
Abstract
F17464, a highly potent preferential D3 antagonist, is a novel compound in development for schizophrenia treatment. This phase II, double-blind, randomized, placebo-controlled, parallel-group study in five European countries evaluated the efficacy and safety of F17464, 20 mg twice daily, versus placebo over 6 weeks in patients with acute exacerbation of schizophrenia. Change from baseline to Day 43 of the Positive and Negative Syndrome Scale (PANSS) total score was the primary outcome. The data from 134 randomized patients (67 per group) were analyzed (efficacy/safety). Using analysis of covariance (ANCOVA) after last observation carried forward (LOCF) imputation (primary analysis), the PANSS total score reduction was statistically significantly greater for F17464 than placebo treated subjects at endpoint (p = 0.014); using ANCOVA with Multiple Imputation (MI) method, the between-group difference was in favor of F17464 but did not reach statistical significance. Differences in PANSS positive and general psychopathology subscale score, Marder positive factor score, PANSS response, and PANSS resolution criteria were also statistically significant in favor of F17464 (p values < 0.05) using the LOCF method, with similar results as for the primary analysis using the MI method. Treatment-related adverse events (AEs) were reported in 49.3% and 46.3% of patients on F17464 and placebo, respectively. The most common AEs in F17464 group: insomnia, agitation, and increased triglycerides; worsening of schizophrenia/drug ineffective was less frequent in F17464. Interestingly, no weight gain, no extrapyramidal disorder except rare akathisia were observed under F17464. This 6-week trial demonstrated therapeutic efficacy of 40 mg/day F17464 in improving symptoms of acute exacerbation of schizophrenia with a favorable safety profile.
Similar content being viewed by others


Introduction
Schizophrenia affects 0.5–1.1% of adults worldwide and is usually diagnosed in early adulthood. It is a chronic, debilitating psychiatric disorder, characterized by positive and negative symptoms as well as cognitive disturbances, which often persists despite treatment [1,2,3].
Although the etiology of schizophrenia is unknown, extensive evidence indicates that the psychotic symptoms are owing to excessive subcortical dopaminergic activity, whereas reduced dopaminergic tone in prefrontal cortex may account for the cognitive impairment and negative symptoms [4, 5]. The typical first-generation antipsychotics, mainly high affinity D2 receptor antagonists [6] improve positive symptoms, but often cause debilitating adverse effects, including extrapyramidal (EP) symptoms. Second-generation (atypical) antipsychotics have comparable antipsychotic efficacy but much fewer neurologic symptoms [7,8,9], though they cause, to varying degrees, metabolic effects (weight gain, hyperglycemia, hyperlipidemia), increasing cardiovascular risk [10,11,12], and can thus negatively affect treatment adherence [13]. In addition, available antipsychotics do not have satisfactory efficacy for negative and cognitive symptoms [8, 14].
F17464 is a new compound, with a unique pharmacological profile, preferentially binding to D3 receptors (antagonist, inhibitory constant [Ki] = 0.17 nM) versus D2 receptors (weak partial agonist, Ki = 6.5–12.1 nM) and with a comparatively high affinity for 5-HT1A receptors (partial agonist; Ki = 0.16 nM) [15]. The homology of D2 and D3 receptors suggests that D3 selective antagonists may be effective antipsychotics for the treatment of schizophrenia, but by virtue of their anatomic distribution within limbic structures of the ventral striatum (as opposed to the dorsal striatum) [16, 17], they could be expected to cause less motor effects than D2 antagonists. Moreover, preclinical studies suggest that D3 antagonists could correct dopamine tone deficiency in the prefrontal cortex [18] and hereby have the potential to improve cognition [19,20,21].
The affinity of F17464 for 5-HT1A receptors may also support its potential for pro-cognitive effects [22, 23].
Thus, a preferential D3 antagonist has the potential to selectively reduce dopaminergic hyperactivity in subcortical areas and indirectly improve the dopaminergic hypoactivity in cortical areas, whereas stimulating cortical 5-HT1A receptors may improve cognition.
To evaluate its efficacy and safety and potentially favorable clinical profile, we conducted a Proof-of-Concept study to evaluate 40 mg/day of oral F17464 in comparison to placebo over 6 weeks in patients with acute exacerbation of schizophrenia.
Methods
Participants
Eligible patients were 18–64 years old with a diagnosis of schizophrenia undergoing an acute exacerbation with prominent “active phase” symptoms. The diagnosis was confirmed by the Diagnostic and Statistical Manual of Mental Disorders 4th edition (DSM-IV) [24] criteria using the related Mini-International Neuropsychiatric Interview (MINI 6.0) [25]. Maximum two antipsychotics were prescribed for the current acute episode for < 2 weeks. Participants had been diagnosed with schizophrenia for ≥ 1 year and ≤ 5 years; the upper limit was removed in a protocol amendment after recruitment of 71 patients to enable inclusion of a broader population. A Clinical Global Impression Severity (CGI-S) [26, 27] score ≥ 4, a Positive and Negative Syndrome Scale [28] (PANSS) score ≥ 70 and < 120 and a rating of at least 4 (moderate) on ≥ 2 of the following PANSS positive symptoms: delusions, hallucinatory behavior, conceptual disorganization, suspiciousness, persecution were required. Additional inclusion criterion was no significant improvement (< 20% reduction) in PANSS positive score between enrollment and inclusion.
Patients were excluded if they were in their first acute episode of psychosis, currently had predominant negative symptoms or were “refractory”, defined as a lack of significant improvement despite treatment with ≥ 3 different antipsychotics for an adequate duration (≥ 4 weeks) and at adequate dosage during the previous 5 years.
Study design
This international, multicenter, double-blind, randomized, placebo-controlled, parallel-group, fixed-dose design, proof-of-concept Phase II study was conducted in 30 hospitals/clinics in five countries (France (2 centers), Hungary (3), Latvia (4), Romania (12) and Russia (9)). Patients were enrolled in two phases: the first 71 patients completing the study were included in an interim analysis for futility, performed by an Independent Data Monitoring Committee to decide whether to continue the study or to stop it.
The study (NCT02151656) was approved by the appropriate independent Ethics Committees and was conducted in accordance with principles of Good Clinical Practice (ICH-E6) [29] and the Declaration of Helsinki, and applicable national regulations in biomedical research. Participants provided written informed consent prior to participation.
Eligible participants were randomized (1:1) to F17464 or placebo. The computer-generated, internally validated randomization list was established by the Sponsor. At inclusion visit (Day 1) the Investigator allocated a treatment number to each patient (based on his/her chronological order of arrival at this visit). Study blinding was ensured by identical external aspect of the F17464 and placebo capsules and by their identical presentation and administration regimen.
Study treatment was administered orally, twice daily with meals (morning and evening): two 10 mg-capsules (20 mg) were taken per administration, for a total daily dose of 40 mg, and a total duration of 6 weeks. During the study patients had to abstain from alcohol and cannabinoids. As recommended by the European Medicine Agency [30], lorazepam and oxazepam were authorized benzodiazepines (BZDs) as rescue medications (for anxiety, agitation, or insomnia) permitted for ≤ 7 consecutive days and ≥ 4 h prior to efficacy assessments. Prescribed authorized BZDs taken at a stable dose for ≥ 3 months before the study and treatment for EP symptoms were also permitted.
Study assessments
Participation involved 10 visits over the study, inpatient screening (Days −7 to −2), including enrollment, baseline assessments and wash-out of previous antipsychotic drugs (within 7 days before first study drug administration), and a 6-week treatment period with patients hospitalized up to Day 22, and then discharged if they had a Clinical Global Impression of Improvement (CGI-I) score ≤ 3 (“at least minimally improved”), which was re-evaluated daily up to Day 29 as necessary. Visits were performed on Days 1 (randomization), 4, 8, 15, 22, 29 ± 2, 36 ± 2, and 43 ± 2 (end of treatment). Follow-up (FU) visit was to be performed for up to 7 days after the last study drug intake for safety assessments.
Primary efficacy based on the PANSS total score was assessed at each visit by trained and certified personnel with standardization of the inter-rater scoring using the Structured Clinical Interview for the PANSS [31]. An independent external expert performed blinded PANSS reviews for the quality control of the primary criterion measurements. CGI-S was evaluated at enrollment and on Days 1, 22, 43, and FU and CGI-I was evaluated at each post-baseline visit after starting treatment.
Blood sampling was performed for plasma F17464 concentration measurement (11 samples per patient between Day 8 and Day 43), including pre-dose samples at each visit and 5 post-dose samples after morning study drug administration on Day 15. Blinded plasma prolactin levels were measured at baseline, Days 8, 15, 22, 43, and FU.
Safety was assessed by analysis of adverse events (AEs), physical examination (including vital signs, weight, body mass index, waist circumference), electrocardiograms (ECGs) and laboratory investigations (hematology and blood chemistry). The following scales specifically assessed EP symptoms (Barnes Akathisia Rating Scale—BARS [32], Simpson-Angus Scale—SAS [33], and Abnormal Involuntary Movement Scale—AIMS [34, 35]), depression (Calgary Depression Scale for Schizophrenia—CDSS [36]) and suicidal ideation (Columbia-Suicide Severity Rating Scale—C-SSRS [37]). Concomitant medications, including antipsychotic medications were recorded.
Efficacy criteria
The primary outcome was change from baseline to Day 43 in PANSS total score. Secondary outcomes included changes from baseline to Day 22 in PANSS total score and to Day 43 in PANSS sub-scores (positive symptoms, negative symptoms, general psychopathology), Marder factors scores [38] and PANSS response (20, 30, and 50%), time to first sustained PANSS response, PANSS resolution (rating of ≤ 3 for delusions, unusual thought content, hallucinatory behavior, conceptual disorganization, mannerism/posturing, blunted affect, social withdrawal, and lack of spontaneity), CGI-S and CGI-I scores at Day 43.
Statistical analysis
Sample size was based on the number of patients needed to detect a difference between groups (standard deviation [SD]) of 8 (19) to give an effect size of 0.42 [39]: 142 patients were required to achieve 80% power with a significance level set to 5% in a one-sided condition.
For the primary criterion, the null one-sided hypothesis tested was H01 = F17464 is not superior to placebo, versus H11 = F17464 is superior to placebo, and if H01 was rejected, a second hypothesis was tested: H02 = there is no difference between groups, versus H12 = F17464 is different to placebo. The primary analysis was performed on the full analysis set (FAS; patients receiving ≥ 1 drug dose with PANSS total score measured at baseline and at least once by Day 43) using an analysis of covariance (ANCOVA) model after last observation carried forward (LOCF) imputation of missing values, with treatment group as fixed factor; PANSS total score at baseline and country were covariates. Supportive analyses were conducted on the per-protocol (PP) set (FAS patients without major protocol deviation and receiving study drug for ≥ 7 days) using the primary ANCOVA model and on the FAS using an ANCOVA combined with Rubin’s multiple imputation (MI) [40]. A treatment by country interaction analysis was also performed.
For secondary criteria, the changes from baseline to Day 22 in total PANSS score and to Day 43 in PANSS subscales scores and each Marder factor score were analyzed as for the primary criterion (LOCF and MI). The PANSS response criteria and PANSS resolution criterion were analyzed using logistic regression with LOCF imputation and using a MI-logistic analysis.
Time to first sustained PANSS response was analyzed using survival curves according to the Kaplan–Meier method and compared between groups using the Gehan test. CGI-S and CGI-I scores were analyzed using logistic regression (LOCF imputation) and the Cochran–Mantel–Haenszel test; PANSS total score and CGI-S at baseline and country were covariates.
Pharmacokinetic (PK) parameters (including maximum plasma concentration (Cmax), time to maximum plasma concentration (tmax) and area under the plasma concentration time curve between two drug administrations (AUCτ)) were determined on the PK set (patients receiving ≥ 1 drug dose and with ≥ 1 PK sample) on Day 15.
Prolactin levels (values and changes from baseline) were analyzed on the safety population. The numbers of patients with hyperprolactinaemia (>20 ng/mL for males and > 25 ng/mL for females) were recorded and categorized by levels of severity [41].
Results
After the study completion, one study center was found to have violated Good Clinical Practice (GCP) standards. Consequently, data from this center (10 randomized patients) were excluded from the database for the final analysis and are therefore also excluded from this manuscript.
Patients
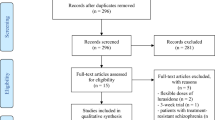
Between 27 August 2014 and 22 December 2015, 134 patients were randomized (67 each to F17464 or placebo) and received the study treatment, 81 completed 6 weeks of treatment (Fig. 1).

Study flow chart. AE: adverse event, excl: excluding, PK: pharmacokineticsor
More patients on placebo versus F17464 discontinued treatment (44.8% versus 34.3%). This difference in attrition rates began in the first week of the out-patient treatment phase and was maintained throughout. Main treatment discontinuation reasons were lack of efficacy and AE ‘worsening of schizophrenia’ (29.9% with placebo and 16.4% with F17464, respectively).
Demographic and baseline disease characteristics were generally similar across treatments; 59.0% of patients were male and the mean (SD) age was 37.3 (11.5) years: most patients had the diagnosis of paranoid schizophrenia (94.8%), with a mean (SD) time since diagnosis of 5.60 (5.05) years and a mean (SD) PANSS total score at baseline of 88.9 (9.1) (Table 1).
More patients in the F17464 versus placebo group were “moderately ill” on the CGI-S (53.7% versus 31.3%); with mirror percentages for the categories “markedly ill” or “severely ill” having fewer patients in the F17464 group versus placebo.
Efficacy
All 134 randomized and treated patients were included in the FAS; 20 were excluded from the PP set (10 each on placebo or F17464) owing to major protocol deviations (19 patients) or insufficient exposure (1 patient) (Fig. 1). Global compliance was similar with placebo (98.9%) and F17464 (97.9%). Unless otherwise stated results with LOCF imputation of missing values are presented.
PANSS total score
For the primary endpoint, change from baseline to Day 43 in PANSS total score, the observed mean (SD) change was −16.2 (16.0) on F17464 versus −11.5 (14.4) on placebo, giving an adjusted between-group difference (standard error; SE) of −6.2 (2.5), statistically significant in both the one-sided test (95% CI: [-α; −2.07], p < 0.01) and two-sided test (95% CI: [−11.08; −1.27], p < 0.02) (Fig. 2). There was no significant treatment by country interaction but a statistically significant country effect (p < 0.003); the highest treatment effect was for Russia (adjusted mean (SE) −9.2 (4.36)). The primary results were supported by analysis on the PP set (−6.6 (2.5), p < 0.01 in both tests). For the ANCOVA with MI the between-group difference (SE) of −4.4 (3.2) was in favor of F17464 but did not reach statistical significance.

Mean (SEM) PANSS total score change from baseline over time after LOCF Imputation (FAS *p < 0.05; **p < 0.01)
The mean reduction in total PANSS score in the F17464 group was greater than in the placebo group at Day 15 and the between-group difference was statistically significant from Day 22 (p < 0.02) (Fig. 2).
For PANSS response, more patients with F17464 (26.9%) versus placebo (14.9%) showed a 30% improvement at Day 43: the difference was statistically significant (p < 0.05), with an odds ratio (OR) estimate for treatment effect 2.6-fold higher with F17464 (95% CI: 1.05; 6.53); the treatment effect with the MI-ANCOVA analysis was in favor of F17464 (OR 2.2-fold higher with F17464, 95% CI: 0.90; 5.28), but not statistically significant (p < 0.10). Similar results were obtained for 20% improvement (49.3% versus 32.8% patients with F17464 and placebo, respectively, p < 0.02) with an OR 2.7-fold higher with F17464 (95% CI: 1.25; 5.95). The treatment effect with MI-ANCOVA analysis (OR 2.2-fold higher with F17464, 95% CI: 1.03; 4.71) was also statistically significant (p = 0.046). A 50% response was observed in 3 patients with F17464 and none with placebo.
A shorter time to first sustained PANSS response was observed for Fl7464 versus placebo; the probability of a sustained 20 or 30% PANSS response was 15–20 and 10–15% higher with F17464 than placebo, respectively, from Day 21 onwards (Fig. 3). These results indicate that once the symptoms reduction was achieved, it was maintained until the end of treatment. PANSS resolution at Day 43 was 53.7% with F17464 and 35.8% with placebo: the difference was statistically significant (p < 0.05) with an OR 2.1-fold higher with F17464 (95% CI: 1.01; 4.24).

Time to first sustained PANSS 20% (a) and 30% (b) response (LOCF)—KM (FAS)
Regarding the use of BZDs, allowed as rescue medication, there was no difference in the number of patients who used BZDs (59.7% with placebo; 58.2% with F17464), the median percentage of days of BZD intake relative to the treatment duration was higher in the placebo group (60.1%) than the F17464 group (51.1%). This did not jeopardize the ability to the study to show superiority of F17464 over placebo.
PANSS subscales scores
Statistically significant decreases for F17464 vs. placebo from baseline to Day 43 in PANSS positive score (p = 0.03; non-parametric test) and PANSS general psychopathology score (adjusted mean difference (SE) of −3.6 (1.3), p < 0.01) were observed. The decrease for F17464 vs. placebo from baseline to Day 43 in PANSS-negative score (mean reduction of −2.0 vs. −1.0) did not reach statistical significance.
Analyses of changes from baseline to Day 43 in the five PANSS Marder factors (negative, positive, cognitive, excitation, and depression) showed a greater mean reduction with F17464 than placebo, which was statistically significant for the Marder positive factor (adjusted mean difference (SE) of −2.1 (0.8), p < 0.02). The treatment effect for each PANSS score and Marder factor with the MI-ANCOVA analysis was in favor of F17464 but not statistically significant.
A post hoc analysis using the Wallwork factors of the PANSS items [42], confirmed a statistically significant difference in favor of F17464 on the positive factor and in addition on a factor corresponding to cognitive function (Disorganization/Concrete factor: P2, N5, G11), (adjusted mean difference (SE): −0.85 (0.40), p = 0.03).
CGI-S and CGI-I
On day 43, more patients with F17464 than placebo were considered improved on CGI-S categories “mildly” or “borderline” mentally ill together, either on the FAS (56.7% versus 37.3%), or on the observed values (74% and 46%) in each group, respectively, and on CGI-I responders with “very much” or “much” improved categories together (43.3% versus 29.9%, respectively). Although the between-group difference was clinically relevant, it did not reach the statistical significance probably owing to the imbalance baseline distribution of the different categories, which did not favor F17464 group (globally less severe than placebo), whereas the treatment effect was higher in the subgroup of “markedly” and “severely” ill patients (results not presented).
Pharmacokinetics
On Day 15 when steady-state was reached: mean Cmax was 2682 ng/mL, with high interindividual variability (86%, owing to one patient with a very low plasma level although evaluated as sufficiently compliant) and mean AUCτ was 30917 h.ng/mL, with moderate-to-high interindividual variability (47%).
Safety
Mean (SD) exposure was similar for both groups: 31.7 (12.6) days with placebo and 33.5 (12.9) days with F17464. The mean duration of hospitalization was 21.04 (5.34) days in the F17464 group and 21.55 (5.84) days in the placebo group.
The incidence of treatment-emergent AEs (TEAEs) was slightly higher for patients receiving F17464 (70.1%) than placebo (61.2%) (Table 2); the incidence of treatment-related TEAEs was similar between groups (49.3% with F17464; 46.3% with placebo). The most common TEAEs (>5% patients) with a higher incidence (>2 patients’ difference) with F17464 than placebo were reported under the preferred terms of schizophrenia (reported term: “worsening of schizophrenia”), insomnia (9.0%; 19.4%), agitation (6.0%; 10.4%) and blood triglycerides increased (3.0%; 7.5%) with F17464 and placebo, respectively. Of note, akathisia was reported in 3 (4.5%) patients with F17464 and no patients with placebo and was reversible (per BARS scores) while continuing treatment. Drug ineffective and anxiety were reported with a lower incidence with F17464 than placebo. As there was no strict definition for reporting the inefficacy-related events (schizophrenia or drug ineffective), it was considered relevant to pool data for these two AEs, giving an incidence of lack of efficacy-related TEAEs of 19.4% with F17464 and 32.8% with placebo.
With both F17464 and placebo, most TEAEs were mild (31.3% vs. 22.4%) or moderate (29.9% in both groups) in severity.
During study treatment, a similar proportion of patients had ≥ 1 authorized benzodiazepine intake (58.2% with F17464; 59.7% with placebo).
The incidences of serious AEs and AEs leading to treatment discontinuation were lower with F17464 than placebo (Table 2). All serious AEs were lack of efficacy-related AEs (14.9% and 22.4% on F17464 and placebo, respectively). the main reason for treatment discontinuation was lack of efficacy or AE “worsening of schizophrenia” (16.4% and 29.9%, respectively). In addition, treatment was discontinued in the F17464 group for one patient with hepatic enzyme increased and one with suicide ideation, depression, and anxiety, and in the placebo group for one patient with blood bilirubin increased, one with vomiting and one with EP disorder. All these events resolved with corrective treatments after stopping the study drug.
There were no clinically relevant differences between groups in laboratory results, ECGs, blood pressure or heart rate abnormalities reported as AEs, physical examinations or when considering abnormal scores obtained in the rating scales (BARS, AIMS, SAS, CDSS, or C-SSRS). Most of abnormalities were either isolated or resolved whilst continuing treatment.
High prolactin values at baseline were recorded for 29.9% and 49.3% patients with F17464 and placebo, respectively. During the study, values decreased with placebo (by 26–34 ng/mL in females and 6–13 ng/mL in males on average), returning to normal values in almost all patients with abnormal baseline values (69.7%). With F17464, prolactin levels increased in males (by 11–12 ng/mL on average) and were stable up to the end of treatment; in females they increased at Days 8 and 15 (by 13 and 12 ng/mL on average, respectively) before returning to near baseline levels at Days 22 and 43.
Changes from normal prolactin values at baseline (≤ 25 ng/mL and ≤20 ng/mL for females and males, respectively) to clinically relevant last values on treatment (>75 ng/mL), were reported with F17464 for three females only and in no female patients on placebo. No hyperprolactinemia-related symptoms were reported during the short exposure to treatment.
Discussion
This 6-week, placebo-controlled study in acutely exacerbated patients with schizophrenia demonstrated the antipsychotic efficacy of F17464, a new preferential D3 antagonist antipsychotic agent.
The NEWMEDS merged the largest data set of individual patient data from 29 randomized placebo-controlled studies of second-generation antipsychotics conducted in adult schizophrenia patients by five large pharmaceutical companies [43]. The study patient population was similar to the NEWMEDS population, with respect to schizophrenia type and severity of the episode (based on PANSS total score), but there were more female patients in this study (41% versus 27% in the NEWMEDS population) [43].
A statistically significant reduction of the PANSS total score after 6 weeks of treatment (primary criterion) was observed with F17464 versus placebo in the FAS and confirmed in the PP set. The MI-based method, assuming a random missingness process that cannot be verified, confirmed the quantitative results of the primary criterion, but did not show a statistical difference between groups; this could be owing to a higher placebo response than in the primary analysis. There was no significant treatment by country interaction, which confirms the primary analysis model validity. The observed low variability could be explained by the efforts to ensure PANSS assessment quality, including training, regular retraining and certification of raters, and blinded quality checks performed by an external reviewer, leading to remediation sessions. In addition, BZDs, taken as rescue medication by a similar proportion of patients in both groups, did not jeopardize the ability to the study to show superiority of F17464 over placebo. The reduction in PANSS total score with F17464 versus placebo was observed after 2 weeks and was statistically significant from 3 weeks onwards. Once the symptoms reduction was achieved, it was maintained until the end of treatment. The effect of F17464 on the PANSS positive and general psychopathology scores, the Marder positive factor of PANSS and on PANSS response and resolution rates confirmed the superiority of F17464 over placebo (secondary efficacy analyses). Trends in favor of F17464 were observed for other Marder factors (cognitive, depression and excitation). There was no statistically significant difference between groups in PANSS negative score and Marder negative factor: this was expected as the study population (acute exacerbation; patients with predominant negative symptoms were not eligible) and study design (short 6-week duration) would not be adequate for testing a potential effect on negative symptoms [30]. A post hoc analysis was performed using the Wallwork factors of the PANSS items; considering the disorganization factor which includes mainly items reflecting cognitive abilities, it was significantly more improved with F17464 than with placebo, indicating a beneficial effect on cognitive functions.
Although results of the CGI-S and CGI-I scores indicated clinically relevant lower severity and increased treatment response after treatment with F17464 versus placebo, differences were not statistically significant. The discrepancy in CGI-S versus PANSS results may be explained by the unbalanced distribution across CGI-S classes at baseline (patients were globally less severe with F17464 than placebo) combined with the higher treatment effect observed in “markedly” and “severely” ill patients. It has been indeed well-established that, the greater the baseline severity, the greater the magnitude of the differences is between active treatment and placebo [44].
In a PET-scan study performed in healthy volunteers, F17464 at a single dose of 15 or 30 mg robustly (> 80%) occupied D3 receptors with little (< 20%) D2 receptor occupancy [15]. These data suggest that the positive results reported here are accompanied by a full D3 receptors occupancy.
In total, 39.6% patients prematurely discontinued study treatment (34.3% with F17464 and 44.8% with placebo) owing to lack of efficacy/worsening of schizophrenia, AEs, or withdrawal of consent. These results are comparable with those obtained in controlled studies with second-generation antipsychotics [43, 45]. The higher incidence of treatment discontinuation with placebo was visible from Day 28 onwards, corresponding to patient hospital discharge.
The short wash-out period of previous antipsychotics could explain the high prolactin levels at baseline (29.9% with F17464 and 49.3% with placebo) and the general decrease observed during the study with placebo. F17464 was associated with an increase in prolactin levels during the study and hyperprolactinemia was more frequent and more marked in female patients (clinically relevant last on treatment values (> 75 ng/mL) but asymptomatic, reported in only three females). Antipsychotics are known to increase prolactin levels, generally in a dose-dependent manner, in both healthy controls and patients with schizophrenia, with greatest effects in females [46].
The overall incidence of TEAEs was slightly higher with F17464 than placebo; the most common TEAEs were related to lack of efficacy and were more frequent in the placebo group (19.4% on F17464 and 32.8% on placebo). TEAEs reported more frequently with F17464 included insomnia, agitation, blood triglycerides increased and akathisia, a known antipsychotic class effect, which was nevertheless rare and reversible whilst continuing treatment. TEAEs reported more frequently with placebo included anxiety and EP disorder.
Among SAEs and AEs leading to treatment discontinuation (both of lower incidence with F17464), most were related to lack of efficacy.
Overall the efficacy of F17464 in improving symptoms in patients with acute exacerbation of schizophrenia was demonstrated versus placebo over 6 weeks. F17464 exhibited a favorable safety profile, with no weight gain, no EP disorder except rare akathisia, supporting the hypothesis that a D3 preferential antagonist is an effective antipsychotic and could provide incremental therapeutic benefits for patients with schizophrenia as compared with currently marketed medications.
Funding and disclosure
Institut de Recherche Pierre Fabre provided funding for the study and editorial support for the manuscript.
I Bitter was the International Coordinating Investigator for the study. F Gaudoux, M Groc, C Delsol, L Barthe, C Fabre, M Fagard, A Montagne, and F Tonner are employees of the Institut de Recherche Pierre Fabre (IRPF); V Brunner, R Chavda, and P Sokoloff were employees of IRPF at the time of their involvement in the study.
I Bitter reports personal fees, in the past 3 years, from Angelini, EGRIS (European Group into Research in Schizophrenia), Eli Lilly, Janssen/Janssen-Cilag, Gedeon Richter, and Servier outside the submitted work.
J. Lieberman had, in the past 3 years, grant/research with Alkermes, Boehringer Ingelheim, Lilly/DeNovo, Taisho Pharmaceutical R&D, Inc., Teva, and was member of advisory boards of Intracellular Therapies, and Pierre Fabre, however, he received no direct financial compensation or salary support for participation in these research, consulting, or advisory board activities. He authored a patent with Repligen, and he received royalties from SHRINKS: The Untold Story of Psychiatry.
References
Andreasen NC, Olsen S. Negative v positive schizophrenia. Definition and validation. Arch Gen Psychiatry. 1982;39:789–794.
Regier DA, Narrow WE, Rae DS, Manderscheid RW, Locke BZ, Goodwin FK. The de facto US mental and addictive disorders service system. Epidemiologic catchment area prospective 1-year prevalence rates of disorders and services. Arch Gen Psychiatry. 1993;50:85–94.
Saha S, Chant D, Welham J, McGrath J. A systematic review of the prevalence of schizophrenia. PLoS Med. 2005;2:e141.
Laruelle M. Dopamine transmission in the schizophrenic brain. Schizophrenia, 2nd Edition. Steven R. Hirsch, Daniel R. Weinberger (eds). Blackwell Science Ltd, 2003.
McGowan S, Lawrence AD, Sales T, Quested D, Grasby P. Presynaptic dopaminergic dysfunction in schizophrenia: a positron emission tomographic [18F]-fluorodopa study. Arch Gen Psychiatry. 2004;61:134–142.
Schotte A, Janssen PF, Gommeren W, Luyten WH, Van Gompel P, Lesage AS, et al. Risperidone compared with new and reference antipsychotic drugs: in vitro and in vivo receptor binding. Psychopharmacol (Berl). 1996;124:57–73.
Correll CU, Leucht S, Kane JM. Lower risk for tardive dyskinesia associated with second-generation antipsychotics: a systematic review of 1-year studies. Am J Psychiatry. 2004;161:414–425.
Leucht S, Corves C, Arbter D, Engel RR, Li C, Davis JM. Second-generation versus first-generation antipsychotic drugs for schizophrenia: a meta-analysis. Lancet. 2009;373:31–41.
Pakpoor J, Agius M. A review of the adverse side effects associated with antipsychotics as related to their efficacy. Psychiatr Danub. 2014;26:273–284.
American Diabetes Association, American Psychiatric Association, American Association of Clinical Endocrinologists, North American Association for the Study of Obestiy. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27:596–601.
Breden EL, Liu MT, Dean SR. Metabolic and cardiac side effects of second generation antipsychotics: what every clinician should know. J Pharm Pract. 2009;22:478–488.
Rojo LE, Gaspar PA, Silvia H, et al. Metabolic syndrome and obesity among users of second-generation antipsychotics: a global challenge for modern psychopharmacology. Pharmacol Res. 2015;101:74–85.
Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353:1209–1223.
Buchanan RW. Persistent negative symptoms in schizophrenia: an overview. Schizophr Bull. 2007;33:1013–1022.
Sokoloff P, Abi-Dargham A, Slifstein M, Martel JC, Heusler P, Leriche L, Girgis RR, et al. Translational imaging activities supporting the development of F17464: a new antipsychotic with preferential D3 antagonist/5-HT1A partial agonist properties. Neuropsychopharmacology. 2016;41:S234.
Meador-Woodruff JH, Scott P, Damask BS, et al. Dopamine receptor mRNA expression in human striatum and neocortex. Neuropsychopharmacology. 1996;15:17–29.
Gurevich EV, Joyce JN. Distribution of dopamine D3 receptor expressing neurons in the human forebrain: comparison with D2 receptor expressing neurons. Neuropsychopharmacology. 1999;20:60–80.
Sokoloff P, Le Foll B. The dopamine D3 receptor, a quarter century later. Eur J Neurosci. 2017;45:2–19.
Millan MJ, Fone K, Steckler T, Horan WP. Negative symptoms of schizophrenia: clinical characteristics, pathophysiological substrates, experimental models and prospects for improved treatment. Eur Neuropsychopharmacol. 2014;24:645–92.
Gross G, Drescher K. The Role of Dopamine D3 Receptors in Antipsychotic Activity and Cognitive Functions. Handb Exp Pharmacol. 2012;213:167–210.
Nakajima S, Gerretsen P, Takeuchi H, Caravaggio F, Chow T, Le Foll B, et al. The potential role of dopamine D3 receptor neurotransmission in cognition. Eur Neuropsychopharmacol. 2013;23:799–813.
Newman-Tancredi A, Kleven MS. Comparative pharmacology of antipsychotics possessing combined dopamine D2 and serotonin 5-HT1A receptor properties. Psychopharmacol (Berl). 2011;216:451–473.
Sumiyoshi T, Higuchi Y, Uehara T. Neural basis for the ability of atypical antipsychotic drugs to improve cognition in schizophrenia. Front Behav Neurosci. 2013;7:1–8.
Diagnostic and statistical manual—text revision (DSM-IV) Washington DC, American Psychiatric Association, 1994.
Sheehan DV, Lecrubier Y, Sheehan KH, et al. “The Mini-International Neuropsychiatric Interview (M.I.N.I.): the development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10”. J Clin Psychiatry. 1998;59:22–33.
Guy W. Clinical Global Impression scale [CGI]. ECDEU assessment manual for psychopharmacology. Rev. Rockville, MD: U.S. National Institute of Health, Psychopharmacology Research Branch; 1976;125–126.
Busner J, Targum SD. The clinical global impressions scale: applying a research tool in clinical practice. Psychiatry (Edgmont). 2007;4:28–37.
Kay SR, Fiszbein A, Opler LA. The positive and negative syndrome scale (PANSS) for schizophrenia. Schizophr Bull. 1987;13:261–276.
Good Clinical Practice (GCP) Guideline. ICH-E6, 1996.
Committee for Medicinal Products for Human Use (CHMP). Guideline on clinical investigation of medicinal products, including depot preparations in the treatment of schizophrenia. In: EMA (ed). VolEMA/CHMP/40072/20120 Rev.1, 2012.
Opler LA, Kay SR, Lindenmayer JP, Fiszbein A. The Structured Clinical Interview for the Positive and Negative Syndromes of Schizophrenia. New York, NY: MultiHealth Systems; 1992.
Barnes TR. A rating scale for drug-induced akathisia. Br J Psychiatry. 1989;154:672–676.
Simpson GM, Angus JW. A rating scale for extrapyramidal side effects. Acta Psychiatr Scand Suppl. 1970;212:11–19.
Guy W. Abnormal involuntary movement scale [AIMS]. ECDEU assessment manual for psychopharmacology. Rev. . Rockville, MD: U.S. National Institute of Health, Psychopharmacology Research Branch; 1976:534–537.
Rush JA. Handbook of psychiatric measures. American Psychiatric Association. 2000:166–168.
Addington D, Addington J, Schissel B. A depression rating scale for schizophrenics. Schizophr Res. 1990;3:247–251.
Posner K, Brent D, Lucas C, Gould M, Stanley B, Brown G et al. Columbia Suicide Severity Rating Scale (C-SSRS). Research Foundation for Mental Hygiene, Inc. Vol Version 1/14/09, 2008.
Marder SR, Davis JM, Chouinard G. The effects of risperidone on the five dimensions of schizophrenia derived by factor analysis: combined results of the North American trials. J Clin Psychiatry. 1997;58:538–546.
FDA - Center for Drug Evaluation and Research website. NDA for lurasidone—Clinical Review, 2010.
Rubin DB. Multiple imputation for nonresponse in surveys. New York, NY: Wiley; 1987.
Serri O, Chik CL, Ur E, Ezzat S. Diagnosis and management of hyperprolactinemia. CMAJ. 2003;169:575–581.
Wallwork RS, Fortgang R, Hashimoto R, Weinberger DR, Dickinson D. Searching for a consensus five-factor model of the Positive and Negative Syndrome Scale for schizophrenia. Schizophr Res. 2012;137:246–50.
Rabinowitz J, Werbeloff N, Caers I, Mandel FS, Stauffer V, Menard F, et al. Determinants of antipsychotic response in schizophrenia: implications for practice and future clinical trials. J Clin Psychiatry. 2014;75:e308–316.
Furukawa TA, Levine SZ, Tanaka S, Goldberg Y, Samara M, Davis JM, Cipriani A, Leucht S. Initial severity of schizophrenia and efficacy of antipsychotics - participant-level meta-analysis of 6 placebo-controlled studies. JAMA Psychiatry. 2015;72:14–21.
Kemmler G, Hummer M, Widschwendter C, Fleischhacker WW. Dropout rates in placebo-controlled and active-control clinical trials of antipsychotic drugs: a meta-analysis. Arch Gen Psychiatry. 2005;62:1305–1312.
Peuskens J, Pani L, Detraux J, De Hert M. The effects of novel and newly approved antipsychotics on serum prolactin levels: a comprehensive review. CNS Drugs. 2014;28:421–453.
Acknowledgements
We acknowledge the study participants, the investigators, Silvia Gatti-McArthur for data interpretation; Jean-Claude Martel, Pierre Fabre Laboratories, and Hannah Bartrum, Scinopsis (Fréjus, France) for editorial support.
Author information
Authors and Affiliations
Contributions
IB, the international study coordinating investigator, was involved in the design of the study, in the data/results interpretation, and was involved in the conduct of the study from implementation to discussion of clinical questions. JL as a member of Pierre Fabre Advisory Board for F17464 project reviewed the study protocol, interpreted the study results, and contributed to manuscript development. FG and PS were responsible for the study concept and contributed to study implementation. MG was involved in data analysis and interpretation. RC was involved in drafting the study protocol and clinical aspects of the study conduct. CD, CF, and MF were responsible for the study overall management from its set-up to its closure to ensure quality, patient safety, and data and analyses reliability. LB and VB were involved throughout the study in the pharmacokinetics objective (study design, data collection, data analysis and interpretation). AM was involved in data interpretation and drafting the study report. FT was involved in designing the study, leading clinical aspects of the study conduct and data interpretation. All authors were involved in the decision to submit the manuscript for publication and reviewed and approved its scientific content.
Corresponding author
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bitter, I., Lieberman, J.A., Gaudoux, F. et al. Randomized, double-blind, placebo-controlled study of F17464, a preferential D3 antagonist, in the treatment of acute exacerbation of schizophrenia. Neuropsychopharmacol. 44, 1917–1924 (2019). https://doi.org/10.1038/s41386-019-0355-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-019-0355-2
This article is cited by
-
Amisulpride augmentation therapy improves cognitive performance and psychopathology in clozapine-resistant treatment-refractory schizophrenia: a 12-week randomized, double-blind, placebo-controlled trial
Military Medical Research (2022)
-
Existing and emerging pharmacological approaches to the treatment of mania: A critical overview
Translational Psychiatry (2022)
-
Progress and Pitfalls in Developing Agents to Treat Neurocognitive Deficits Associated with Schizophrenia
CNS Drugs (2022)
-
Developments in Biological Mechanisms and Treatments for Negative Symptoms and Cognitive Dysfunction of Schizophrenia
Neuroscience Bulletin (2021)
-
Trace Amine-Associated Receptor 1 as a Target for the Development of New Antipsychotics: Current Status of Research and Future Directions
CNS Drugs (2021)
