
Abstract
Nasal spray formulations of naloxone, a mu-opioid receptor (MOR) antagonist, are currently used for the treatment of opioid overdose. They may have additional therapeutic utility also in the absence of opioid agonist drugs, but the onset and duration of action at brain MORs have been inadequately characterized to allow such projections. This study provides initial characterization of brain MOR availability at high temporal resolution following intranasal (IN) naloxone administration to healthy volunteers in the absence of a competing opioid agonist. Fourteen participants were scanned twice using positron emission tomography (PET) and [11C]carfentanil, a selective MOR agonist radioligand. Concentrations of naloxone in plasma and MOR availability (relative to placebo) were monitored from 0 to 60 min and at 300–360 min post naloxone. Naloxone plasma concentrations peaked at ~20 min post naloxone, associated with slightly delayed development of brain MOR occupancy (half of peak occupancy reached at ~10 min). Estimated peak occupancies were 67 and 85% following 2 and 4 mg IN doses, respectively. The estimated half-life of occupancy disappearance was ~100 min. The rapid onset of brain MOR occupancy by IN naloxone, evidenced by the rapid onset of its action in opioid overdose victims, was directly documented in humans for the first time. The employed high temporal-resolution PET method establishes a model that can be used to predict brain MOR occupancy from plasma naloxone concentrations. IN naloxone may have therapeutic utility in various addictions where brain opioid receptors are implicated, such as gambling disorder and alcohol use disorder.
Similar content being viewed by others

Introduction
Naloxone is well established as an antidote for the reversal of respiratory depression in the management of opioid overdose. Recently, formulations of naloxone have become available for intranasal (IN) administration, as the US Food and Drug Administration (FDA) approved 2 and 4 mg doses of IN naloxone for the emergency treatment of known or suspected opioid overdose [1].
Of the opioid receptor subtypes present in the central nervous system (CNS), naloxone binds with the greatest affinity to mu-opioid receptors (MOR) [2]. When administered in usual doses and in the absence of opioid agonists, it exhibits essentially no pharmacologic activity [3].
In addition to management of opioid overdose, IN naloxone may have therapeutic utility in other conditions. In particular, IN naloxone has been proposed for the treatment of pathological gambling (PG), a disorder currently recognized as a “behavioral addiction” (Diagnostic and Statistical Manual of Mental Disorders, Fifth edition). The biological, psychological, social, and cultural background factors behind PG are largely similar to those behind other addiction disorders [4]. Thus, the principles used in the treatment of alcohol dependence may also be largely applied to the treatment of PG. To date, two orally administered opioid antagonist drugs that are used in the treatment of alcohol dependence, naltrexone and nalmefene, have been investigated in subjects with PG [5,6,7,8,9,10]. The results of placebo-controlled trials have so far failed to demonstrate statistically significant differences in outcomes between the treatment arms. These disappointing results, together with a growing body of evidence for the pivotal role of the brain’s opioidergic systems in addictions [11,12,13], highlight the need to investigate the possible utility of IN naloxone in the treatment of PG.
As a possible explanation for their lack of efficacy in PG, naltrexone and nalmefene have rather long durations of action; the drugs remain for a long time in the systemic circulation (nalmefene’s elimination half-life in plasma is 12.5 h, naltrexone’s half-life is 4 h; [14]), and MOR occupancy in the brain is maintained at high levels even longer than one would predict from plasma concentrations [15, 16]. Possible advantages offered by transient MOR antagonism would be the ensuing targeted reduction in MOR availability, and the possibility to maintain important physiological reward functions unimpaired when addiction cues are not present. Gambling disorder as a behavioral addiction is characterized by a very rapid onset of craving that can arise without any significant external stimulus. In this context, for a pathological gambler, a self-administered and short-acting MOR antagonist might be capable of dampening the reward and thus reduce the risk of gambling. For accurate cue-targeted timing of the drug therapy, IN naloxone administration might be useful both because of the rapid entry of the drug into the systemic circulation and subsequently into the CNS, and for its short elimination half-life in plasma (2 h; [17]).
Pharmacokinetic (PK) investigations of novel IN formulations of naloxone indicate that the drug is rapidly absorbed into the bloodstream from the nasal mucosa, with peak plasma concentrations in plasma reached in 20–30 min [17, 18]. Efficient MOR blockade in the brain after IN naloxone has been documented in non-human primates with positron emission tomography (PET) imaging [19], but without detailed information about the time course. Clinical evidence obtained from opioid overdose victims treated with IN naloxone suggests rapid entry of the drug into the CNS, as over 70% of successful rescues occurred in <5 min after dosing [1, 20].
Based on these findings, we hypothesized that IN naloxone readily enters the brain and promptly occupies MORs to a sufficient extent to attain targeted antagonistic effects, while its duration of action would be significantly shorter than that of the orally administered opioid antagonists nalmefene and naltrexone. To test this hypothesis, we characterized the early (0–60 min) and late (300–360 min) phases of brain MOR occupancy in healthy, young male volunteers following IN administration of two doses of naloxone (2 and 4 mg) approved for the treatment of opioid overdose.
Materials and methods
Subjects
Fourteen healthy, young (mean age 25 years, range 20–31 years) male subjects participated after written informed consent; eight in Part 1 of the study, assessing emergence of brain MOR occupancy (0–60 min after dosing of IN naloxone), and six in Part 2 of the study, assessing its disappearance (300–360 min after dosing). Inclusion criteria included good general health, body weight 60–90 kg, and body mass index 18–28 kg m−2. Exclusion criteria included intake of any medication that could affect the outcome of the study, within 2 weeks prior to the tracer administration (2 months for enzyme-inducing drugs like rifampicin or carbamazepine), or <5 times the half-life of the medication, history of drug abuse or positive result in urine drug screen, high alcohol consumption, and regular smoking. Subject candidates with high scores (>2) on the Finnish version of the South Oaks Gambling Screen (SOGS-R) were excluded [21]. The study protocol was approved by the Ethics Committee of South-West Finland Hospital District and the Finnish Medicines Agency (Fimea). Relevant regulations and guidance for biomedical research involving human subjects, such as the Declaration of Helsinki and GCP were adhered to. The study was registered with the EudraCT registration number 2015-002681-23.
Study outline
The study was double blind and placebo controlled, consisting of two parts, each with different participants. Each participant received one IN dose of naloxone (2 mg, n = 7, or 4 mg, n = 7; the 4 mg doses were delivered by administering one 0.1 mL puff into each nostril; 2 mg into either the left or right nostril) and one placebo treatment in a balanced, randomized order. Doses (spray formulation of naloxone hydrochloride developed by Lightlake Therapeutics Inc., UK, 20 mg mL−1, or matching placebo) were given to supine subjects with a Pfeiffer Bidose liquid device. The two sessions were held at least 5 days apart in order to eliminate possible treatment-related effects on MOR availability. In Part 1 of the study, the participants received their IN doses of placebo and naloxone (2 mg, n = 4, or 4 mg, n = 4) during the PET scan, 20 min after the [11C]carfentanil tracer injection. PET scan data were collected until 60 min after the IN drug administration. In Part 2 of the study, placebo and naloxone (2 mg, n = 3, or 4 mg, n = 3) were administered 5 h before [11C]carfentanil and the start of PET scanning.
Structural magnetic resonance imaging (MRI) of the brain was conducted to exclude subjects with anatomical brain abnormalities and to collect anatomical reference data for PET data analysis. T1-weighted MRI scans were acquired using a 3 T scanner (Philips Ingenuity TF PET/MR, Philips Medical Systems, Cleveland, OH, USA).
PK assessments
Venous blood samples were collected for determination of naloxone concentrations in plasma. Samples were collected prior to dosing and at 3, 6, 9, 12, 15, 20, 25, 30, 40, 50, 60, 80, and 100 min, as well as at 2, 3, 4, and 6 h after IN dosing (see Supplementary Methods for sample preparation and analysis). The lower limit of quantitation was 0.10 ng mL−1.
Non-compartmental analysis with WinNonlin® Phoenix 6.4 software (Pharsight Corporation, Mountain View, CA, USA) was used to determine the PK parameters peak concentration (Cmax), time to peak (Tmax), area under the concentration by time curve (AUC) to 6 h (AUC0–6), AUC to the last measurable concentration (AUC0–t), elimination half-life (t1/2), and AUC extrapolated to infinity (AUC0–∞). Terminal time points were selected based on best-fit criteria and review of the selected points. The linear trapezoidal rule was used to estimate AUC.
PET assessments
The PET tracer, [11C]carfentanil, was produced [22] at the radiochemistry laboratory of Turku PET Center on the day of PET imaging. The target radioactivity of each administered bolus injection was 500 MBq, and the carfentanil mass was not allowed to exceed 2.0 µg per injected dose. PET data were acquired with a High-Resolution Research Tomograph (HRRT, Siemens Medical Solutions, Knoxville, TN, USA), and image reconstruction and preprocessing of the data were conducted as reported earlier [23] (see Supplementary Information for details).
Automated regions-of-interest (ROIs) were generated with the help of individual T1 MRI anatomical data and FreeSurfer software (version 5.3; http://surfer.nmr.mgh.harvard.edu/; [24]), and using Statistical Parametric Mapping (SPM8; Wellcome Institute, London, UK; [25]) to generate atlas-based subcortical ROIs, resulting in 60 ROIs [26]. An aggregate average ROI was generated over the regions listed in Table 1 to best reflect the global effect of naloxone in the brain. All analyzed ROIs are additionally reported as Supplementary Information. The lateral occipital cortex served as a reference region, being practically devoid of MORs [27].
ROI-based binding potential as estimated using reference tissue models (BPND; [28]) was considered as the primary outcome parameter in PET modeling. Multilinear reference tissue modeling (MRTM) with fixed k2′ [29, 30] was conducted for BPND estimation in placebo scans and late (Part 2) naloxone scans, while an additional time-dependent term [31] was required for successful modeling of early (Part 1) naloxone scans, as indicated by model selection criteria (Supplementary Information). Model fits derived with the most compatible model were generally of high quality (cf. Figure 1c), whereas visually apparent deviations were observed when poorly complying models were applied for the early naloxone scans (data not shown). Subsequently, dynamic BPNDs were calculated in early naloxone scans as described before [32], yielding dynamic receptor occupancies (RO(t)) as defined in Eq. (1),
where t is the time of BPND estimation under the naloxone treatment. Dynamic receptor occupancies were analyzed to determine peak receptor occupancy (ROmax), and time to reach half of ROmax (tRO/2). Late naloxone scans were analyzed for static receptor occupancy using Eq. (1) without time dependence.

a Plasma naloxone concentrations (mean ± SEM) after intranasal (IN) naloxone administration in healthy male volunteers (n = 7 for 4 mg and n = 7 for 2 mg doses). b Mu-opioid receptor (MOR) occupancies (%) (mean ± SEM) after IN naloxone administration in healthy male volunteers (n = 4 (3) for 4 mg and n = 4 (3) for 2 mg doses in 0–60 min (300–360 min) time intervals) as estimated from [11C]carfentanil-derived BPND data under the placebo and naloxone conditions (see Methods), using linearized-parametric neurotransmitter positron emission tomography (PET) (lp-ntPET) analysis (see Methods). c, d [11C]carfentanil PET-derived standardized time-activity course (TAC) data during the placebo and naloxone conditions in one individual subject (101) after 4 mg dosing of naloxone. Solid lines depict the radioactivity observations from the reference region (occipital cortex), and circles the ones from a relevant brain ROI with high MOR availability (the limbic striatum, average of both sides), and dashed lines depict the best lp-ntPET model fits to the data. Application of the nasal spray formulation occurred 20 min after [11C]carfentanil administration and initiation of PET data acquisition (vertical arrow). Appearance of MOR occupancy by naloxone is observable as a perturbation of the [11C]carfentanil signal pattern in the naloxone scan as compared to the expected pattern observed during the placebo scan
Relationships between PK and PD data
The relationship of the time courses of naloxone concentrations in plasma and brain MOR occupancy was investigated by plotting (GraphPad Software Inc., La Jolla, CA, USA) the log-linear receptor occupancy estimates and drug concentrations in plasma according to each in-scan PK sampling time point (3, 6, 9, 12, 15, 20, 25, 30, 40, 50, and 60 min). Departure of the plots of the early time points from the log-linear regression line of the last (60 min post naloxone) measurement was interpreted as hysteresis in brain MOR occupancy, while the earliest measurement time point exhibiting similar log-linear relationship with the 60 min data was deemed as the start time of equilibrium between plasma naloxone concentrations and brain MOR occupancy.
The drug concentration associated with half-maximal effect (EC50) was estimated using GraphPad by fitting the plasma naloxone concentrations (C) at all in-scan measurement time points after equilibrium (see above) to the estimated occupancy (RO):
where RO′ represents the model-derived maximal occupancy. Theoretically, RO′ should be fixed at 100%, but to account for some uncertainty in the RO estimates, a non-fixed RO′ parameter was employed.
Statistical analysis
The small number of observations per naloxone dose level and time point precluded formal statistical testing of the primary PK and PET (PD) variables. The results of PK and PET data analysis are presented in terms of individual values and descriptive statistics including mean and standard deviation (SD) or median and range calculated with Microsoft Excel (Microsoft Office Professional Plus 2010, Version 14.0).
Results
Naloxone is rapidly absorbed into the bloodstream after IN spray dosing
Naloxone was readily absorbed after IN administration. Its peak concentration in plasma was reached in 15 to 80 min (median Tmax 25 min) after the 2 mg dose and in 12 to 100 min (median Tmax 15 min) after the 4 mg dose (Table 2 and Fig. 1a). Subjects 107 and 108 (Table 2) showed significantly (>3-fold difference from median) delayed absorption of naloxone, but they were, nevertheless, included in the subsequent analysis if not otherwise mentioned. The means (SD) of the observed Cmax concentrations in plasma after 2 and 4 mg IN naloxone were 1.83 (0.70) and 4.33 (3.24)ngmL−1, respectively; the means (SD) of AUC0–t were 166 (55) and 329 (202) min ng mL−1; and the means (SD) of AUC0–∞ were 180 (57) and 349 (208) min ng mL−1, suggesting dose proportionality of the achieved concentrations in plasma. The means (SD) of the elimination half-lives (t1/2) after 2 and 4 mg IN naloxone were 58 (16) and 76 (22) min, respectively, indicating that naloxone was rapidly cleared after IN administration.
IN spray dosing of naloxone results in rapid and sustained displacement of [11C]carfentanil
Within the first 60 min after IN naloxone administration, a marked decline was observed in [11C]carfentanil binding (see Fig. 1c), while administration of placebo failed to elicit any observable perturbation in the [11C]carfentanil signal (see Fig. 1d); this was also confirmed by model selection criteria (Supplementary Information).
Maximum MOR occupancy by naloxone was observed within the first 60 min in a dose-dependent manner, in the range of 54–82% following 2 mg, and in the range of 71–96% following 4 mg doses (Table 2). Receptor occupancy was maintained at these high levels until the end of the 60-min observation period (Fig. 1b). The time taken to achieve 50% occupancy (tRO/2) ranged from 11 to 14 min after 2 mg and from 5 to 13 min after 4 mg doses (Table 2), indicating rapid displacement of [11C]carfentanil from MORs after IN naloxone. Markedly lower MOR occupancy by naloxone was observed at 300–360 min after IN naloxone administration—ranging from 7 to 22% following 2 mg, and from 0 to 20% after dosing of 4 mg (Table 2), suggesting relatively rapid clearance of naloxone from the brain. Based on 60 and 360 min post-dose receptor occupancies, the half-life of disappearance of MOR occupancy was estimated to be ~100 min. There were no clear differences between the brain ROIs in either the time course or magnitude of MOR occupancy (Supplementary Information), supporting the use of a compound ROI for subsequent analysis.
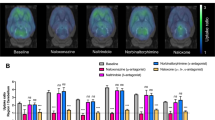
Average spatially normalized maps of BPPlacebo and BPNaloxone, pooled according to the dose level and time after administration, and the corresponding MOR occupancy images (Fig. 2) confirmed global brain MOR blockade following IN naloxone; this had almost disappeared by 5.5 h after drug administration.

Average BPND images (top) of [11C]carfentanil overlaid on an magnetic resonance imaging (MRI) template image, after placebo (first column; n = 14), after 4 mg of naloxone at 0–60 min after administration (second column; n = 4), after 2 mg of naloxone at 0–60 min after administration (third column; n = 4), and after 2 or 4 mg of naloxone at 300–360 min after administration (last column; n = 6). Corresponding occupancy (%) images relative to the placebo condition are shown in the bottom row
Naloxone plasma concentrations predict brain MOR occupancy in a time-dependent manner
The log-linear relationships between naloxone plasma concentrations and MOR occupancy are presented in Fig. 3a. In this analysis, data were pooled from both dose levels and grouped by the post-dose time points of PK and MOR occupancy data collection. Figure 3a demonstrates that plasma naloxone concentrations consistently predicted the brain MOR occupancy, while a departure from the 60 min regression line was evident at sampling time points earlier than 30 min. This observation was supported by a marked rightward shift in the MOR occupancy time course as compared to the time course of naloxone concentrations in plasma, as depicted in Fig. 3b. This was particularly evident in those subjects with fast naloxone absorption, while in those subjects with slow naloxone absorption, the time courses of naloxone in plasma and MOR occupancy in the brain were in good synchrony. Nevertheless, data collected before 30 min after IN naloxone were excluded from the dose-occupancy model calculation shown in Fig. 3c, which yielded an average EC50 estimate of 0.70 ± 0.12 ng mL−1 and an average RO’ estimate of 105 ± 5%.

a Log-linear relationships between naloxone plasma concentrations and mu-opioid receptor (MOR) occupancy (%) data, pooled by the time of measurement. Symbols represent individual data points from the indicated post dose time and lines represent log-linear fit results. b Plasma naloxone concentrations (mean, left axis, solid lines) after intranasal (IN) naloxone (2 mg, n = 6, gray line or 4 mg, n = 6, black line) dosing in healthy male volunteers, and the corresponding occupancy (%) estimates (mean, right axis, lines; broken lines). c MOR occupancy determined as a function of the naloxone plasma concentration at all post-dose time points later than 30 min. The solid line represents the best fit to the two-parameter model indicating an estimated plasma drug concentration associated with half-maximal effect (EC50) of 0.70 ± 0.12 ng mL−1
Discussion
Here, we provide preliminary evidence of fast systemic uptake of naloxone after IN administration in a small dose volume (0.1 mL), associated with a rapid displacement of the high-affinity MOR agonist radioligand [11C]carfentanil in the brain. The ensuing MOR occupancy by naloxone reached high average levels of 67 and 85%, respectively, following IN administration of clinically relevant (2 mg and 4 mg) doses. A slight lag of ~20 min was observed in maximal MOR occupancy relative to the naloxone concentrations in plasma, even if [11C]carfentanil displacement started to emerge promptly after naloxone administration, in accord with the clinical observations of opioid overdose patients being resuscitated within minutes after IN naloxone [20]. It should, however, be noted that our study was not designed to explore the reversion of opioid overdosing, and thus no direct inferences should be made to such conditions. The presence of high concentrations of opioid agonists would be expected to shorten the duration of brain MOR occupancy by naloxone.
If the results obtained from two outlier subjects with slow uptake of naloxone into the circulation are not considered, the present PK results are well in line with those of two earlier PK studies on IN naloxone [17, 18]. For most participants (12 out of 14), the time to reach peak naloxone concentrations in plasma was in the range of 15–30 min, that is, in good concordance with the earlier reports of Tylleskar et al. [18] (range 11–24 min) and Krieter et al. [17] (range 12–60 min). The delayed peak times of 80 and 100 min observed in two participants could be explained by delayed absorption due to sub-optimal delivery of the dose too close to the orifice of the nostril, with subsequent slow absorption. This interpretation is supported by the non-discordant total AUCs of these two outliers (Table 2). Furthermore, total AUCs suggested dose proportionality between the 2 and 4 mg doses, in good concordance with earlier findings [17, 18].
The employed time-sensitive PET analysis technique allowed the assessment of brain MOR occupancy by naloxone at unprecedentedly high temporal resolution. The minute-level temporal resolution of the BPND estimates yielded nearly instantaneous depiction of brain MOR occupancy by naloxone, as compared to average estimates of MOR occupancy in conventional PET studies that typically span over 1–2 h post drug. Nevertheless, the peak occupancies observed in the present study were well in line with earlier estimates [19, 33]. The present estimate of 67% peak MOR occupancy after 2 mg of IN naloxone was in accord with an average MOR occupancy of 80% measured after 2 mg of intravenous (IV) naloxone in humans at 5–65 min post naloxone [33], allowing for higher expected occupancy after IV drug delivery. In non-human primates, Saccone et al. [19] reported 56–66% average MOR occupancy at 10–70 min after IN delivery of naloxone (0.032 mg kg−1), also in good concordance with the present results when considering that peak occupancy is expected to be higher compared to the average occupancy over this time interval. Yet, it should be noted that the anesthetics (ketamine and sevoflurane) administered to the non-human primate study subjects of Saccone et al. [19] may have directly or indirectly affected MOR availability, possibly complicating the comparison.
The present experimental results add evidence to the hypothesized behavior of naloxone in the brain after IN dosing in the presence of negligible agonist concentrations, thus supporting its use as a fast-acting and transient MOR antagonist in the treatment of behavioral addictions such as PG. A growing body of evidence has linked personality traits predisposing to addictions with relative differences in brain MOR availability [34, 35], and individuals with PG have shown blunted opioidergic responses to pharmacological stimuli [36]. These findings indicate that clinical benefit could be derived from opioid antagonist treatment in PG. Thus, even if the long-lasting MOR antagonists naltrexone and nalmefene have failed to show consistent clinical efficacy in PG [5,6,7,8,9,10], targeted IN administration of naloxone may prove clinically feasible because of its ease of delivery, fast brain uptake and relatively short duration of MOR blockade, as shown in the present study. Neuropharmacological treatments of PG have gained increasing interest after a reclassification of PG under the “Substance-related and addictive disorders” section of DSM-5, and future studies are expected to shed light on the neurochemical underpinnings of this particular behavioral addiction that might serve to improve our understanding of other related disorders. In particular, converging evidence highlighting the role of MORs in human emotions (see [37] for a review), in regulation of food intake [38], in eating disorders [22, 23] and even in the pleasure related to physical exercise [39] underlines the importance of better understanding of the function and dysfunction of this system in order to develop new efficacious treatments for various addictions.
At present, IN naloxone is being investigated as treatment of PG in a placebo-controlled, double-blinded study in similar doses (2 or 4 mg) as in the present study, although not using exactly the same nasal spray device (ClinicalTrials.gov Identifier: NCT03430180). Regardless of the outcome of the ongoing clinical trial, it might be motivated to extend the present work to PG populations and larger cohorts in the future, to further investigate the robustness of IN drug delivery and to elaborate the required naloxone dosage for desired MOR occupancy levels in the brain. Nevertheless, pending these experiments, a preliminary dose-occupancy model with an EC50 = 0.70 ± 0.12 ng mL−1 naloxone concentration in plasma was established in the present study, which together with naloxone plasma concentration data derived from future studies may fairly robustly predict the attained brain MOR occupancy levels.
Funding and disclosure
The Ministry of Social Affairs and Health, Helsinki, Finland, funded the study (§52 Appropriation of the Lotteries Act). The Ministry had no role in the study design, analysis, or interpretation of the results presented here or in any phase of the publication process. The study was also supported by an unrestricted grant from Opiant Pharmaceuticals Inc., to the National Institute for Health and Welfare, Helsinki, Finland. HA is principal investigator in a clinical trial evaluating IN naloxone as treatment of PG (ClinicalTrials.gov Identifier: NCT03430180). HA has no conflict of interest in relation to this manuscript. AA was employed by Opiant Pharmaceuticals, Inc. during the course of these studies. RC is a full-time employee and shareholder of Opiant Pharmaceuticals Inc. MS is an employee and shareholder and ZL and MP are employees of CRST Ltd., a clinical research organization that was contracted by the National Institute for Health and Welfare to perform the current study. The other authors declare no competing interests.
References
Skolnick P. On the front lines of the opioid epidemic: rescue by naloxone. Eur J Pharmacol. 2018;835:147–53.
Martin WR. Naloxone. Ann Intern Med. 1976;85:765–8.
Handal KA, Schauben JL, Salamone FR. Naloxone. Ann Emerg Med. 1983;12:438–45.
Hodgins DC, Stea JN, Grant JE. Gambling disorders. Lancet. 2011;378:1874–84.
Kim SW, Grant JE, Adson DE, Shin YC. Double-blind naltrexone and placebo comparison study in the treatment of pathological gambling. Biol Psychiatry. 2001;49:914–21.
Kovanen L, Basnet S, Castrén S, Pankakoski M, Saarikoski ST, Partonen T, et al. A randomised, double-blind, placebo-controlled trial of as-needed naltrexone in the treatment of pathological gambling. Eur Addict Res. 2016;22:70–79.
Toneatto T, Brands B, Selby P. A randomized, double-blind, placebo-controlled trial of naltrexone in the treatment of concurrent alcohol use disorder and pathological gambling. Am J Addict. 2009;18:219–25.
Grant JE, Kim SW, Hartman BK. A double-blind, placebo-controlled study of the opiate antagonist naltrexone in the treatment of pathological gambling urges. J Clin Psychiatry. 2008;69:783–9.
Grant JE, Odlaug BL, Potenza MN, Hollander E, Kim SW. Nalmefene in the treatment of pathological gambling: multicentre, double-blind, placebo-controlled study. Br J Psychiatry. 2010;197:330–1.
Grant JE, Potenza MN, Hollander E, Cynningham-Williams R, Nurminen T, Smits G, et al. Multicenter investigation of the opioid antagonist nalmefene in the treatment of pathological gambling. Am J Psychiatry. 2006;163:303–12.
Williams TM, Daglish MR, Lingford-Hughes A, Taylor LG, Hammers A, Brooks DJ, et al. Brain opioid receptor binding in early abstinence from opioid dependence: positron emission tomography study. Br J Psychiatry. 2007;191:63–69.
Williams TM, Davies SJC, Taylor LG, Daglish MRC, Hammers A, Brooks DJ, et al. Brain opioid receptor binding in early abstinence from alcohol dependence and relationship to craving: an [11C]diprenorphine PET study. Eur Neuropsychopharmacol. 2009;19:740–8.
Heinz A, Reimold M, Wrase J, Hermann D, Croissant B, Mundle G, et al. Correlation of stable elevations in striatal μ-opioid receptor availability in detoxified alcoholic patients with alcohol craving: a positron emission tomography study using carbon 11-labeled carfentanil. Arch Gen Psychiatry. 2005;62:57–64.
Meyer MC, Straughn AB, Lo MW, Schary WL, Whitney CC. Bioequivalence, dose-proportionality, and pharmacokinetics of naltrexone after oral administration. J Clin Psychiatry. 1984;45:15–19.
Ingman K, Hagelberg N, Aalto S, Någren K, Juhakoski A, Karhuvaara S, et al. Prolonged central mu-opioid receptor occupancy after single and repeated nalmefene dosing. Neuropsychopharmacology. 2005;30:2245–53.
Rabiner EA, Beaver J, Makwana A, Searle G, Long C, Nathan PJ, et al. Pharmacological differentiation of opioid receptor antagonists by molecular and functional imaging of target occupancy and food reward-related brain activation in humans. Mol Psychiatry. 2011;16:826–35.
Krieter P, Chiang N, Gyaw S, Skolnick P, Crystal R, Keegan F, et al. Pharmacokinetic properties and human use characteristics of an FDA-approved intranasal naloxone product for the treatment of opioid overdose. J Clin Pharmacol. 2016;56:1243–53.
Tylleskar I, Skulberg AK, Nilsen T, Skarra S, Jansook P, Dale O. Pharmacokinetics of a new, nasal formulation of naloxone. Eur J Clin Pharmacol. 2017;73:555–62.
Saccone PA, Lindsey AM, Koeppe RA, Zelenock KA, Shao X, Sherman P, et al. Intranasal opioid administration in rhesus monkeys: PET imaging and antinociception. J Pharmacol Exp Ther. 2016;359:366–73.
Avetian GK, Fiuty P, Mazzella S, Koppa D, Heye V, Hebbar P. Use of naloxone nasal spray 4 mg in the community setting: a survey of use by community organizations. Curr Med Res Opin. 2018;34:573–6.
Lesieur HR, Blume SB. The South Oaks Gambling Screen (SOGS): a new instrument for the identification of Pathological gamblers. Am J Psychiatry. 1987;144:1184–8.
Karlsson HK, Tuominen L, Tuulari JJ, Hirvonen J, Parkkola R, Helin S, et al. Obesity is associated with decreased mu-opioid but unaltered dopamine D2 receptor availability in the brain. J Neurosci. 2015;35:3959–65.
Majuri J, Joutsa J, Johansson J, Voon V, Alakurtti K, Parkkola R, et al. Dopamine and opioid neurotransmission in behavioral addictions: a comparative PET study in pathological gambling and binge eating. Neuropsychopharmacology. 2017;42:1169–77.
Desikan RS, Ségonne F, Fischl B, Quinn BT, Dickerson BC, Blacker D, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage. 2006;31:968–80.
Ashburner J, Friston KJ. Unified segmentation. Neuroimage. 2005;26:839–51.
Johansson J, Ajalin R, Tuisku J, Karjalainen T, Joutsa J, Scheinin H. Quantification of mu-opioid receptor binding with [11C]Carfentanil PET: a comparison of ROI-methods using test-retest analysis. J Cereb Blood Flow Metab. 2017;37(SI):210–1.
Hirvonen J, Aalto S, Hagelberg N, Maksimow A, Ingman K, Oikonen V, et al. Measurement of central μ-opioid receptor binding in vivo with PET and [11C]carfentanil: a test–retest study in healthy subjects. Eur J Nucl Med Mol Imaging. 2009;36:275–86.
Innis RB, Cunningham VJ, Delforge J, Fujita M, Gjedde A, Gunn RN, et al. Consensus nomenclature for in vivo imaging of reversibly binding radioligands. J Cereb Blood Flow Metab. 2007;27:1533–9.
Ichise M, Liow JS, Lu JQ, Takano A, Model K, Toyama H, et al. Linearized reference tissue parametric imaging methods: application to [11C]DASB positron emission tomography studies of the serotonin transporter in human brain. J Cereb Blood Flow Metab. 2003;23:1096–112.
Wu Y, Carson RE. Noise reduction in the simplified reference tissue model for neuroreceptor functional imaging. J Cereb Blood Flow Metab. 2002;22:1440–52.
Normandin MD, Schiffer WK, Morris ED. A linear model for estimation of neurotransmitter response profiles from dynamic PET data. Neuroimage. 2012;59:2689–99.
Sander CY, Hooker JM, Catana C, Normandin MD, Alpert NM, Knudsen GM, et al. Neurovascular coupling to D2/D3 dopamine receptor occupancy using simultaneous PET/functional MRI. Proc Natl Acad Sci USA. 2013;110:11169–74.
Kim S, Wagner HN, Villemagne VL, Kao PF, Dannals RF, Ravert HT, et al. Longer occupancy of opioid receptors by nalmefene compared to naloxone as measured in vivo by a dual-detector system. J Nucl Med. 1997;38:1726–31.
Karjalainen T, Tuominen L, Manninen S, Kalliokoski KK, Nuutila P, Jääskeläinen IP, et al. Behavioural activation system sensitivity is associated with cerebral mu-opioid receptor availability. Soc Cogn Affect Neurosci. 2016;11:1310–6.
Love TM, Stohler CS, Zubieta JK. Positron emission tomography measures of endogenous opioid neurotransmission and impulsiveness traits in humans. Arch Gen Psychiatry. 2009;66:1124–34.
Mick I, Myers J, Ramos AC, Stokes PRA, Erritzoe D, Colasanti A, et al. Blunted endogenous opioid release following an oral amphetamine challenge in pathological gamblers. Neuropsychopharmacology. 2016;41:1742–50.
Nummenmaa L, Tuominen L. Opioid system and human emotions. Br J Pharmacol. 2018;175:2737–49.
Tuulari JJ, Tuominen L, Boer F, de, Hirvonen J, Helin S, Nuutila P, et al. Feeding releases endogenous opioids in humans. J Neurosci. 2017;37:0976–17.
Saanijoki T, Tuominen L, Tuulari JJ, Nummenmaa L, Arponen E, Kalliokoski K, et al. Opioid release after high-intensity interval training in healthy human subjects. Neuropsychopharmacology. 2018;43:246–54.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Johansson, J., Hirvonen, J., Lovró, Z. et al. Intranasal naloxone rapidly occupies brain mu-opioid receptors in human subjects. Neuropsychopharmacol. 44, 1667–1673 (2019). https://doi.org/10.1038/s41386-019-0368-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-019-0368-x
This article is cited by
-
Dopamine release in human associative striatum during reversal learning
Nature Communications (2024)
-
Clinical Pharmacokinetics and Pharmacodynamics of Naloxone
Clinical Pharmacokinetics (2024)
-
Opioid antagonism in humans: a primer on optimal dose and timing for central mu-opioid receptor blockade
Neuropsychopharmacology (2023)
-
Re-examining Naloxone Pharmacokinetics After Intranasal and Intramuscular Administration Using the Finite Absorption Time Concept
European Journal of Drug Metabolism and Pharmacokinetics (2023)
-
Drug characteristics derived from kinetic modeling: combined 11C-UCB-J human PET imaging with levetiracetam and brivaracetam occupancy of SV2A
EJNMMI Research (2022)
