychiatry Clin Neurosci. 2019;269:741–52.
Zhang Y, Wu C, Chang H, Yan Q, Wu L, Yuan S, et al. Genetic variants in oxytocin receptor gene (OXTR) and childhood physical abuse collaborate to modify the risk of aggression in chinese adolescents. J Affect Disord. 2018;229:105–10.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B. 1995;57:289–300.
Rice F, Harold GT, Boivin J, van den B, Hay DF, Thapar A. The links between prenatal stress and offspring development and psychopathology: disentangling environmental and inherited influences. Psychol Med 2010;40:335–45.
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. 2018.
Bates D, Maechler M, Bolker B, Walker S. Fitting linear mixed-effects models using lme4. J Stat Softw. 2015;67:1–48.
Zhang Z, Ersoz E, Lai C, Todhunter RJ, Tiwari HK, Gore MA, et al. Mixed linear model approach adapted for genome-wide association studies. Nat Genet 2010;42:355–60.
Price AL, Zaitlen NA, Reich D, Patterson N. New approaches to population stratification in genome-wide association studies. Nat Rev Genet. 2010;11:459–63.
Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet 2006;2:e190.
Kennedy AE, Ozbek U, Dorak MT. What has GWAS done for HLA and disease associations? Int J Immunogenet. 2017;44:195–211.
van Donkelaar MMJ, Hoogman M, Pappa I, Tiemeier H, Buitelaar JK, Franke B, et al. Pleiotropic contribution of MECOM and AVPR1A to aggression and subcortical brain volumes. Front Behav Neurosci. 2018;12:61.
The Autism Spectrum Disorders Working Group of The Psychiatric Genomics Consortium. Meta-analysis of GWAS of over 16,000 individuals with autism spectrum disorder highlights a novel locus at 10q24.32 and a significant overlap with schizophrenia. Mol Autism 2017;8:21.
Bosia M, Pigoni A, Zagato L, Merlino L, Casamassima N, Lorenzi C, et al. ADDing a piece to the puzzle of cognition in schizophrenia. Eur J Med Genet. 2016;59:26–31.
Woolfrey KM, Srivastava DP, Photowala H, Yamashita M, Barbolina MV, Cahill ME, et al. Epac2 induces synapse remodeling and depression and its disease-associated forms alter spines. Nat Neurosci 2009;12:1275–84.
Fukumoto K, Tamada K, Toya T, Nishino T, Yanagawa Y, Takumi T. Identification of genes regulating GABAergic interneuron maturation. Neurosci Res 2017;134:18–29.
Purkayastha P, Malapati A, Yogeeswari P, Sriram D. A review on GABA/glutamate pathway for therapeutic intervention of ASD and ADHD. Curr Med Chem. 2015;22:1850–9.
Ripke S, Neale BM, Corvin A, Walters JTR, Farh KH, Holmans PA, et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014;511:421–7.
Cima M, Raine A, Meesters C, Popma A. Validation of the Dutch Reactive Proactive Questionnaire (RPQ): differential correlates of reactive and proactive aggression from childhood to adulthood. Aggress Behav 2013;39:99–113.
Ueno K, Ackermann K, Freitag CM, Schwenck C. Assessing Callous–Unemotional Traits in 6- to 18-Year-Olds: Reliability, Validity, Factor Structure, and Norms of the German Version of the Inventory of Callous–Unemotional Traits. Assessment. 2019; In press.
Kohannim O, Hibar DP, Stein JL, Jahansha N, Hua X, Rajagopalan P, et al. Discovery and replication of gene influences on brain structure using LASSO regression. Front Neurosci 2012;6:115.
Kim E, Lee SH, Lee KS, Cheong HK, Namkoong K, Hong CH, et al. AMPK γ2 subunit gene PRKAG2 polymorphism associated with cognitive impairment as well as diabetes in old age. Psychoneuroendocrinology 2012;37:358–65.
McCracken JT, McGough J, Shah B, Cronin P, Hong D, Aman MG, et al. Risperidone in children with autism and serious behavioral problems. N. Engl J Med. 2002;347:314–21.
Cohn MD, Veltman DJ, Pape LE, Van Lith K, Vermeiren RRJM, Van Den Brink W, et al. Incentive processing in persistent disruptive behavior and psychopathic traits: A functional magnetic resonance imaging study in adolescents. Biol Psychiatry 2015;78:615–24.
Murray L, Shaw DS, Forbes EE, Hyde LW. Reward-related neural correlates of antisocial behavior and callous–unemotional traits in young men. Biol Psychiatry Cogn Neurosci Neuroimaging. 2017;2:346–54.
Wright N, Hill J, Pickles A, Sharp H. Callous-unemotional traits, low cortisol reactivity and physical aggression in children: findings from the Wirral Child Health and Development Study. Transl Psychiatry 2019;9:79.
Popma A, Vermeiren R, Geluk CAML, Rinne T, van den Brink W, Knol DL, et al. Cortisol moderates the relationship between testosterone and aggression in delinquent male adolescents. Biol Psychiatry 2007;61:405–11.
Acknowledgements
We are very grateful to all families who participated in the NeuroIMAGE-study, and the whole NeuroIMAGE-team, including interviewers, technicians, scientists, clinicians, volunteers, and managers of all involved organizations and facilities, for recruitment and collection, and preprocessing of the data used in this study. We are very grateful to the Donders Institute for Brain, Cognition and Behavior of the Radboud University Nijmegen for granting us access to the high-performance computing environment required to conduct this study.
Author information
Authors and Affiliations
Contributions
I.H.R., A.D., and P.J.H. designed the study. I.H.R. wrote a manuscript draft and conducted the statistical analyses. M.K. assisted with the data analysis and interpretation of results. A.D., M.K., S.V.F., J.O., J.K.B., and P.J.H. provided both general feedback and discussed specific methodological, clinical, or theoretical issues related to the study. All authors discussed the results and implications as well as contributed and approved the final version of the manuscript.
Corresponding author
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Ruisch, I.H., Dietrich, A., Klein, M. et al. Aggression based genome-wide, glutamatergic, dopaminergic and neuroendocrine polygenic risk scores predict callous-unemotional traits. Neuropsychopharmacol. 45, 761–769 (2020). https://doi.org/10.1038/s41386-020-0608-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-020-0608-0
This article is cited by
-
Psychopathy
Nature Reviews Disease Primers (2021)
-
Association of serotonin system-related genes with homicidal behavior and criminal aggression in a prison population of Pakistani Origin
Scientific Reports (2021)
-
Genetic Moderation of the Association Between Early Family Instability and Trajectories of Aggressive Behaviors from Middle Childhood to Adolescence
Behavior Genetics (2021)
-
Oxytocin receptor gene polymorphism (rs53576) and digit ratio associates with aggression: comparison in seven ethnic groups
Journal of Physiological Anthropology (2020)
