
Abstract
Genome-wide association studies (GWAS) have identified hundreds of genetic variants associated with schizophrenia (SCZ). However, prioritizing risk variants and regulatory elements for follow-up functional studies remains a major challenge. Therefore, we performed an integrated analysis to identify variants who affect methylation levels of nearby genes and contribute to the risk of SCZ, and to explore the potential role of these variants in SCZ pathogenesis. First, we used the Summary data-based Mendelian Randomization (SMR) method to integrate GWAS and methylation quantitative trait loci data. Then, the SNP-methylation combinations as associated with SCZ were replicated across multiple samples. Totally, we identified and replicated 14 and one SNP-methylation combinations in blood and brain tissues, respectively, that significantly associated with SCZ. Furthermore, our expression quantitative trait loci analysis, differential methylation analysis, neuroimaging genetics, and cognitive genetics analysis consistently supported the potential roles of these 15 SNPs in the pathogenesis of SCZ. Finally, using the convergent functional genomics method, we prioritized three risk SNPs, including rs3765971 (RERE, PSMR = 3.87 × 10−8), rs55742290 (ARL6IP4, PSMR = 1.50 × 10−7), and rs7293091 (CENPM, PSMR = 5.09 × 10−7), may represent promising risk variants in SCZ. These convergent lines of evidence suggest that three risk variants may be involved in the pathogenesis of SCZ. Further investigation of the roles of these variants in the pathogenesis of SCZ is warranted.
Similar content being viewed by others


Introduction
Recent approaches to understanding the pathogenesis of schizophrenia (SCZ) have focused on describing the genetic contribution to SCZ. A recent large-scale genome-wide association study (GWAS) by the Psychiatric Genomics Consortium (PGC) identified 108 independent loci exhibiting a genome-wide significant association with SCZ [1]. Nevertheless, owing to linkage disequilibrium, identified risk loci usually contain many highly linked genetic variants that exhibit similar association significance. Therefore, it is a major challenge to pinpoint causal genes involved in SCZ pathogenesis and to elucidate the exact genetic mechanisms underlying SCZ.
Considering that most identified risk variants reside in noncoding regions of the genome, it is likely that these identified risk variants confer risk for SCZ by modulating gene regulation, for instance, through methylation and altered expression [2]. Recent studies have incorporated data on genetic variants associated with DNA methylation (methylation quantitative trait loci, mQTL) or gene expression (expression quantitative trait loci, eQTL) into results from GWAS for complex traits to help identify putative causal variants in a particular genomic region, as well as to provide evidence, suggesting which genes might be influenced by this variant [3]. These studies could integrate different omics data to gain further insights into the mechanisms of genetic variants and causally associated disease. In addition, recent studies have successfully identified genetic variants associated with DNAm, reporting that they appear to be enriched in SCZ GWAS risk loci [4, 5].
To identify genes whose methylation levels are affected by SCZ risk variants and to explore the potential role of these genes in SCZ pathogenesis, we incorporated data from different levels, including SCZ GWAS data, eQTL, mQTL, differential gene methylation patterns in SCZ patients and controls, association with human brain structure, and association with cognitive performance. Through the convergent functional genomics (CFG) method, we prioritize three risk SNPs may represent promising causal variants in SCZ.
Materials and methods
Methodological overview
To identify genes whose methylation levels are affected by SCZ risk variants and to explore the potential role of these genes in SCZ pathogenesis, we performed integrative analyses by integrating data from different levels, including genetic associations from large-scale GWAS, mQTL, eQTL, differential gene methylation in SCZ patients and controls, and association with hippocampal volume and cognitive function (Fig. 1). Our integrative analyses showed that several identified genes may represent promising risk genes for SCZ. This study used data of several public data sets. All the samples used in the study were described in Table 1, including the age, sex, and ethnic distributions of each cohort along with sample sizes and diagnoses included. For further specifics on the respective ethics approvals, we refer to the single study publications. All data are available in public databases as detailed in the methods section.

Based on the hypothesis that SCZ risk variants are enriched among mQTL, we systematically integrated SCZ GWAS and genome-wide mQTL data with the SMR software tool. The top genes identified by SMR were then replicated in a larger scale SCZ sample and mQTL data sets. Moreover, we explored the genetic mechanisms, using eQTL analysis, differential methylation analysis in SCZ patients and controls, association with human brain structure, and association with cognitive performance. Finally, we used the convergent functional genomics method to prioritize SCZ risk genes. SNP, single-nucleotide polymorphism; SCZ, schizophrenia; DNAm, DNA methylation; SMR, summary data-based Mendelian randomization; mQTL, methylation quantitative trait loci; eQTL, expression quantitative trait loci GWAS, genome-wide association study.
Discovery mQTL and SCZ GWAS data sets
In the discovery phase, to keep homogeneity in the genetic background, we used SCZ GWAS and mQTL data from individuals of European ancestry. For SCZ GWAS, we utilized the PGC SCZ GWAS data set for individuals of European ancestry, consisting of 35,476 SCZ patients and 46,839 healthy controls [1]. Genome-wide SNP associations were download from the PGC website (http://www.med.unc.edu/pgc/). Considering the high linkage disequilibrium and complexity of the major histocompatibility complex region, we excluded SNPs in this region (chr6:28477797–33448354; GRCh37.p13 version). The blood mQTL data were from the study of McRae et al. [6]. In brief, blood mQTL data originated from two samples, including Brisbane Systems Genetics Study (BSGS, n = 614) and Lothian Birth Cohorts (LBC, n = 1366). DNA were extracted using standard procedures. All individuals were of European ancestry [6]. Meanwhile, brain mQTL data were from a study by Hannon et al. in which mQTLs were characterized in a large collection (n = 166) of human fetal brain samples [7].
Integration of SCZ GWAS and mQTL data
We investigate the association between DNAm and SCZ owing to a shared variant at a locus by an integrated method called Summary data-based Mendelian Randomization (SMR), which utilizes a Mendelian randomization approach to test for joint association in GWAS and mQTL data [3]. In our SMR analysis, DNAm is the exposure (X), SCZ is the outcome (Y), and the top cis-mQTL that is strongly associated with DNAm is used as the instrumental variable (Z). Equivalently, it is an analysis to test whether the effect of Z on Y is mediated by X (a model of Z → X → Y). The significant SMR results could reflect pleiotropic or linkage model. A pleiotropic model reflects gene methylation and SCZ risk are associated owing to a single shared genetic variant. Meanwhile, a linkage model is that there are two or more distant genetic variants in LD-affecting gene methylation and SCZ risk independently. However, linkage was of less-biological interest. Therefore, we used the heterogeneity in dependent instruments (HEIDI) test to distinguish pleiotropy model from linkage model. The HEIDI test compares the profile of association for nearby coinherited variants in GWAS and mQTL data to assess whether the signals are dissimilar. If the HEIDI test is not significant, the identified GWAS and mQTL signals are likely to be driven by the same genetic variant, and overlap can be incidental as a result of pleiotropy. Therefore, to account for multiple testing, we adjusted p-SMR values using the Bonferroni approach. SNPs passed SMR and HEIDI tests were inferred as plausible causal variants. The set of GWAS associated genes was defined as genes with a Bonferroni-corrected p-SMR significant at a threshold of < 0.05 and a p-HEIDI > 0.05.
mQTL data for replication analysis
We also used four well-characterized databases for replication analysis of blood and brain mQTL associations. (1) The first blood mQTL data were from the ARIES mQTL database [8]. Samples were drawn from the Avon Longitudinal Study of Parents and Children. In this study, we only used mQTL data of mothers in middle age (n = 742) [8]. (2) The second blood mQTL data set were from the first- and second-stage data of the study by Hannon et al. [9]. This study performed a two-stage epigenome-wide association study. First- and second-stage samples were from the University College London (n = 639) and Aberdeen (n = 665) case–control samples, respectively. (3) The first brain mQTL data were from a study by Jaffe et al. [7] wherein they characterized mQTL data in the prefrontal cortex from 335 nonpsychiatric controls and 191 SCZ patients [7]. (4) The second brain mQTL data were from a study by Ng et al. [10]. Samples were collected from healthy participants in the Religious Orders Study and the Rush Memory and Aging Project (ROSMAP), two longitudinal studies of aging designed by the same group of investigators. ROSMAP mQTL data were generated from the dorsolateral prefrontal cortex of 468 individuals [10].
SCZ GWAS data for replication analysis
Replication analyses on SCZ used summary statistics from a large-scale SCZ GWAS of the PGC and CLOZUK study [11]. After excluding related and overlapping samples of PGC and CLOZUK study, it comprised of 40,675 cases and 64,643 controls [11]. Genome-wide SNP associations were download from the website of Walters group data repository (http://walters.psycm.cf.ac.uk). Meta-analyses across all samples were conducted using a fixed-effects model with inverse-variance weighting. If the SNPs identified by SMR achieve the genome-wide significance level (P < 5 × 10−8) in the replication analysis, they are likely to be risk variants. Then, we would perform follow-up analyses based on these validated SNPs.
Methylomic pattern of SCZ risk CpG sites
To explore whether SCZ risk CpG sites identified by SMR integrative analysis were differentially methylated in SCZ patients compared with controls, we analyzed publicly available blood (GSE80417 [12] and GSE84727 [12]) and brain (GSE74193 [7]) methylation data sets. DNA methylation data was preprocessed including: quality control on samples, probes filtering, normalization, and converted to M-values. To avoid technical inference, batch effect was corrected by “Combat”. The algorithm ‘projectCellType’ in minfi R package was used to perform cell correction [13]. Differentially methylated probes were accessed by a multiple linear regression model using “limma”, with age and gender as covariates [14]. The P values were adjusted using the Benjamini–Hochberg method.
eQTL analysis
We explored the effects of identified SNPs on expression in eQTL studies using two eQTL databases as follows. (1) Blood eQTL data were from the eQTLGen database. The eQTLGen Consortium has been set up to identify the downstream consequences of trait-related genetic variants. The eQTLGen consortium incorporates 37 data sets, with a total of 31,684 individuals [15]. (2) Brain eQTL data were from the Brain-eMeta data set. The brain eQTL database was from the brain eQTL analysis by Qi et al. (n = 1194) [16]. To increase the power of detecting brain eQTLs, the authors performed a meta analysis using eQTL data from the GTEx [17], CommonMind Consortium [18], and ROSMAP [10].
Association of SCZ risk genes with hippocampal volume and cognitive function
Previous studies have shown that hippocampus volume was smaller in patients with SCZ compared with control and SCZ risk variants were associated with hippocampal volume [19,20,21,22]. Thus, to examine the associations between SCZ risk variants and hippocampal volume, we used a recent GWAS of hippocampal volume conducted by two independent consortia: Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) and Enhancing Neuro Imaging Genetics through Meta Analysis (ENIGMA), comprised of 33,536 individuals of European ancestry [23]. Recent studies have shown that SCZ risk variants were associated with cognitive function in either SCZ patients or healthy control [24, 25]. Therefore, we used two large-scale GWAS data sets to investigate the associations between SCZ risk variants and cognitive function. The first GWAS examined the association between educational attainment and genetic variants and was conducted by the Social Science Genetic Association Consortium (SSGAC, N = 766,345) [26]. The second GWAS was from the Centre for Cognitive Ageing and Cognitive Epidemiology (CCACE) (http://www.ccace.ed.ac.uk/node/335), which performed a study to identify genetic loci influencing cognitive function, including general cognitive function (N = 282,014), reaction time (N = 330,069), and verbal numerical reasoning (N = 168,033) [27].
Prioritization of SCZ risk genes
To prioritize SCZ risk genes, we used the CFG [28] method to combine the above-mentioned evidence. In brief, the CFG method uses multiple layers of evidence to prioritize SCZ risk genes. In this study, evidence used for scoring was as follows: (1) SMR analysis, (2) differential gene methylation (DGM) analysis, (3) eQTL analysis, (4) hippocampal volume association analysis, and (5) cognitive function association analysis. All cross-validating lines of evidence were weighted equally, receiving a maximum of 1 point each, other than the evidence from SMR, DGM, and eQTL (0.5 point if the results were from blood tissues; 1 point if the results are from human postmortem brain). Thus, the maximum possible CFG score for each gene is 5. Furthermore, we limited each line of evidence at 1 point, regardless of how many different results support that line of evidence, to avoid potential ‘popularity’ biases [28]. A higher total score suggests that more evidence from different analyses supports this gene as a SCZ risk gene.
Results
SMR integrative analysis
To explain the logic of the present study, a flow chart describing the analytical methods and movement of genetic variants from one stage of analysis to the next is shown in Fig. 1. First, to identify genes whose changes in DNAm level may confer risk for SCZ, we systematically integrated genetic associations (a total of 15,358,497 SNPs) from the PGC GWAS of SCZ, including 33,640 patients and 43,456 controls, with blood (n = 1980) and brain mQTL (n = 166) [6] data, respectively. After quality control, associations between 86,655 probes from blood mQTL and 7728 probes from brain mQTL were used in the integrated analysis. It should be noted that all probes included in the analysis had at least a cis-mQTL at PmQTL < 5 × 10−8. For each probe, the top associated cis-mQTL was used as the instrument for SMR testing. To control the genome-wide type I error rate, we used Bonferroni correction to account for multiple testing, which resulted in a genome-wide significance level of P < 5.3 × 10−7 (i.e., 0.05/nSMR, with nSMR = 94,383 being the total number of SMR tests using blood and brain mQTL data sets).
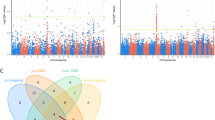
Using the SMR statistical inference method to match the signature of genes from blood and brain mQTL with patterns of association in GWAS, we identified 168 and 33 SCZ susceptibility loci in blood and brain, respectively, whose methylation levels may affect SCZ risk. If a single variant is associated with both phenotype and DNAm, the HEIDI P value will be non-significant. We conducted the HEIDI test and retained for further investigation only those probes with little evidence of heterogeneity PHEIDI ≥ 0.05. After the application of the HEIDI test, 65 gene-trait associations in blood (n = 60) and brain (n = 5) tissues significantly affected risk for SCZ (Fig. 2; Supplementary Table 1).

a SMR test for blood mQTL. b SMR test for brain mQTL. Shown on each y axis are the −log10 (P values) from SMR tests. The red line represents the genome-wide significance level (P < 5.3 × 10−7).
Replication of mQTL effects
Given the myriad confounders in the single mQTL database, we validated the above SNP-DNAm associations using three blood and two brain mQTL data sets. In the blood mQTL analysis, of 60 identified SNP-methylation associations, 15 were validated across all three blood mQTL databases and survived after Bonferroni correction (P < 0.05/n = 2.73 × 10−4, with n = 183 the total number of blood eQTL tests using three data sets; Supplementary Table 2). In the replication analysis of brain mQTL, only one SNP-methylation (rs876701-cg22675791) association was validated in two independent follow-up brain mQTL databases (P < 0.05/n = 5 × 10−3, with n = 10 the total number of brain eQTL tests using two data sets; Supplementary Table 3). Among these SNPs, the SNP rs2743467 affected the DNA methylation levels of two cytosine-phosphate-guanine (CpG) sites in blood tissue, including cg15557168 and cg05082376. Therefore, 15 SNPs (14 SNPs in blood and one SNP in brain) are validated across independent databases and likely to be authentic mQTL SNPs (Supplementary Tables 2, 3).
Replication of GWAS results
Given the replication of significant associations between the 15 risk SNPs and methylation levels of nearby genes, we opted to further analyze these associations with regard to SCZ risk using a meta analysis of PGC and CLOZUK studies (40,675 cases and 64,643 controls) [29]. All the 15 SNPs were validated within the replication samples and attained a genome-wide significance level (P < 5 × 10−8; Supplementary Table 4). Therefore, these 15 SNPs are likely to be risk variants. To investigate the potential roles of risk genes in the pathogenesis of SCZ, we subsequently performed follow-up analyses based on these validated SNP-DNAm combinations, including eQTL analysis, differential methylation/expression analysis, neuroimaging genetics analysis, and cognitive genetics analysis.
Identified SCZ risk SNPs affect expression levels of nearby genes
Considering that DNAm might regulate the expression of nearby genes, we examined the associations between the 15 identified SNPs and expression levels of nearby genes using public blood and brain eQTL databases, respectively. In blood eQTL analysis, all the 14 SNPs identified by SMR analysis in blood were significantly associated with expression level of nearby genes after multiple testing (P < 3.50 × 10−4, i.e., 0.05/n with n = 143; n being the total number of blood eQTL tests; Supplementary Table 5). In brain eQTL analysis, the SNP rs3765971 identified by SMR analysis in brain was significantly associated with expression levels of nearby gene RERE (P < 2 × 10−3, i.e., 0.05/n with n = 25; n being the total number of brain eQTL tests; Supplementary Table 6).
Then, to identify risk genes whose DNAm level changes may affect gene expression, we further applied the SMR approach to assess for pleiotropic associations between DNAm sites and gene expression in blood and brain, respectively. First, we performed SMR analysis using blood eQTL [15] and mQTL [6] data. Totally, 12 DNAm sites in blood were associated with 25 gene expressions at an experimental-wise significance level (PSMR < 1.35 × 10−4, i.e., 0.05/mSMR, with mSMR = 371; mSMR being the total number of SMR tests; Supplementary Table 7). Of the genes that passed the SMR test, 34 DNAm-gene combinations were not rejected by the HEIDI test (PHEIDI > 0.05; Supplementary Table 7). Similarly, we performed SMR analysis using brain eQTL (n = 1194) [16] and mQTL (n = 1980) data. However, no DNAm site was associated with gene expression in brain.
Methylation analysis of SCZ risk CpG sites in patients and controls
SMR infers disease-associated genes under the assumption that DNAm of the candidate genes are altered in patients. Therefore, we performed methylation analysis of the identified CpG sites in SCZ patients and healthy controls using blood and brain GEO data sets, respectively. For risk DNAm sites identified by SMR analysis, three sites shown significantly differential methylation levels across two different GEO databases (Supplementary Table 8) [12], including cg00376283 (PGSE80417 = 4.54 × 10−3; PGSE84727 = 1.36 × 10−5; ABCB9), cg21663219 (PGSE80417 = 2.01 × 10−3; PGSE84727 = 2.08 × 10−14; TNFRSF13C), and cg21782813 (PGSE80417 = 2.81 × 10−3; PGSE84727 = 4.04 × 10−6; MAD1L1) across two blood GEO data sets. Therefore, they are likely to be risk CpG sites of SCZ.
Effects of SCZ risk SNPs on hippocampal volumes and cognitive functions
Among 15 risk SNPs, the SNP rs3765971 was significantly associated with hippocampal volumes (P = 2.58 × 10−4; Supplementary Table 9) after Bonferroni correction (P < 3.33 × 10−3, i.e., 0.05/n, with n = 15). In addition, after Bonferroni correction (P < 6.67 × 10−4, i.e., 0.05/n, with n = 75, calculating with 15 SNPs and 5 cognitive domains), six risk SNPs (rs10460394, rs2675960, rs1107592, rs1790129, rs55742290, and rs2036534) were significantly associated with general cognitive function, three SNPs (rs1727293, rs4148863, and rs7293091) were significantly associated with reaction time, five SNPs (rs2675960, rs1107592, rs55742290, rs2036534, and rs7293091) were significantly associated with verbal numerical reasoning, seven SNPs (rs10460394, rs2675960, rs1107592, rs876701, rs55742290, rs2036534, and rs7293091) were significantly associated with cognitive performance, and four SNPs (rs2675960, rs1107592, rs55742290, and rs2036534) were significantly associated with educational attainment (Supplementary Table 9).
Prioritization of SCZ risk genes
Based on the above findings, we used the CFG method [28] to prioritize identified genes (Table 2). Through integrating evidence at different levels, we found that three SNP-DNAm-gene combinations, rs3765971-cg00546117-RERE, rs7293091-cg21663219-TNFRSF13C, and rs55742290-cg00376283-ABCB9, have the highest CFG scores 2.5, strongly suggesting that they are promising candidates for SCZ risk.
Discussion
Considering that SCZ is an environmentally influenced and complex disease driven by both genetic and epigenetic variation, understanding genetic control of DNAm may highlight regulatory components involved in SCZ pathogenesis. To evaluate putative causal influences of DNAm in SCZ, we used the SMR approach to systematically integrate genome-wide SNP association results from SCZ and mQTL data. Ultimately, we identified and validated 15 risk SNPs, which change DNAm levels of nearby genes and contribute to risk for SCZ. This provides compelling evidence that DNA methylation might have a mediating role for effects at these loci. In addition, to investigate the potential roles of these risk SNPs in the pathogenesis of SCZ, we incorporated data from different levels, including eQTL analysis, differential methylation analysis, and brain structure and cognitive function association analysis. Using the CFG method, we prioritized the risk SNPs and identified three promising candidates for SCZ risk, including rs55742290-cg00376283-ABCB9, rs3765971-cg00546117-RERE, rs7293091-cg21663219-TNFRSF13C and. Our analysis provides opportunities to infer plausible regulatory mechanisms at these risk loci for SCZ that could be prioritized in future functional studies.
For the SNP rs55742290 located in ARL6IP4 gene, it was genome-wide significantly associated with the risk of SCZ. In addition, the SCZ risk allele [C] of SNP rs55742290 in ARL6IP4 gene was also associated with decreased DNAm levels of the cg00376283 site in ABCB9. Moreover, rs55742290 was also the top eQTL in blood; the C allele of this SNP is associated with the high expression levels of C12orf65 gene. Using the SMR method, our results suggested that the C allele of rs55742290 potentially upregulates the transcription of C12orf65 by reducing the methylation level at cg00376283. Subsequently, using Haploreg v4 database [30], we found that the risk SNP rs55742290 located in promoter region and might affect the binding of the transcription initiation complex, resulting in dysregulation of gene expression. Moreover, we found that the SCZ risk allele [C] of SNP rs55742290 was also associated with dysfunction of cognitive performances using several public GWAS databases. Leveraging all the information above, we proposed a potential mechanism in which the risk allele [C] of genetic variant rs55742290 at the promoter of ARL6IP4 gene decreased the DNAm level of cg00376283, which disrupts the binding of repressor(s), increasing the expression of the C12orf65 gene, affecting cognitive performances and increasing the risk of SCZ (Fig. 3).

When the promoter region is highly methylated, which prevents binding of transcription initiation complex (RNA polymerase II, transcription factor protein and mediator proteins) to the promoter region, will form a decrease the transcription. However, when the methylation level of the promoter region is low, transcription initiation complex can more efficiently bind to the promoter, which increases the transcription of C12orf65 genes.
Intriguingly, the risk SNP rs7293091 share similar mechanism, that is, the risk genetic variant rs7293091 affects DNAm levels, dysregulates expression of nearby genes, disrupts cognitive performance, and increases the risk of SCZ. Notably, in the previous SCZ GWAS [1], the SNP rs7293091 in CENPM gene was not genome-wide significantly associated with SCZ (P = 5.33 × 10−8). It might not be found in previous GWAS owing to the lack of power. Our SMR analysis identified this novel functional SNP rs7293091. Hence, the SMR method has identified some of the missing heritability of GWAS data and might contribute to the identification of new genetic factors underlying SCZ.
At the RERE locus, we detected one DNAm site, cg00546117, tagging RERE that was significantly associated with SCZ. The CpG site cg00546117 is in the enhancer regions of the RERE gene according to chromatin state annotations from the Haploreg v4 database [30]. In SMR analysis, the regression coefficient of SCZ on DNAm was − 0.06, indicating that a genetic increase in log2 transformed DNAm of RERE by one standard deviation decreased SCZ by 0.06 standard deviation. Furthermore, the SNP rs3765971 affected expression levels of the RERE gene in both blood and brain tissues. Moreover, expression levels of the RERE gene in the brain were relatively high across developmental stages. The risk allele C of SNP rs3765971 was significantly associated with smaller hippocampal volume and longer reaction time, in accordance with the conventional view that risk alleles generally lead to smaller hippocampal volume and poorer cognition. Taking all the evidence together, we hypothesize a potential mechanism in which the risk allele C of the genetic variant rs3765971 in the RERE gene decreases DNAm levels, upregulates expression of the RERE gene, disrupts cognitive performance, and therefore, increases the risk of SCZ.
These examples above provide hypotheses for how SNPs may affect the SCZ risk through regulatory pathways. However, these putative candidates need to be further validated in functional experiments.
For the other identified SNPs, we identify that they affect the DNAm levels of nearby genes and contribute to the risk of SCZ. In addition, previous studies also indicate that these genes might have a role in the pathogenesis of SCZ. For example, RFTN2 is a member of the raftlin family and mediates clathrin-dependent internalization of toll-like receptor 4 (TLR4) in dendritic cells [31]. Moreover, some studies indicate that TLR4 may play a significant role in neurodevelopment and plasticity [32]. Considering the correlation between RFTN2 and TLR4, RFTN2 may also have a role in SCZ. Therefore, several lines of evidence suggest a plausible regulatory mechanism for the RFTN2 locus, and further functional exploration of RFTN2 is needed. MAD1L1 encodes MAD1 (mitotic arrest deficient-like 1) and contributes to cell cycle control through the regulation of mitosis. GWAS have suggested that it is a susceptibility gene associated with bipolar disorder and SCZ, being involved in reward system functions in healthy adults [33, 34]. The DGKZ gene belongs to the family of diacylglycerol kinase enzymes, which are central to a wide range of signal transduction pathways of potential relevance to neuropsychiatric disorders [35]. PITPNM2 encodes a phosphatidylinositol transfer protein with limited functional information. Whole-exome sequencing of individuals has implicated rare risk variants of PITPNM2 in bipolar disorder [36]. Therefore, the potential role of these genes in the pathogenesis of SCZ requires further exploration.
This study also has several limitations. First, although our findings provide new insights into the genetic mechanism of SCZ, the susceptibility loci were identified in samples of European ancestry. These identified variants might not be associated with SCZ in other ethnic groups. Validation studies in other populations are necessary, not only to investigate whether the identified loci can be generalized to the other ethnicities but also to identify new susceptibility loci for SCZ. Second, in the integrative analysis on SCZ GWAS and mQTL data, the sample size of mQTL data were relatively small. As such, it is possible that some genes that may contribute to SCZ risk but did not meet the genome-wide significance level could have been missed. To ensure a more comprehensive and higher quality mQTL data, the mQTL data of discovery and replication stages could be meta-analyzed. However, the results might be significantly affected by different data process of DNA methylation, such as signal intensities analysis, DNAm normalization, and quality control. Therefore, we validate our findings across several samples. The biases in SNP mQTL associations due to sample size are likely to be very modest. Third, a significant finding of pleiotropic effect from SMR can be explained as a SNP influencing SCZ by modifying a DNAm site (e.g., biological pleiotropy), or it can be explained that the variant is influencing both SCZ and the DNAm site independently (e.g., spurious pleiotropy) [37]. However, the SMR and HEIDI approach could not separate biological pleiotropy from spurious pleiotropy. Therefore, further investigation is needed to examine whether the genetic variant is casually associated with SCZ through affecting the DNAm level.
In summary, our integrative study identifies and validates 15 SCZ risk SNPs, whose DNAm levels may contribute to SCZ risk. The integrated analysis links SCZ risk SNPs of a large-scale GWAS to specific genes, providing a possible mechanistic explanation for genetic variations and SCZ susceptibility.
Funding and disclosure
This study was funded by the National Key Technology R&D Program of China (2016YFC1307000), National Basic Research Program of China (973 Program, 2015CB856404), Clinical Medicine Plus X—Young Scholars Project of Peking University (PKU2018LCXQ008), National Natural Science Foundation of China (81825009, 81221002, and 81901358), Natural Science Foundation of Shandong Province (ZR2019BH001), Medical and Health Science, and Technology Development Plan of Shandong Province (2018WS457), and Supporting Fund for Teachers’ research of Jining Medical University (JYFC2018JS010). All authors report no conflicts of interest.
References
Schizophrenia Working Group of the Psychiatric Genomics C. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–7.
Edwards SL, Beesley J, French JD, Dunning AM. Beyond GWASs: illuminating the dark road from association to function. Am J Hum Genet. 2013;93:779–97.
Zhu Z, Zhang F, Hu H, Bakshi A, Robinson MR, Powell JE, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet. 2016;48:481–7.
Bell JT, Tsai PC, Yang TP, Pidsley R, Nisbet J, Glass D, et al. Epigenome-wide scans identify differentially methylated regions for age and age-related phenotypes in a healthy ageing population. PLoS Genet. 2012;8:e1002629.
Shi J, Marconett CN, Duan J, Hyland PL, Li P, Wang Z, et al. Characterizing the genetic basis of methylome diversity in histologically normal human lung tissue. Nat Commun. 2014;5:3365.
Mcrae A, Marioni RE, Shah S, Yang J, Powell JE, Harris SE, et al. Identification of 55,000 Replicated DNA Methylation QTL. Sci Rep. 2017;8:17605.
Jaffe AE, Gao Y, Deep-Soboslay A, Tao R, Hyde TM, Weinberger DR, et al. Mapping DNA methylation across development, genotype and schizophrenia in the human frontal cortex. Nat Neurosci. 2016;19:40–7.
Gaunt TR, Shihab HA, Hemani G, Min JL, Woodward G, Lyttleton O, et al. Systematic identification of genetic influences on methylation across the human life course. Genome Biol. 2016;17:61.
Hannon E, Dempster E, Viana J, Burrage J, Smith AR, Macdonald R, et al. An integrated genetic-epigenetic analysis of schizophrenia: evidence for co-localization of genetic associations and differential DNA methylation. Genome Biol. 2016;17:176.
Ng B, White CC, Klein HU, Sieberts SK, McCabe C, Patrick E, et al. An xQTL map integrates the genetic architecture of the human brain’s transcriptome and epigenome. Nat Neurosci. 2017;20:1418–26.
Pardiñas AF, Holmans P, Pocklington AJ, Escott-Price V, Ripke S, Carrera N, et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat Genet. 2018;50:381–9.
Hannon E, Dempster E, Viana J, Burrage J, Smith AR, Macdonald R, et al. An integrated genetic-epigenetic analysis of schizophrenia: evidence for co-localization of genetic associations and differential DNA methylation. Genome Biol. 2016;17:176–6.
Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30:1363–9.
Smyth GK limma: Linear Models for Microarray Data. In: Gentleman R, Carey V, Huber W, Irizarry R, Dudoit S, (editors.) Bioinformatics and Computational Biology Solutions Using R and Bioconductor. Springer New York; 2005. p. 397–420.
Võsa U, Claringbould A, Westra H-J, Bonder MJ, Deelen P, Zeng B, et al. Unraveling the polygenic architecture of complex traits using blood eQTL meta-analysis. bioRxiv. 2018:447367.
Qi T, Wu Y, Zeng J, Zhang F, Xue A, Jiang L, et al. Identifying gene targets for brain-related traits using transcriptomic and methylomic data from blood. Nat Commun. 2018;9:2282.
Kubicek S, Dimond A. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;13:307–8.
Fromer M, Roussos P, Sieberts SK, Johnson JS, Kavanagh DH, Perumal TM, et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat Neurosci. 2016;19:1442–53.
Hýža M, Kuhn M, Češková E, Ustohal L, Kašpárek T. Hippocampal volume in first-episode schizophrenia and longitudinal course of the illness. World J Biol Psychiatry. 2016;17:429–38.
Dietsche B, Kircher T, Falkenberg I. Structural brain changes in schizophrenia at different stages of the illness: a selective review of longitudinal magnetic resonance imaging studies. Aust N Z J Psychiatry. 2017;51:500–8.
Jiang Y, Luo C, Li X, Duan M, He H, Chen X, et al. Progressive reduction in gray matter in patients with schizophrenia assessed with mr imaging by using causal network analysis. Radiology. 2018;287:633–42.
Shepherd AM, Laurens KR, Matheson SL, Carr VJ, Green MJ. Systematic meta-review and quality assessment of the structural brain alterations in schizophrenia. Neurosci Biobehav Rev. 2012;36:1342–56.
Hibar DP, Adams HHH, Jahanshad N, Chauhan G, Stein JL, Hofer E, et al. Novel genetic loci associated with hippocampal volume. Nat Commun. 2017;8:13624.
McIntosh AM, Gow A, Luciano M, Davies G, Liewald DC, Harris SE, et al. Polygenic risk for schizophrenia is associated with cognitive change between childhood and old age. Biol psychiatry. 2013;73:938–43.
Lencz T, Knowles E, Davies G, Guha S, Liewald DC, Starr JM, et al. Molecular genetic evidence for overlap between general cognitive ability and risk for schizophrenia: a report from the Cognitive Genomics consorTium (COGENT). Mol psychiatry. 2014;19:168–74.
Lee JJ, Wedow R, Okbay A, Kong E, Maghzian O, Zacher M, et al. Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat Genet. 2018;50:1112–21.
Davies G, Lam M, Harris SE, Trampush JW, Luciano M, Hill WD, et al. Study of 300,486 individuals identifies 148 independent genetic loci influencing general cognitive function. Nat Commun. 2018;9:2098.
Ayalew M, Le-Niculescu H, Levey DF, Jain N, Changala B, Patel SD, et al. Convergent functional genomics of schizophrenia: from comprehensive understanding to genetic risk prediction. Mol psychiatry. 2012;17:887–905.
Lam M, Chen C-Y, Li Z, Martin A, Bryois J, Ma X, et al. Comparative genetic architectures of schizophrenia in East Asian and European populations. Nat Genet. 2019;51:1670–8.
Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic acids Res. 2012;40:D930–4.
Tatematsu M, Yoshida R, Morioka Y, Ishii N, Funami K, Watanabe A, et al. Raftlin controls lipopolysaccharide-Induced TLR4 internalization and TICAM-1 signaling in a cell type-specific manner. J Immunol. 2016;196:3865–76.
Okun E, Griffioen KJ, Mattson MP. Toll-like receptor signaling in neural plasticity and disease. Trends Neurosci. 2011;34:269–81.
Ikeda M, Takahashi A, Kamatani Y, Okahisa Y, Kunugi H, Mori N, et al. A genome-wide association study identifies two novel susceptibility loci and trans population polygenicity associated with bipolar disorder. Mol psychiatry. 2018;23:639–47.
Ruderfer DM, Fanous AH, Ripke S, McQuillin A, Amdur RL, Gejman PV, et al. Polygenic dissection of diagnosis and clinical dimensions of bipolar disorder and schizophrenia. Mol psychiatry. 2014;19:1017–24.
Moskvina V, Craddock N, Holmans P, Nikolov I, Pahwa JS, Green E, et al. Gene-wide analyses of genome-wide association data sets: evidence for multiple common risk alleles for schizophrenia and bipolar disorder and for overlap in genetic risk. Mol psychiatry. 2009;14:252–60.
Lescai F, Als TD, Li Q, Nyegaard M, Andorsdottir G, Biskopsto M, et al. Whole-exome sequencing of individuals from an isolated population implicates rare risk variants in bipolar disorder. Transl psychiatry. 2017;7:e1034.
Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014;23(R1):R89–98.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Yu, H., Cheng, W., Zhang, X. et al. Integration analysis of methylation quantitative trait loci and GWAS identify three schizophrenia risk variants. Neuropsychopharmacol. 45, 1179–1187 (2020). https://doi.org/10.1038/s41386-020-0605-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-020-0605-3
