
Abstract
GLI1 fusions involving ACTB, MALAT1, and PTCH1 genes have been recently reported in a subset of malignant soft tissue tumors with characteristic monomorphic nested epithelioid morphology and frequent S100 positivity. However, we encountered a group of morphologically similar soft tissue tumors lacking the canonical GLI1 gene fusions and sought to investigate their genetic abnormalities. A combined approach including RNA sequencing, targeted exome sequencing and FISH methodologies were used to identify potential novel genetic abnormalities. Ten patients (five females, five males) with an age range of 4–65 years (median 32.5) were identified. Tumors were located in the soft tissues of the limbs, trunk and head and neck, with one each in the tongue and lung. Histologically, tumors revealed ovoid to epithelioid cells arranged in a distinctive nested-trabecular pattern, separated by thin septa and a delicate vascular network. Two cases showed areas of increased nuclear pleomorphism and focal fascicular spindle cell growth. Four tumors showed a high mitotic count (≥15/10 HPFs), with necrosis seen in three of them. Lymphovascular invasion was noted in two cases. No consistent immunoprofile was detected, with positivity for CD56 (six cases), S100 (four cases), SMA (two cases), and pan-CK (one case). FISH showed GLI1 (12q13.3) gene amplification in all 10 cases, with co-amplification of CDK4 (12q14.1) in nine (90%) and MDM2 (12q15) in eight (80%) cases. Targeted exome sequencing performed in three cases confirmed the GLI1, CDK4, and MDM2 co-amplification. Only one case showed the presence of both GLI1 break-apart and amplification, although no gene partner was detected. Our findings suggest that GLI1 amplification, often associated with co-amplifications of CDK4 and MDM2 genes, may represent an alternative genetic mechanism of GLI1 oncogenic activation akin to GLI1 fusions, defining the pathogenesis of an emerging group of malignant soft tissue tumors with a distinctive nested growth pattern and variable immunoprofile.
Similar content being viewed by others

Introduction
GLI1 gene fusions were first described in ‘pericytomas with t(7;12) translocation’ resulting in ACTB-GLI1 gene fusion. The tumors had a monomorphic ovoid cytomorphology arranged in a distinctive perivascular distribution and showed immunoreactivity for smooth muscle actin and laminin, suggestive of ‘pericytic’ differentiation [1]. Subsequently, additional cases with a similar morphology and genetic alteration were reported, including one case each in the bone and stomach [2, 3]. GLI1 gene fusions were later described in two unrelated tumors: plexiform fibromyxoma and gastroblastoma, both occurring within the gastric wall [4,5,6]. More recently, our group identified GLI1 fusions involving various gene partners (ACTB, MALAT1, and PTCH1) in a subset of malignant soft tissue tumors with a characteristic monomorphic nested epithelioid morphology and frequent S100 immunoreactivity [7]. As we have encountered a group of soft tissue tumors with similar morphologic features but lacking GLI1 gene fusions, in this study we applied a combined molecular methodology approach to identify their genetic alterations.
Methods
Patient selection
Archival material and personal consult files of the senior author (CRA) were searched for cases resembling the histologic features of the recently described soft tissue tumor entity characterized by GLI1 fusions [7], but lacking the canonical gene fusions by FISH or other methodologies. Specifically, we selected tumors with a monomorphic cytomorphology composed of round, epithelioid to ovoid cells, with scant to moderate amount of cytoplasm and arranged in a distinctive nested growth pattern, separated by delicate fibrous septa containing an arborizing capillary network. The study group was analyzed for demographic information, anatomic site, tumor size, and morphologic features, including cell type (round, spindle, and mixed), degree of cellularity, type and amount of stromal component, nuclear features, mitotic activity, and presence of necrosis. Available immunohistochemical stains were reviewed and additional work-up was performed in retrospect based on the molecular results. The clinical follow-up information was obtained from review of the electronic medical records and from contacting referring pathologists and clinicians. The study was approved by the Institutional IRB.
Fluorescence in situ hybridization (FISH)
Formalin-fixed paraffin-embedded tissues were available in each case for FISH analysis. FISH for GLI1 gene abnormalities (break-apart, amplification) was performed on all cases, using a set of five flanking custom bacterial artificial chromosomes (BAC) (three centromeric, two telomeric), as detailed in Supplementary Table 1. As GLI1 and DDIT3 are neighboring genes separated only by 50 kb, the telomeric bacterial artificial chromosomes applied for GLI1 also interrogated DDIT3 gene. Subsequent FISH assays were performed for MDM2 and CDK4 gene copy number alterations. Five cases were also tested for HMGA2 gene abnormalities. Samples were considered positive for amplification when one of the following signal patterns of amplification was observed: HSR-homogeneous-stained-region, DM-double-minutes, ring chromosomes, and MDA-multiple dot-like amplicons in various sizes.
If GLI1 rearrangement was identified, additional FISH for the various fusion partners reported, including ACTB1, MALAT1, and PTCH1 was performed [7]. FISH was performed on 4-µm thick formalin-fixed paraffin-embedded (FFPE) tissue sections. Custom probes were made by bacterial artificial chromosomes clones (Supplementary Table 1) flanking the target genes, according to UCSC genome browser (http://genome.ucsc.edu) and obtained from BACPAC sources of Children’s Hospital of Oakland Research Institute (Oakland, CA; http://bacpac.chori.org). DNA from each bacterial artificial chromosome was isolated according to the manufacturer’s instructions. The bacterial artificial chromosome clones were labeled with fluorochromes by nick translation and validated on normal metaphase chromosomes. The slides were deparaffinized, pretreated, and hybridized with denatured probes. After overnight incubation, the slides were washed, stained with DAPI, mounted with an antifade solution, and then examined on a Zeiss fluorescence microscope (Zeiss Axioplan, Oberkochen, Germany) controlled by Isis 5 software (Metasystems, Newton, MA).
Targeted RNA sequencing (RNA-seq) and analysis
RNA was extracted from formalin-fixed paraffin-embedded tissue using Amsbio’s ExpressArt FFPE Clear RNA Ready kit (Amsbio LLC, Cambridge, MA) in two cases. Fragment length was assessed with an RNA 6000 chip on an Agilent Bioanalyzer (Agilent Technologies, Santa Clara, CA). RNA-seq libraries were prepared using 20–100 ng total RNA with the Trusight RNA Fusion Panel (Illumina, San Diego, CA). Each sample was subjected to targeted RNA sequencing on an Illumina MiSeq at eight samples per flow cell (approximately 3 million reads per sample). All reads were independently aligned with STAR(ver 2.3) and BowTie2 against the human reference genome (hg19) for Manta-Fusion and TopHat-Fusion analysis, respectively. The GLI1 mRNA expression level was evaluated and compared to those of other samples analyzed on the same targeted RNA sequencing platform, including three GLI1-fusion-positive soft tissue tumors, and three dedifferentiated liposarcomas (one with GLI1-co-amplifications and two without GLI1 amplifications).
MSK-IMPACT assay
Details of the MSK-IMPACT assay have been previously published [8]. Briefly, MSK-IMPACT is a comprehensive molecular profiling assay that involves hybridization capture and deep sequencing of all exons and selected introns of up to 468 oncogenes and tumor-suppressor genes, allowing the detection of point mutations, small and large insertions or deletions, and rearrangements. In addition to capturing all coding regions of the genes, the assay also captures >1000 intergenic and intronic single-nucleotide polymorphisms (tiling probes), interspersed homogenously across the genome, aiding the accurate assessment of genome-wide copy number. In total, the probes target approximately 1.2 megabases of the human genome.
Results
Clinicopathologic findings
Ten patients were identified, including five females and five males, with an age range of 4–65 years (median 32.5). (Table 1) Tumors were located in the thigh [2], shoulder, elbow, forearm, finger, back, neck, tongue, and lung. Histologically, tumors revealed a monomorphic appearance of round to epithelioid cells arranged in nests, cords, and reticular patterns, often surrounded by a rich capillary network. The lesional cells showed scant to moderate amount of amphophilic to light eosinophilic or clear cytoplasm, uniform round nuclei with fine chromatin and inconspicuous nucleoli (Figs. 1–3). A myxoid background was noted in one case, where the tumor cells were arranged in a reticular pattern (case 3). Two cases showed areas of increased nuclear pleomorphism and focal fascicular spindle cell growth, in relative sharp contrast from the epithelioid nests (Fig. 2). One case showed an unusual pattern with tumor cells arranged in rosette-like pattern (Fig. 3). Five tumors showed a low mitotic activity (2-5/10 HPFs), one tumor showed mitotic activity of 10 per 10 HPF, while the remaining four had a high mitotic count (≥15/10 HPFs). Necrosis was seen in three tumors, all of which showed increased mitotic activity. Lymphovascular invasion was seen in two cases (Fig. 2).

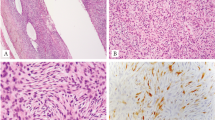
Pathologic and FISH findings of GLI1-amplified tumors. A-B: Images from Case 1 (4/F, shoulder). A well-circumscribed lesion with distinct lobulated architecture (A) showing monomorphic ovoid cells arranged in a cords separated by delicate capillaries (B). C-I: Images from Case 2 (10/M, finger) of a superficial dermal nodule with a nodular growth pattern (C), in areas protruding into dilated vascular spaces (D) and showing tumor cells with clear cytoplasm in a nested arrangement (E). Immunohistochemical stains for CDK4 (F) and MDM2 (G) were strongly positive. FISH studies showing (H) amplification and break-apart of GLI1 gene (green- 5’-centromeric separated from red-3’-telomeric, arrow; the latter red signal being also being amplified) and (I) Co-amplifications of CDK4 and MDM2 genes (orange-MDM2, red-CDK4, arrow)

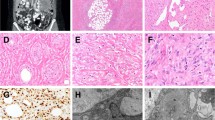
Morphologic and immunohistochemical spectrum of GLI1-amplified tumors. A–C Case 6 (39/M, neck) showed a biphasic histology with the epithelioid nested growth (A) in abrupt transition with the spindle cell component arranged in short fascicles (B); immunohistochemical stain for SMA (C) showing diffuse positivity. D–F Case 7 (51/F, back) showing (D) bland epithelioid cells arranged in single files in a collagenous stroma. Other areas showed more solid growth with increased atypia and mitotic activity (E). Immunohistochemical stain for S100 (F) showing patchy strong positivity. G–I Images from Case 8 (54/F, elbow) showing distinctive multinodular growth (G), which on high power reveals a tight nested growth of uniform epithelioid cells (H). Immunohistochemical stain for CDK4 (I) showing strong and diffuse positivity

Morphologic and immunohistochemical spectrum of GLI1-amplified tumors. Case 4 (23/F, thigh) showing tumor lobules with cribriform or distinctive rosette-like pattern, separated by a fibrotic stroma (A), with high power also showing cord-like and trabecular pattern (B); C–D Case 5 (26/F, lung) showing tumor cells in a trabecular pattern with intervening sinusoidal pattern. Immunohistochemical stain for CD56 (D) showing positivity. E– I Case 10 (65/M, tongue) showed a compact growth, with vague nested growth of epithelioid cells separated by fibrotic stroma; tumor cells were uniform and showed clear cytoplasm (E). Diffuse positivity for MDM2 (F), CDK4 (G) and STAT6 (H) immunohistochemical stains was noted. FISH showed GLI1 gene (green-centromeric, red-telomeric) amplification (arrows) (I)
Immunohistochemical stains showed no consistent immunoprofile, (Supplementary Table 2) although most cases showed CD56 positivity, which was seen in six of the 10 tumors (Fig. 3). S100 staining was focally positive in four of the 10 cases (Fig. 2). Smooth muscle actin positivity was seen in two and keratin in one of the 10 cases. CDK4 positivity was seen in five cases tested, MDM2 was positive in four of the five cases tested (Figs. 1–3), while STAT6 positivity was seen in two of eight cases tested (Fig. 3).
GLI1 gene amplification detected by FISH and targeted next-generation sequencing
FISH and/or targeted sequencing studies (MSK-IMPACT) showed GLI1 (12q13.3) gene amplification in all 10 cases. Co-amplification of neighboring genes CDK4 (12q14.1) was noted in nine (90%) and co-amplification of MDM2 (12q15) was seen in eight (80%) cases. Targeted exome sequencing was performed in three cases, confirming the GLI1, CDK4, and MDM2 gene amplifications. Case 2, in addition to the GLI1 amplification, showed a GLI1 gene break-apart. (Fig. 1) However, none of the known GLI1 gene partners (ACTB, MALAT1, and PTCH1) were rearranged by FISH and tissue was insufficient for RNA-seq analysis. One case (case 4) showed only GLI1 gene amplification without co-amplification of CDK4 and/or MDM2 genes (Table 1). HMGA2 on 12q14.3 was tested by FISH in five cases (cases #1–3, 8, 10) and showed co-amplification.
No other significant copy number alterations were identified in the three cases on which targeted exome sequencing was performed. In addition to the 12q regional amplifications, case 6 showed gains/low level amplifications of the 8p11.2 region, case 7 showed gains in chromosome 18 and case 4 did not show any other copy number alterations.
GLI1-amplified tumors showed similar GLI1 upregulation to tumors with GLI1 gene fusions
RNA-seq, performed on two cases, showed overexpression of GLI1 mRNA in both cases (Fig. 4). The upregulated levels of GLI1 mRNA overexpression were similar to those seen in the three GLI1-fusion-positive tumors (involving ACTB and PTCH1 gene partners) [7], and higher than the mRNA levels seen in a dedifferentiated liposarcoma with GLI1/MDM2/CDK4 co-amplifications (Fig. 4), analyzed on the same platform. In contrast, two dedifferentiated liposarcomas showing only MDM2/CDK4 amplifications had low GLI1 mRNA expression. Based on the MSK-IMPACT results of the three GLI1-amplified tumors, the average fold change for GLI1 gene was 5.7.

GLI1 mRNA upregulation in tumors with GLI1 genetic abnormalities. A high GLI1 mRNA overexpression was present in cases with both GLI1 amplifications (cases’ 3 and 8) and GLI1 fusions (3 cases) [7]. One dedifferentiated liposarcoma with GLI1/CDK4/MDM2 co-amplification showed a lower level of upregulation. In contrast 2 dedifferentiated liposarcomas with MDM2/CDK4 amplification and other sarcoma types showed no increased GLI1 mRNA levels. Higher levels of STAT6 and HMGA2 expression were noted in cases with GLI1 amplification suggesting their co-amplification gene status; in contrast to tumors with GLI1 fusions. The genomic positions and relationship of the genes located on the 12q13-15 locus (lower panel)
A subset of dedifferentiated liposarcomas shows concurrent GLI1/MDM2/CDK4 amplifications
As dedifferentiated liposarcomas are characterized by recurrent MDM2 and CDK4 co-amplifications, we investigated the incidence of GLI1 amplification in these tumors by reviewing the MSK-IMPACT data. To date, 42 of 184 (24.7%) dedifferentiated liposarcomas showed amplifications of GLI1 gene, along with CDK4 and MDM2 gene amplifications, with the average fold change for GLI1 amplification was 5.9, which was significantly lower than the 10.9 average fold change for CDK4 gene amplification and 13.0 for MDM2 gene amplification (Supplementary Fig. 2). Upon review, none of these 42 dedifferentiated liposarcomas with GLI1 gene co-amplifications showed histologic overlap to that of our study group. However, during case selection of the study group, we came across five unusual cases of dedifferentiated liposarcoma, which showed, at least in part, a striking morphologic similarity to our current cohort, all of which showed GLI1 gene co-amplification by FISH (Supplementary Table 3). Most of these cases were sent in consultation with a presumed diagnosis of GLI1-fusion-positive tumor. Three of these cases occurred in the retroperitoneum and two in the paratesticular area. One patient (DDLS1) initially presented with a finger mass that showed ovoid to spindle cells in a reticular pattern, in a myxoid background. Targeted RNA sequencing showed the presence of co-amplifications of GLI1, CDK4, and MDM2 genes, which were further confirmed by FISH. Within a short time span (2 months), imaging work-up revealed lung and retroperitoneal masses, resection of which showed features of dedifferentiated liposarcoma, with areas of well differentiated liposarcoma component, while the dedifferentiated component showing similar nested morphology to that seen on the initial finger lesion (Supplementary Fig. 1). The remaining cases (DDLS2-DDLS5), presenting in the retroperitoneum and paratesticular areas, showed a predominant dedifferentiated component composed of primitive epithelioid to ovoid cells arranged in a nested and trabecular growth, in a rich vascular background, reminiscent of the features described in the study group cases. Areas of well-differentiated liposarcoma component were quite minimal in all cases (less than 5%) and were often composed of scattered atypical multinucleated stromal cells infiltrating the adjacent soft tissues (Supplementary Fig. 1).
Follow-up
Follow-up information was available on three patients (cases# 4, 6, and 7), ranging from 16 to 36 months. Case 4 had no evidence of disease after 36 months. Case 6 developed lung metastasis from a neck primary and was alive with disease at 26 months follow-up. Case 7 developed a local recurrence following a marginal resection and had no evidence of disease at 16 months. Four cases were recent cases and no follow-up data were available on the remaining two patients.
Discussion
GLI1-fusion-positive tumors were initially reported by Dahlen et al. in 2004 in the context of an unusual tumor entity designated as ‘pericytoma with t(7;12) translocation’, resulting in an ACTB-GLI1 gene fusion [1]. Three of their five cases were located in the tongue, with one each in the stomach and calf. A handful of subsequent similar cases were reported in the bone [2] and stomach [3]. These tumors had a uniform cytomorphology of spindle cells with scant clear cytoplasm and ovoid to tapered nuclei, with a distinct perivascular distribution around thin-walled arborizing vessels. Based on these morphologic features and consistent actin immunoreactivity, these tumors were thought to have a pericytic line of differentiation, although distinct and likely not related to the more common perivascular/pericytic tumors such as glomus tumor/myopericytoma [9, 10]. None of these cases were reported to show immunoreactivity for cytokeratin or S100 protein, and all seemed to follow a benign clinical course, without evidence of metastatic potential.
Subsequently, an identical MALAT1-GLI1 gene fusion was identified in two distinct gastric mesenchymal neoplasms, namely: gastric plexiform fibromyxoma [5] and gastroblastoma [4]. Plexiform fibromyxoma occurs in young patients and is usually composed of a multinodular growth of bland myofibroblast-like cells, associated with a fibromyxoid stroma and a rich capillary network. This tumor is positive for smooth muscle actin and follows a benign course [5, 6]. Gastroblastoma, in contrast, is a biphasic neoplasm occurring in young adults, composed of a fascicles or solid sheets of primitive spindle cells, and clusters or cords of epithelial cells occasionally forming glandular structures with small lumens or rosette-like structures [11]. These tumors show positivity for cytokeratin in all cases, patchy smooth muscle actin and CD56 in some cases. Gastroblastomas do have a metastatic potential, with two patients documented with metastatic disease to the liver, lymph node, and peritoneum [4].
In a recent study [7], we reported recurrent GLI1 gene fusions involving a number of gene partners in a unique and possibly novel soft tissue tumor characterized by a distinctive epithelioid nested phenotype and frequent, but not consistent, S100 immunoreactivity. The initial study included six cases from four females and two males, with a wide age range at diagnosis (16–70 years) and spanning various anatomic locations. Most cases showed ACTB or MALAT1 gene rearrangements, while one case showed a novel PTCH1-GLI1 gene fusion. Microscopically, all tumors showed monomorphic round to epithelioid cells in a distinctive nested growth pattern, separated by delicate stroma and capillary network. Although some tumors were reminiscent of glomus tumor, the main differential diagnosis, also suggested by the diffuse and strong S100 expression in four of the cases, was a soft tissue myoepithelial tumor. Moreover, this novel soft tissue tumor was associated with a malignant clinical course, three patients developing lymph node metastasis and one recurred distantly with lung metastasis.
The current study was triggered by a number of cases, which shared a similar morphology to the phenotype described in soft tissue tumors with GLI1 fusions [7], but lacking these defining genetic abnormalities by FISH. Thus, we have identified 10 cases, which show GLI1 amplifications, detected by a number of genetic approaches, resulting in GLI1 upregulation. All except one lacked GLI1 gene rearrangements by FISH. One case showed an unbalanced GLI1 break-apart and amplification; however, FISH was negative for all the GLI1 known fusion partners. The patients had a wide age range (4–65 years) at diagnosis, with a median age of 32.5 years, presenting with tumors at various locations, including soft tissues of the limbs, trunk, head, and neck, and one case each in the tongue and lung.
Morphologically, GLI1-amplified tumors showed similarities to the previously described GLI1-fusion-positive tumors [1, 7]. Although the overriding morphology noted was that of monomorphic ovoid to epithelioid cells in a nested to trabecular pattern, with intervening thin septa with delicate capillary network, this current subset showed a wider histological spectrum, spanning from morphologically banal to overtly malignant. Among the additional architectural patterns noted, one case showed distinct rosette-like formation, reminiscent of a primitive neuroepithelial phenotype. Two cases showed a biphasic histology, with the epithelioid component being arranged in the typical nested architecture, in abrupt transition with a bland spindle cell component arranged in a fascicular pattern. Two cases showed a nested epithelioid growth with predominant clear cytoplasm. Four cases showed increased mitotic activity, with three of them also displaying areas of necrosis.
Overall, tumors with GLI1 gene abnormalities from various anatomic locations have failed thus far to show a consistent immunoprofile. In the initial study [1], t(7;12)-positive pericytomas showed smooth muscle actin positivity, with no expression of S100 and CD34. MALAT1-GLI1-positive gastric plexiform fibromyxoma show only smooth muscle actin positivity, while being negative for S100 [5]. In contrast, gastroblastomas are positive for Cytokeratin and CD56, with only patchy smooth muscle actin staining and no S100 expression [4]. Finally, the recently described malignant epithelioid soft tissue tumors with various GLI1 gene fusions [7], S100 was diffusely positive in four of the six cases, without expression for other ancillary markers. Furthermore, in the current study, no consistent immunoprofile was identified. CD56, a relatively non-specific marker, was the most consistently positive immunostain, seen in six of the 10 tumors. S100 staining was seen in four of the 10 cases, often as a focal and patchy reactivity. Smooth muscle actin was positive in two of the 10 cases and cytokeratin staining was observed in one case. As expected from the level of gene amplification, CDK4 was positive in five cases tested and MDM2 stain showed diffuse nuclear positivity in four of the five cases tested.
The defining genetic feature of this study group was the presence of GLI1 gene amplification. GLI1 amplification was first reported in gliomas in the 1990s [12, 13], resulting in the gene being designated ‘glioma-associated oncogene 1’. Amplification of GLI1 has also been reported to occur rarely in childhood sarcomas [14]. In the study by Roberts et al., 29 cell lines derived from childhood rhabdomyosarcomas and osteosarcomas were studied for GLI1 amplification. Only one cell line each derived from a rhabdomyosarcoma and an osteosarcoma showed amplification of GLI1. The histologic images illustrated in their study were suboptimal for comparison with our study group. In a different study [15], 40 adult sarcomas were studied for GLI1 amplification and none showed evidence of this genetic alteration. From a query of our institutional targeted exome sequencing database, GLI1 amplification was noted in less than 1% of all the tumors analyzed (~30,000 tumors). Among the different subgroups, GLI1 amplification is most frequently seen in soft tissue sarcomas (6%) followed by glioma (4%), melanoma (2%), and lung adenocarcinoma (1%). Among the soft tissue sarcomas with GLI1 amplification, liposarcomas (well-differentiated/dedifferentiated) are the most common (85%), followed by equal frequency of intimal sarcoma (5%) and tumors that were classified as Sarcoma, NOS (5%).
As the GLI1 gene is located within an oncogene-rich region, the 12q13-15 locus, it is not surprising that a significant number of our cases showed an amplicon composed of GLI1, DDIT3, CDK4, and MDM2 co-amplified genes. DDIT3 gene, located adjacent to the GLI1 gene, showed amplification in all of the 10 cases (being interrogated based on our bacterial artificial chromosome probe design for the GLI1 telomeric probe). HMGA2, located in the same region, was also tested on four cases and showed amplification. Co-amplifications of GLI1 and CDK4, with or without MDM2 were first reported by Khatib et al. in rare sarcoma cell lines [16]. Co-amplification of CDK4 and MDM2 is the defining genetic event in a number of bone and soft tissue tumors, such as well differentiated/ dedifferentiated liposarcomas [17], intimal sarcomas [18], low-grade central osteosarcoma [19], and parosteal (juxtacortical) osteosarcomas [20]. The incidence and functional role of GLI1 co-amplification in this spectrum of neoplasms has not been investigated to date. Based on our institutional targeted exome sequencing IMPACT database, one-fourth of well-differentiated/dedifferentiated liposarcomas showed evidence of GLI1 gene co-amplification. GLI1 is located on 12q13.3, less than 100 KBs from the DDIT3 gene, 300 KBs from the CDK4 (12q14.1) and STAT6 (12q13.3) genes. MDM2 is located at 12q15, about 11 Mb from the GLI1/CDK4 gene cluster (Fig. 4). Due to this positional proximity, the amplicon of most cases tested included GLI1/CDK4 and MDM2 genes. Based on strong STAT6 expression in two cases, it also suggests that STAT6 gene might be involved in the co-amplification in a subset of cases. Although the significance of the co-amplified genes in this region remains uncertain, the similarity in morphology of the study cases to the GLI1 gene fusion-positive tumors and the GLI1 overexpression suggests that GLI1 represents the pathogenetic driver of our study group tumors.
One case in our study (case 2) showed both rearrangement (break-apart) and amplification of the GLI1 gene. Given the morphologic overlap among the GLI1-amplified and the GLI1-fusion-positive cases, the molecular findings in this case represent an important pathogenetic link between the two groups, suggesting that alternative genetic events involving the GLI1 gene could lead to tumors with similar clinicopathologic features. This phenomenon of rearrangement and amplification involving a single oncogene has been observed previously in myoepithelial tumors of the salivary gland and soft tissues with HMGA2 rearrangements, where subsequent amplification of HMGA2 gene might be related to increased genomic instability or tumor progression [21,22,23].
Although none of the dedifferentiated liposarcomas harboring GLI1/CDK4/MDM2 gene co-amplification from our IMPACT database showed any morphologic overlap with the current cohort, we have encountered a small subset of highly unusual dedifferentiated liposarcomas, which closely resembled the features of the study group, being sent in consultation as presumed GLI1-fusion-positive sarcomas. However, all except one case had the typical clinical presentation of a dedifferentiated liposarcoma (retroperitoneum, paratesticular) and showed at least focal areas of sclerosing well-differentiated liposarcoma component, recognized mostly in retrospect after the genetic abnormalities of 12q13-15 amplifications were noted. It is tempting to speculate that at least in some cases, the presence of GLI1 gene amplifications might be responsible of the primitive nested epithelioid phenotype seen in these tumors, which might be shared by a wider spectrum of pathologic entities.
GLI1 encodes a member of the Kruppel family of zinc finger proteins, which is activated by the sonic hedgehog signal transduction cascade and regulates stem cell proliferation. Upon activation, the Sonic Hedgehog ligand binds to its receptor Patched, and relieves the repressive activity on Smoothened (SMO), resulting in GLI1 transcription factor accumulation and activation [24]. GLI expression in adult tissues is restricted to the fallopian tube, myometrium, and testis [7]. A GLI1 inhibitor, GANT61, has been studied to target GLI1 overexpression in lymphoma, melanoma, glioma, and pancreatic cancer [25,26,27,28].
Based on the pathologic features, the differential diagnoses that would be considered for our study group is broad and include myoepithelial tumors of soft tissue, glomus tumor/myopericytoma, solitary fibrous tumor and dedifferentiated liposarcoma. Soft tissue myoepithelial tumor is the most likely consideration, based on the tumor morphology. The immunoprofile and FISH analysis helps in excluding this entity. Myoepithelial tumors of soft tissue and defined immunohistochemically by co-expression of S100 and EMA/pan-cytokeratin and harbor EWSR1/FUS gene rearrangements in about half of the cases [29]. The rich vascularity and the perivascular pattern of growth of GLI1 family of tumors is highly reminiscent of glomus tumor. Glomus tumors are consistently positive for actin and show the miR143-NOTCH2 gene fusion in most cases [10]. Malignant solitary fibrous tumor might also be considered in the differential of GLI1-amplified tumors, mainly due to the nested growth pattern shared by the two tumor types and the positivity of STAT6 in a small subset of GLI1-amplified tumors. The immunoreactivity for CD34 and demonstration of NAB2-STAT6 gene fusion can confirm a diagnosis of solitary fibrous tumors. The immunoreactivity for CDK4 and MDM2 brings up the differential of dedifferentiated liposarcoma. The presence of a well-differentiated liposarcoma component and the typical clinical presentation within the retroperitoneum, paratesticular soft tissue, and mediastinum, could help in distinguishing between the two entities. One of our cases (case 10) occurred in the tongue of a 65-year-old male, which raised the alternative consideration of a ‘pericytoma with t(7;12)’, since most of the reports of this entity have been described in the tongue [1]. Our case showed a nested epithelioid neoplasm with round uniform nuclei and clear cytoplasm, delineated by fibrous septa. Immunohistochemically, this case was positive for CDK4, MDM2, and STAT6, while being negative for smooth muscle actin, desmin, S100, and Cytokeratins (Fig. 3). As the emerging spectrum of tumors defined by GLI1 oncogenic activation reveals a variable immunoprofile, it is difficult to draw definitive conclusions regarding their shared pathogenesis. However, based on the existing published data certain morphologic overlap is noted among some of these family members. Further studies are needed to investigate the pathogenetic link between the so-called ‘pericytoma with t(7;12) translocation’ and the evolving new entity of soft tissue tumors with GLI1 fusion/amplification.
In conclusion, we report GLI1 gene amplifications as a potential alternative mechanism of GLI1 activation in a subset of soft tissue tumors with distinctive epithelioid nested phenotype, expanding the spectrum of tumors recently reported as harboring recurrent GLI1 gene fusions. The current cohort showed co-amplifications of CDK4 and MDM2 genes in most cases, while one case showed concurrent unbalanced GLI1 gene rearrangement as well as amplification. In contrast to the GLI1-fusion-positive subset, the current study group displayed a wider morphologic spectrum, ranging from benign to clearly sarcomatous, including a biphasic epithelioid and spindle cell morphology, and overall had an inconsistent immunoprofile. These results open opportunities for targeted therapies in advanced or metastatic cases.
References
Dahlen A, Fletcher CD, Mertens F, Fletcher JA, Perez-Atayde AR, Hicks MJ, et al. Activation of the GLI oncogene through fusion with the beta-actin gene (ACTB) in a group of distinctive pericytic neoplasms: pericytoma with t(7;12). Am J Pathol. 2004;164:1645–53.
Bridge JA, Sanders K, Huang D, Nelson M, Neff JR, Muirhead D, et al. Pericytoma with t(7;12) and ACTB-GLI1 fusion arising in bone. Hum Pathol. 2012;43:1524–9.
Castro E, Cortes-Santiago N, Ferguson LM, Rao PH, Venkatramani R, López-Terrada D. Translocation t(7;12) as the sole chromosomal abnormality resulting in ACTB-GLI1 fusion in pediatric gastric pericytoma. Hum Pathol. 2016;53:137–41.
Graham RP, Nair AA, Davila JI, Jin L, Jen J, Sukov WR, et al. Gastroblastoma harbors a recurrent somatic MALAT1-GLI1 fusion gene. Mod Pathol. 2017;30:1443–52.
Spans L, Fletcher CD, Antonescu CR, Rouquette A, Coindre JM, Sciot R, et al. Recurrent MALAT1-GLI1 oncogenic fusion and GLI1 up-regulation define a subset of plexiform fibromyxoma. J Pathol. 2016;239:335–43.
Miettinen M, Makhlouf HR, Sobin LH, Lasota J. Plexiform fibromyxoma: a distinctive benign gastric antral neoplasm not to be confused with a myxoid GIST. Am J Surg Pathol. 2009;33:1624–32.
Antonescu CR, Agaram NP, Sung YS, Zhang L, Swanson D, Dickson BC. A distinct malignant epithelioid neoplasm with GLI1 gene rearrangements, frequent S100 protein expression, and metastatic potential: expanding the spectrum of pathologic entities with ACTB/MALAT1/PTCH1-GLI1 fusions. Am J Surg Pathol. 2018;42:553–60.
Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial sloan kettering-integrated mutation profiling of actionable cancer targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn: Jmd. 2015;17:251–64.
Granter SR, Badizadegan K, Fletcher CD. Myofibromatosis in adults, glomangiopericytoma, and myopericytoma: a spectrum of tumors showing perivascular myoid differentiation. Am J Surg Pathol. 1998;22:513–25.
Mosquera JM, Sboner A, Zhang L, Chen CL, Sung YS, Chen HW, et al. Novel MIR143-NOTCH fusions in benign and malignant glomus tumors. Genes Chromosomes Cancer. 2013;52:1075–87.
Miettinen M, Dow N, Lasota J, Sobin LH. A distinctive novel epitheliomesenchymal biphasic tumor of the stomach in young adults (“gastroblastoma”): a series of 3 cases. Am J Surg Pathol. 2009;33:1370–7.
Collins VP. Gene amplification in human gliomas. Glia. 1995;15:289–96.
Wasson JC, Saylors RL 3rd, Zeltzer P, Friedman HS, Bigner SH, Burger PC, et al. Oncogene amplification in pediatric brain tumors. Cancer Res. 1990;50:2987–90.
Roberts WM, Douglass EC, Peiper SC, Houghton PJ, Look AT. Amplification of the gli gene in childhood sarcomas. Cancer Res. 1989;49:5407–13.
Stein U, Eder C, Karsten U, Haensch W, Walther W, Schlag PM. GLI gene expression in bone and soft tissue sarcomas of adult patients correlates with tumor grade. Cancer Res. 1999;59:1890–5.
Khatib ZA, Matsushime H, Valentine M, Shapiro DN, Sherr CJ, Look AT. Coamplification of the CDK4 gene with MDM2 and GLI in human sarcomas. Cancer Res. 1993;53:5535–41.
Dei Tos AP, Marino-Enriquez A, Pedeutour F, Rossi S. Dedifferentiated liposarcoma. In: Fletcher C, Bridge JA, Hogendoorn PC, editors. WHO Classification of Tumours of Soft Tissue and Bone. 4th edn. Lyon: IARC; 2013. p. 37–8.
Bode-Lesniewska B, Debiec-Rychter M. Intimal sarcoma. In: Fletcher C, Bridge JA, Hogendoorn PC, editors. WHO Classification of Tumours of Soft Tissue and Bone. 4th edn. Lyon: IARC; 2013. p. 232–3.
Inwards C, Squire J. Low-grade central osteosarcoma. In: Fletcher C, Bridge JA, Hogendoorn PC, editors. WHO Classification of Tumours of Soft Tissue and Bone. 4th edn. Lyon: IARC; 2013. p. 281–2.
Lazar A, Mertens F. Parosteal osteosarcoma. In: Fletcher C, Bridge JA, Hogendoorn PC, editors. WHO Classification of Tumours of Soft Tissue and Bone. 4th ed. Lyon: IARC; 2013. p. 292–3.
Antonescu CR, Zhang L, Shao SY, Mosquera JM, Weinreb I, Katabi N, et al. Frequent PLAG1 gene rearrangements in skin and soft tissue myoepithelioma with ductal differentiation. Genes Chromosomes Cancer. 2013;52:675–82.
Bahrami A, Perez-Ordonez B, Dalton JD, Weinreb I. An analysis of PLAG1 and HMGA2 rearrangements in salivary duct carcinoma and examination of the role of precursor lesions. Histopathology. 2013;63:250–62.
Katabi N, Ghossein R, Ho A, Dogan S, Zhang L, Sung YS, et al. Consistent PLAG1 and HMGA2 abnormalities distinguish carcinoma ex-pleomorphic adenoma from its de novo counterparts. Hum Pathol. 2015;46:26–33.
Gonnissen A, Isebaert S, Haustermans K. Targeting the Hedgehog signaling pathway in cancer: beyond Smoothened. Oncotarget. 2015;6:13899–913.
Geng L, Lu K, Li P, Li X, Zhou X, Li Y, et al. GLI1 inhibitor GANT61 exhibits antitumor efficacy in T-cell lymphoma cells through down-regulation of p-STAT3 and SOCS3. Oncotarget. 2017;8:48701–10.
Li J, Cai J, Zhao S,Yao K, Sun Y, Li Y, et al. GANT61, a GLI inhibitor, sensitizes glioma cells to the temozolomide treatment. J Exp Clin Cancer Res. 2016;35:184.
Miyazaki Y, Matsubara S, Ding Q, Tsukasa K, Yoshimitsu M, Kosai K, et al. Efficient elimination of pancreatic cancer stem cells by hedgehog/GLI inhibitor GANT61 in combination with mTOR inhibition. Mol Cancer. 2016;15:49.
Vlckova K, Reda J, Ondrusova L, Krayem M, Ghanem G, Vachtenheim J. GLI inhibitor GANT61 kills melanoma cells and acts in synergy with obatoclax. Int J Oncol. 2016;49:953–60.
Antonescu CR, Zhang L, Chang NE, Pawel BR, Travis W, Katabi N, et al. EWSR1-POU5F1 fusion in soft tissue myoepithelial tumors. A molecular analysis of sixty-six cases, including soft tissue, bone, and visceral lesions, showing common involvement of the EWSR1 gene. Genes Chromosomes Cancer. 2010;49:1114–24.
Acknowledgements
The authors would like to thank Jordana Shapiro for preparation of composite figures. They also thank the following pathologists and oncologists who kindly contributed case material and/or clinical follow-up information when available: Dr. Esperanza Villanueva-Siles, Bronx, NY; Dr. Roberto Tirabosco, London, UK; Dr. Henry Tazelaar, Scottsdale, AZ, USA. This study was supported by P50 CA140146-01 (SS, CRA); P50 CA217694 (SS, CRA, NPA); P30-CA008748 (CRA, NPA); Kristen Ann Carr Foundation (CRA); Cycle for Survival (CRA).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Agaram, N.P., Zhang, L., Sung, YS. et al. GLI1-amplifications expand the spectrum of soft tissue neoplasms defined by GLI1 gene fusions. Mod Pathol 32, 1617–1626 (2019). https://doi.org/10.1038/s41379-019-0293-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-019-0293-x
This article is cited by
-
Molecular pathology in diagnosis and prognostication of head and neck tumors
Virchows Archiv (2024)
-
Spindle Cell Tumors of the Sinonasal Tract: A Diagnostic Update with Focus on Ancillary Workup
Head and Neck Pathology (2024)
-
Molecularly defined sinonasal malignancies: an overview with focus on the current WHO classification and recently described provisional entities
Virchows Archiv (2024)
-
Spezielle mesenchymale Tumoren der Kopf-Hals-Region: Neues aus der WHO 2022
Die Pathologie (2023)
-
Non-cutaneous syncytial myoepitheliomas are identical to cutaneous counterparts: a clinicopathologic study of 24 tumors occurring at diverse locations
Virchows Archiv (2023)

{kind=link}
{kind=link}