
Abstract
Oncogenic fusions are rare in colorectal carcinomas, but may be important for prognosis and therapy. An effective strategy for screening targetable oncogenic fusions in colorectal carcinomas is needed. Here, we investigate molecular genetic alterations in colorectal carcinomas based on their DNA mismatch repair status, and to effectively screen for targetable oncogenic fusions in colorectal carcinomas. In this retrospective study, the initial cohort included 125 consecutive mismatch repair-deficient and 238 randomly selected mismatch repair-proficient colorectal carcinomas diagnosed between July 2015 and December 2017 at Peking Union Medical College Hospital. Targeted sequencing was performed. MLH1 promoter hypermethylation analysis was further employed for subgrouping dMMR colorectal carcinomas. Clinicopathological characteristics, molecular features, and survival outcome of colorectal carcinomas harboring oncogenic fusions were assessed. A multicenter cohort comprised of 227 colorectal carcinomas with dual loss of MLH1/PMS2 was used to validate the efficacy of the proposed screening strategy for oncogenic fusions. Of the 363 patients in the initial cohort, 11(3.0%) harbored oncogenic fusions and were all mismatch repair-deficient colorectal carcinomas with hypermethylated MLH1 and wild-type BRAF and KRAS, comprising 55% (11/20) of this subgroup. These patients with oncogenic fusions showed poorer 3-year cancer-specific survival compared with other Stage III/IV mismatch repair-deficient colorectal carcinoma patients (40% vs. 97%), and significantly higher CD274(PD-L1) expression in tumor cells compared with other dMMR colorectal carcinoma patients (46% vs. 6.1%, P < 0.001). An easy-to-perform and cost-efficient strategy for screening targetable fusions was proposed based on the current molecular testing algorithms for colorectal carcinomas, and validated in an independent multicenter cohort. In conclusion, oncogenic fusions were highly enriched and frequently detected in mismatch repair-deficient colorectal carcinomas with MLH1 hypermethylation and wild-type BRAF and KRAS, and were associated with poor prognosis and high tumor CD274(PD-L1) expression.
Similar content being viewed by others


Introduction
Colorectal carcinoma is a globally prevalent cancer and the third and fifth leading cause of cancer deaths in the United States and China, respectively [1, 2]. Colorectal tumors at the same tumour, node and metastasis (TNM) classification of malignant tumors stage are known to show different clinical outcomes, possibly due to intrinsic molecular heterogeneity. Approximately, 10–15% of colorectal carcinomas are deficient in DNA mismatch repair resulting in microsatellite instability, which is an important prognostic and predictive factor [3].
Mismatch repair-deficient colorectal carcinomas have been assigned to three subgroups based on etiology. The onset of sporadic mismatch repair-deficient colorectal carcinomas is primarily caused by epigenetic hypermethylation of the MLH1 promoter region resulting in transcriptional silencing of MLH1 in tumors with the CpG island methylator phenotype [4]. The second subgroup consists of inherited mismatch repair-deficient colorectal carcinomas mainly caused by Lynch syndrome and is the most common form of hereditary colorectal carcinomas. They are characterized by the presence of germline deleterious mutations in any of the mismatch repair genes (MLH1, MSH2, MSH6, and PMS2) or alterations in the EPCAM gene that affects MSH2 function [5]. The third subgroup consists of mismatch repair-deficient tumors that cannot be explained by either mismatch repair genetic mutations or MLH1 promoter hypermethylation and are referred to as “other mismatch repair-deficient” or “Lynch-like syndrome” colorectal carcinomas [6]. The potential causes for the onset of these tumors are varied and include unidentified germline mutations in mismatch repair or mismatch repair-related genes and somatic mosaicism or double mutations in mismatch repair genes [7]. The three molecular subgroups of mismatch repair-deficient colorectal carcinomas have distinct implications for future cancer risk and clinical management strategies for patients and their relatives. Sporadic mismatch repair-deficient colorectal carcinomas with MLH1 promoter hypermethylation is the most common subgroup and account for the majority of mismatch repair-deficient colorectal carcinomas [8, 9]. These tumors share many characteristics with Lynch syndrome-associated mismatch repair-deficient colorectal carcinomas, but are more likely to be diagnosed in older women and frequently arise via the serrated neoplasia pathway [10, 11]. They are known to be closely, but not exclusively, associated with the BRAF V600E mutation [12, 13]. Approximately 30% of MLH1-hypermethylated mismatch repair-deficient colorectal carcinomas with a wild-type BRAF gene harbor KRAS mutations and develop via the conventional adenoma pathway, which suggests resistance to anti-epidermal growth factor receptor (EGFR) therapy [14]. The notable heterogeneity of the mismatch repair-deficient colorectal carcinoma subgroups indicates that a comprehensive genetic analysis of these tumors might enable the identification of potential molecular targets for tailored therapy.
In this study, we used targeted sequencing to explore the molecular features of mismatch repair-deficient colorectal carcinoma subgroups. We found an enrichment of recurrent targetable oncogenic fusions in mismatch repair-deficient colorectal carcinomas with a hypermethylated MLH1 promoter. The mismatch repair-deficient colorectal carcinomas harboring oncogenic fusions displayed distinct molecular features and significantly CD274 (higher programmed death-ligand 1, PD-L1) expression. Based on our results, we proposed an easy to use, cost-effective strategy for screening these potentially targetable oncogenic fusions in colorectal carcinomas, and validated it in an independent cohort. Our proposed screening strategy can be incorporated into the routine molecular tests for colorectal carcinomas and shows promise for use in targeted therapy and immunotherapy.
Materials and methods
Patient selection
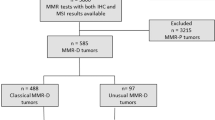
This retrospective study used data from pathology archives of the Peking Union Medical College Hospital collected between July 2015 and December 2017. Patients who underwent surgical resection of the primary colorectal tumor without receiving neoadjuvant therapy were considered eligible. Patients with intact immunohistochemical staining of all four mismatch repair proteins were classified as “mismatch repair-proficient” and those with no IHC expression of any mismatch repair protein were classified as “mismatch repair-deficient”. The discovery cohort from Peking Union Medical College Hospital consisted of consecutive patients with mismatch repair-deficient colorectal carcinoma (n = 125) and randomly selected patients with mismatch repair-proficient colorectal carcinoma (n = 238), who were followed-up to August 2018. We used systematic sampling as a random sampling technique to select mismatch repair-proficient colorectal carcinoma cases. In brief, all eligible mismatch repair-proficient colorectal carcinoma cases (n = 1138) comprised the sampling frame in chronological order. The sampling interval was calculated as k = 1138/250 = 4.6. The random starting point was selected as a non-integer between 0 and 4.6. Each non-integer selected was rounded up to the next integer. After excluding samples with poor DNA quality, 238 cases were finally included. A validation cohort consisting of 227 patients with colorectal carcinomas harboring dual loss of MLH1/PMS2 from three other institutions was concurrently tested with the same inclusion criteria used for the discovery cohort. The validation cohort was comprised of 203 mismatch repair-deficient colorectal carcinomas presented with loss of MLH1/PMS2 expression from three academic institutes during the period between July 2015 and December 2017: Cancer Hospital Chinese Academy of Medical Sciences, Beijing, China (n = 115), Nanfang Hospital of Southern Medical University, Guangzhou, China (n = 66), and Xuanwu Hospital Capital Medical University, Beijing, China (n = 22). This study was approved by ceding review to the Peking Union Medical College Hospital Institutional Review Board.
Targeted sequencing
Targeted sequencing was performed using hybrid capture-based targeted next-generation sequencing as previously described [15, 16]. In brief, DNA from formalin-fixed paraffin-embedded colorectal tumors and matched normal tissues were extracted and sheared. Barcoded libraries were hybridized to our customized panel of 1021 genes containing whole exons and selected introns of 288 genes and selected regions of 733 genes (Supplementary eTable 1). The libraries were sequenced to a uniform median depth ( > 500 × ) and assessed for somatic variants including single nucleotide variants, small insertions and deletions, copy number alterations, and gene fusions/rearrangements. Microsatellite instability status, loss of heterozygosity, and tumor mutation burden were analyzed. To calculate tumor mutation burden, the number of somatic, coding, nonsynonymous single nucleotide variants, and insertions and deletions mutations per megabase (Muts/Mb) of genome examined was defined. tumor mutation burden levels were divided into three categories: low (tumor mutation burden-low, 1–5 Muts/Mb), intermediate (tumor mutation burden-medium, 6–19 Muts/Mb), and high (tumor mutation burden-high, ≥ 20 Muts/Mb). The microsatellite instability status of next-generation sequencing data were inferred using MSIsensor (v0.2), which reported the percentage of unstable somatic microsatellites through Chi-square test on predefined microsatellite regions covered by our panel. Default parameters were used. Loss of heterozygosity in mismatch repair genes was called by analysis of the variant allele fraction using a local algorithm modified from a previously reported method. Loss of heterozygosity was established when: (1) the variant allele fraction for a mutation was > 80% higher than the average variant allele fraction for somatic mutations in the tumor, and (2) verified by analysis of shifts in expected variant allele fraction for germline polymorphisms within the same gene region. Possible loss of heterozygosity was called when the variant allele fraction for a mutation was between 40–80% higher than the average variant allele fraction for somatic mutations in the tumor.
The average sequencing depth for target regions of tumor samples was 1026 × and 99.0% of the average coverage for targeted regions was >200 × , which were suitable for variant calling and microsatellite instability analysis (Supplementary eTable 2).
MLH1 promoter hypermethylation analysis
All patients with colorectal carcinoma that showed loss of MLH1 expression and no mismatch repair germline mutations were analyzed for MLH1 promoter hypermethylation. Bisulfide modification was applied to DNA specimens using the EZ DNA Methylation Kit (Zymo Research, USA) according to the manufacturer instructions. One microgram of bisulfite converted genomic DNA from each sample was eluted in 18 μl elution buffer. The bisulfite-treated DNA was amplified with the methylation-specific primer set: 5ʹ-AATTAATAGGAAGAGCGGATAGC-3ʹ and 5ʹ-CCTCCCTAAAACGACTACTACCCG-3ʹ for methylated MLH1 promoter and 5ʹ-TGAATTAATAGGAAGAGTGGATAGT-3ʹ and 5ʹ-TCCCTCCCTAAAACAACTACTACCCA-3ʹ for unmethylated MLH1 promoter. The representative methylation-specific PCR results and the interpretation criteria is shown in Supplementary eFig. 1.
Immunohistochemical staining
Immunohistochemical staining was performed on 4-μm formalin-fixed paraffin-embedded tissue sections using a BenchMark ULTRA autostainer, version 12.3 (Ventana Medical Systems, USA) and the following antibodies in accordance with the manufacturers’ recommendations: ALK (D5F3, Cell Signaling Technology, USA), CD274(PD-L1) (SP263, Roche Applied Science, USA), PDCD1(PD-1) (UMAB199, Beijing Zhongshan Golden Bridge Biotechnology, China). CD274(PD-L1) immunoreactivity in tumor cells was assessed and divided into the following subcategories: membranous with or without cytoplasmic staining of any intensity in < 1%, 1–5%, 6–50%, and > 50% of tumor cells. CD274(PD-L1) staining in the peritumoral immune compartment was considered positive if punctuate or linear membrane staining was seen in lymphocytes or macrophages in association with the tumor and was graded as negative, minimal, moderate, and brisk. PDCD1(PD-1) staining on tumor-infiltrating lymphocytes was also graded similarly.
Fluorescence in situ hybridization (FISH)
All tissue samples harboring ALK, NTRK1/3, RET, or BRAF fusions that were detected by targeted sequencing were further evaluated by FISH analysis. FISH was performed on 5-μm paraffin sections using the following probe kits according to the manufacture’s protocol: ALK (Vysis ALK break-apart FISH probe kit, Abott Molecular, USA), NTRK1 (SPEC NTRK1 dual color break-apart FISH probe kit, ZytoLight, Germany), NTRK3 (ETV6/NTRK3 dual color dual fusion probe kit, Jinlu Biotechnology, China), RET (Vysis 10q11 RET break-apart FISH probe, Abott Molecular, USA) and ETV6 (Vysis ETV6 break-apart FISH probe kit, Abbott, USA), BRAF (7q34 BRAF break-apart FISH probe, Anbiping Biotechnology, China). At least 50 tumor cells were evaluated for each sample. ALK, RET, NTRK1, BRAF, and ETV6 rearrangements were indicated by isolated red signals and/or split red/green signals (defined by more than one signal diameter apart from each other). ETV6-NTRK3 fusions were indicated by red/green fusion signals. A threshold of 15% nuclei positivity was used to establish the cutoff for positive FISH.
Statistical methods
Continuous variables were presented as mean ± standard deviation, and categorical variables were expressed as percentages. Chi-square test, Fisher’s exact test, or Mann–Whitney test was used when appropriate for comparison between mismatch repair-deficient and mismatch repair-proficient groups. For multiple comparisons between mismatch repair-deficient subgroups, we used the Benjamini-Hochberg procedure to correct the P-values resulting from the Chi-square test, Fisher’s exact test, or Kruskal–Wallis test. Cancer-specific survival was calculated using the Kaplan–Meier method and compared via log-rank test. Statistical processing was performed using SPSS version 24 (SPSS Inc., Chicago, IL, USA) and P < 0.05 (two-sided) was considered statistically significant.
Results
Identification of colorectal carcinomas with oncogenic fusions and development of a screening method
The discovery cohort of 125 patients with mismatch repair-deficient colorectal carcinomas was divided into three groups: the MLH1-hypermethylated group (MLH1-hypermethylated) consisted of 50 patients with loss of MLH1/PMS2 expression (50/125, 40%) who harbored no mismatch repair gene germline mutations but showed MLH1 promoter hypermethylation; the Lynch syndrome-associated group consisted of 48 patients that had pathogenic/likely pathogenic germline mutations in one of the four mismatch repair genes, or combined germline EPCAM-MSH2 deletions (48/125, 38%); and the remaining patients (27/125, 22%) who displayed neither mismatch repair gene germline mutations nor MLH1 promoter hypermethylation were classified into the Lynch-like group [17] (Supplementary eTable 3).
Oncogenic gene rearrangements were identified in 3.0% (11/363) of patients in the discovery cohort. Interestingly, all 11 patients had mismatch repair-deficient colorectal carcinoma and were part of the MLH1-hypermethylated group (11/50, 22%). To further categorize the patients based on the presence of oncogenic fusions, the MLH1-hypermethylated group was divided into two subgroups: MLH1-hypermethylated with fusion (n = 11) and MLH1 hypermethylation without fusion (n = 39) (Supplementary eFig. 2). In the ‘MLH1-hypermethylated with fusion’’ subgroup, two patients showed ALK gene rearrangements: one with known STRN-ALK fusion and another with EML4-ALK fusion with an atypical breakpoint at ALK exon 19. Five patients showed NTRK1 gene rearrangements involving known partners such as TPM3, LMNA, and PLEKHA6. Two patients showed NTRK3-ETV6 fusions and two others showed NCOA4-RET fusions (Fig. 1a). All fusion events were confirmed by FISH analysis (Fig. 1b and Supplementary eFig. 3A). ALK rearrangements were also verified by the Ventana ALK immunohistochemical assay (Fig. 1c).

Identification of oncogenic ALK, NTRK, and RET fusions. a Schematic representations indicate STRN-ALK, EML4-ALK, TPM3-NTRK1, LMNA-NTRK1, PLEKHA6-NTRK1, ETV6-NTRK3, and NCOA4-RET fusions. Arrows indicate the direction of transcription for each gene and arrowheads indicate the breaking points. Exons and introns are represented by colored boxes and lines, respectively. b Representative FISH images confirm genomic rearrangements (original magnification: × 1000), with isolated red signals (arrowheads) indicating ALK rearrangements (#53, STRN-ALK), split red/green signals (white arrows) indicating NTRK1 (#49, TPM3-NTRK1) and RET (#110, NCOA4-RET) rearrangements, and red/green fused signals (yellow arrows) indicating ETV6-NTRK3 fusions (#71). c Immunohistochemical stainings show ALK overexpression in case #53 (STRN-ALK) and #10 (EML4-ALK) (original magnification: × 100, inserts: × 400), indicating the presence of ALK rearrangements. FISH: fluorescence in situ hybridization
Mutation profiles of major colorectal carcinoma driver genes in tumors of 125 mismatch repair-deficient and 238 mismatch repair-proficient patients were summarized in Fig. 2. In the MLH1-hypermethylated group, 20 patients (20/50, 40%) had BRAF V600E mutations and 10 (10/50, 20%) had KRAS mutations, but none harbored NRAS mutations. We found that oncogenic fusions were mutually exclusive with BRAF or KRAS mutations, thus accounting for 55% (11/20) of the MLH1-hypermethylated group with wild-type BRAF and KRAS genes. No oncogenic mutations in PIK3CA and CTNNB1 were seen in 9/11 patients (82%) in the ‘MLH1-hypermethylated with fusion’’ subgroup. Only two patients with TPM3-NTRK1 and ETV6-NTRK3 fusions harbored mutations of PIK3CA (p.C378R) and CTNNB1 (p.S45del), respectively. Based on these results, we proposed a practical strategy for screening oncogenic fusions in colorectal carcinomas (Fig. 3).

Germline and somatic genome alterations. Columns represent individual patients sorted by MMR status and dMMR subgroups. Tracks indicate tumor location, MLH1 hypermethylation status, germline mutations of MMR genes, somatic mutations of driver genes, oncogenic gene fusions, LOH of MMR genes, and POLE and POLD1 mutations. Individual genes are listed by rows and mutations are classified according to the effect on protein sequence. Only well-documented activating mutations were shown for BRAF, KRAS, NRAS, PIK3CA, and CTNNB1. Bar chart below displays TMB of individual patients. dMMR: mismatch repair deficient. pMMR: mismatch repair proficient. LOH: loss of heterozygosity; LS: Lynch syndrome; TMB: tumor mutational burden; dMMR: DNA mismatch repair-deficient; pMMR: DNA mismatch repair proficient

Proposed strategy for screening oncogenic fusions such as ALK, NTRK, and RET rearrangements in CRCs. CRC: colorectal carcinoma; MMR: mismatch repair; FISH: fluorescence in situ hybridization; NGS: next-generation sequencing
Clinicopathological features and disease progression in patients with mismatch repair-deficient colorectal carcinomas
We found significant clinicopathological differences between the mismatch repair-deficient and mismatch repair-proficient groups of patients (eTable 4 in Supplement). Patients with mismatch repair-deficient colorectal carcinomas in the ‘MLH1-hypermethylated with fusion’’ subgroup shared similar clinicopathological features with those in the ‘MLH1-hypermethylated without fusion’’ subgroup. They were older than patients in the LS-associated (69.5 ± 9.3 vs. 48.8 ± 13.4, P < 0.001) and Lynch-like groups (69.5 ± 9.3 vs. 52.7 ± 11.4, P < 0.001). Although they appeared to display a preponderance of right-sided tumors compared with the LS-associated patient group (91% vs. 48%, P = 0.025), the Benjamin-Hochberg correction for multiple comparisons showed that the data was not statistically significant. No significant differences in gender, stage, histological grade, and mucinous differentiation were observed between the three groups of patients with mismatch repair-deficient colorectal carcinoma.
After a median follow-up of 19 months, 27% (3/11) of patients, who were at Stage III, in the ‘MLH1-hypermethylated with fusion’’ subgroup experienced cancer-related death. Among Stage III/IV patients with mismatch repair-deficient colorectal carcinomas, those with oncogenic fusions showed relatively poorer 3-year CSS compared with others (40% vs. 97%) (Fig. 4).

Survival outcomes in patients with Stage III/IV dMMR colorectal cancer. Cancer-specific survival in patients with oncogenic fusions (red line) is compared with those without oncogenic fusions (blue line) using the Kaplan–Meier method. dMMR: DNA mismatch repair deficient
Tumor mutation burden status and PD-1/PD-L1 expression profile
All the 125 patients with mismatch repair-deficient colorectal carcinomas showed high tumor mutation burden. Of the 238 patients with mismatch repair-proficient colorectal carcinomas, 228 (96%) showed either low (80/238, 34%) or medium tumor mutation burden (148/238, 62%). Of the remaining ten patients with mismatch repair-proficient colorectal carcinomas, six had a tumor mutation burden level of more than 100 Muts/Mb due to deleterious somatic mutations in the DNA polymerase epsilon (POLE) proofreading domain (Fig. 2). Mismatch repair-deficient tumors showed significantly higher tumor mutation burden compared with mismatch repair-proficient tumors (66.8 ± 35.1 vs. 15.7 ± 54.9 Muts/Mb, P < 0.001). We found no significant differences in tumor mutation burden between the ‘MLH1-hypermethylated with fusion’’ subgroup and other groups of mismatch repair-deficient colorectal carcinomas (Fig. 5a and eTable 5 in Supplement).

Profiles of biomarkers for immunotherapy. a Comparison of TMB between the dMMR group and pMMR groups of patients with CRC, and between the dMMR subgroups. b Comparison of CD274 (PD-L1) expression in tumor cells, CD274 (PD-L1) expression in tumor-associated immune cells, and PDCD1(PD-1) expression in tumor-infiltrating lymphocytes between the dMMR and pMMR groups, and between the dMMR subgroups. c Representative images showing PDCD1(PD-1)/ CD274(PD-L1) immunohistochemical staining of five ALK, NTRK, or RET rearranged tumors with CD274 (PD-L1) expression in > 50% of the tumor cells. dMMR: mismatch repair deficient; pMMR: mismatch repair proficient; LS: Lynch syndrome
Immunohistochemical staining results of PDCD1(PD-1)/CD274(PD-L1) in patients with mismatch repair-deficient and mismatch repair-proficient colorectal carcinomas were compared in Fig. 5b and eTable 5. Notably, five patients in the ‘MLH1-hypermethylated with fusion’’ subgroup showed CD274(PD-L1) staining in > 50% of tumor cells (Fig. 5c and eFig. 4 in Supplement), which was significantly higher than that in other patients with mismatch repair-deficient colorectal carcinomas (5/11, 46% vs. 7/114, 6.1%, P < 0.001). However, the ‘MLH1-hypermethylated with fusion’’ subgroup showed similar stromal immunoreactivity for CD274(PD-L1) (P = 0.28) and PDCD1(PD-1) (P = 0.52) with other mismatch repair-deficient colorectal carcinoma groups (Fig. 5b).
Validation of the screening method for colorectal carcinomas harboring oncogenic fusions
Of the 227 patients with mismatch repair-deficient colorectal carcinomas lacking MLH1/PMS2 expression in the independent validation cohort, 137 (60%) showed MLH1 promoter hypermethylation. Our screening method identified 44/137 (32%) patients, who lacked the BRAF V600E mutation or the KRAS/NRAS activating mutations, as potential candidates of oncogenic fusions. Targeted sequencing analysis showed that 32% (14/44) of patients displayed oncogenic fusions: 10 patients with ALK, NTRK, or RET rearrangements [SPTBN1-ALK (1/10), TPM3-NTRK1 (4/10), TPR-NTRK1 (2/10), ETV6-NTRK3 (2/10), and GPHN-RET (1/10)]; three patients with BRAF rearrangements; and one patient with FGFR2 rearrangement (Supplementary eFig. 5). All fusion events involving ALK, NTRK, RET, and BRAF were confirmed by FISH analysis (Supplementary eFig. 3B).
Discussion
In this study, we have shown that a majority of mismatch repair-deficient colorectal carcinomas with oncogenic fusions, such as ALK, NTRK, or RET rearrangements, showed hypermethylation of the MLH1 promoter and lacked activating mutations in BRAF and KRAS. In the discovery cohort, patients harboring oncogenic fusions formed one-fifth of the ‘MLH1-hypermethylated’’ subgroup and one-half of ‘MLH1-hypermethylated’’ subgroup containing wild-type BRAF and KRAS genes. Our results suggested a novel molecular subtype of colorectal carcinoma with a characteristic driver gene mutation profile, which showed a significantly higher CD274(PD-L1) expression and might have poorer prognosis compared with other types of colorectal carcinomas. We also developed a valid, practical, and cost-effective method to screen for colorectal carcinoma patients harboring oncogenic fusions.
Kinases activated by gene fusions represent a critical set of oncogenes associated with both solid and hematopoietic malignancies, and their protein products often offer actionable targets for cancer therapy [18]. Previous studies have shown that 0.05–2.5% of patients with colorectal carcinomas have ALK fusions [19,20,21], 0.5–1.5% have NTRK fusions [22, 23], and < 1% have RET fusions [24, 25]. Such a low prevalence of oncogenic fusions in colorectal carcinomas require highly sensitive and cost-effective screening methods, which is challenging for daily clinical practice. Recent studies have suggested an association between high levels of microsatellite instability and increased prevalence of oncogenic fusions in metastatic colorectal carcinomas [26, 27]. However, those studies failed to describe the molecular features of mismatch repair-deficient colorectal carcinomas in detail, such as mismatch repair gene mutations and MLH1 hypermethylation status. Our retrospective and detailed molecular analysis revealed that oncogenic rearrangements in colorectal carcinomas were highly enriched (~50%) in mismatch repair-deficient tumors belonging to the ‘MLH1-hypermethylated’’ subgroup with wild-type BRAF and KRAS genes. Based on our results, we recommend screening for oncogenic fusions such as ALK, NTRK, and RET rearrangements by immunohistochemical staining, FISH, or next-generation sequencing in the ‘MLH1-hypermethylated’’ subgroup of patients with mismatch repair-deficient colorectal carcinomas harboring wild-type BRAF and KRAS genes. We independently validated the efficacy and universal applicability of this screening method in a multicenter cohort. Rare occurrences of oncogenic fusions have also been reported in mismatch repair-proficient colorectal carcinomas [19, 26, 27]. In this study, we did not detect oncogenic fusions in the 238 patients with mismatch repair-proficient colorectal carcinomas, possibly due to the limited sample size. However, our results underscored the rarity of oncogenic fusions in mismatch repair-proficient colorectal carcinomas and highlighted the efficiency of our proposed screening method.
Patients harboring oncogenic kinase fusions are potential candidates for personalized cancer therapy. Studies have shown that colorectal carcinomas harboring ALK fusions respond to ceritinib19193 or entrectinib [28], and colorectal carcinomas with NTRK fusions respond to entrectinib [29]. Larotrectinib, a highly selective tropomyosin receptor kinase (TRK) inhibitor displays strong antitumor activity in many types of NTRK fusion-positive tumors including colorectal carcinomas, which suggests that kinase fusions represent a distinct molecular subgroup of colorectal carcinomas that show a favorable response to targeted therapy [30]. BRAF and FGFR2 fusions, which were detected in our validation cohort, have also been considered as potential targets in solid tumors [31, 32]. We showed that oncogenic fusions were mostly excluded in colorectal carcinomas containing activating mutations in driver genes, such as BRAF, KRAS, NRAS, PIK3CA, and CTNNB1. Although patients with metastatic colorectal carcinomas harboring wild-type BRAF and RAS genes may benefit from anti-EGFR therapy [33, 34], the presence of oncogenic fusions has been shown to predict resistance to anti-EGFR therapy in colorectal carcinomas with wild-type BRAF and RAS genes [35], and verified by several recent studies [27, 36]. Therefore, tailored management strategies that target oncogenic fusions instead of EGFR should be considered in patients with colorectal carcinoma harboring both oncogenic fusions and wild-type BRAF and KRAS genes. The presence of ALK, NTRK, or RET fusions has been shown to be associated with shorter overall survival in patients with metastatic colorectal carcinomas [26, 27]. We found that oncogenic fusions were significantly associated with poor prognosis in patients with Stage III/IV mismatch repair-deficient colorectal carcinomas. Therefore, our screening method for oncogenic fusions may have important therapeutic and prognostic significance, especially for patients with advanced-stage colorectal carcinoma. Given the relatively limited sample size and follow-up, the prognostic significance of oncogenic fusions in advanced-stage mismatch repair-deficient colorectal carcinoma patients needs confirmation in a larger cohort.
The latest colon/rectal cancer guidelines from the National Comprehensive Cancer Network recommend tumor mismatch repair or microsatellite instability testing in all colorectal carcinomas and KRAS/NRAS/BRAF genotyping in all metastatic colorectal carcinomas [37, 38]. Also, the hybrid use of BRAF V600E mutation and MLH1 hypermethylation testing has been established for screening newly diagnosed patients with colorectal carcinomas for Lynch syndrome [39]. With such testing practices in place, our screening method was easy-to-perform and cost-efficient.
Blockade of immune-checkpoint proteins such as PDCD1(PD-1) and CD274(PD-L1) is emerging as a promising approach for anticancer therapy. Mismatch repair or microsatellite instability status [40], CD274(PD-L1) expression by tumor cells [40] and immune stroma [41], tumor-infiltrating lymphocytes [42], tumor mutation burden [43, 44], and tumor neoantigens [45, 46] have all been used as predictive biomarkers to guide checkpoint inhibitor-based immunotherapy and to maximize therapeutic benefit. Anti-PDCD1(PD-1) immune-checkpoint inhibitors such as nivolumab and pembrolizumab have been approved by the US Food and Drug Administration for the treatment of mismatch repair-deficient or microsatellite instability-high solid tumors, including colorectal carcinomas. However, a combination of multiple predictors is likely to be more effective in predicting response to immunotherapy [47]. In this study, we investigated other predictive biomarkers (tumor mutation burden and PDCD1(PD-1)/CD274(PD-L1) expression) for checkpoint inhibitor-based immunotherapy in mismatch repair-deficient colorectal carcinomas. We showed that high microsatellite instability and tumor mutation burden were associated with mismatch repair-deficient colorectal carcinomas, which also displayed increased CD274(PD-L1) expression in tumor cells or PDCD1(PD-1)/CD274(PD-L1) expression in tumor-infiltrating immune cells. Notably, the ‘MLH1-hypermethylated with fusion’’ subgroup showed significantly higher CD274(PD-L1) expression in tumor cells compared with other mismatch repair-deficient colorectal carcinoma subgroups. Therefore, patients with mismatch repair-deficient colorectal carcinomas that harbor oncogenic fusions may be suitable for immunotherapy. Clinical trials are needed to validate this premise [48].
Given the relatively small sample size, limited number of Stage III/IV cases, and short follow-up duration, our results need further validation in a larger multicenter cohort. In addition, tumor neoantigen burden has recently been correlated with increased tumor-infiltrating lymphocytes and positive selection of HLA gene mutations in colorectal carcinomas, and is theoretically believed to be directly linked to response to immunotherapy. Further investigation of microsatellite instability in mismatch repair-deficient colorectal carcinomas and colorectal carcinomas with oncogenic fusion is warranted.
In conclusion, we are the first to show that targetable oncogenic fusions frequently occurred in MLH1-hypermethylated-mismatch repair-deficient colorectal carcinomas harboring wild-type BRAF and KRAS genes. This distinct molecular subtype of colorectal carcinoma was characterized by high tumor CD274(PD-L1) expression and was associated with poor prognosis. An easy-to-perform and cost-effective method for screening targetable oncogenic fusions has the potential to optimize treatment regimens for patients with colorectal carcinoma, especially those at advanced stages of the disease.
References
Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115–32.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30.
Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, Kuebler P, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med. 2005;352:1851–60.
Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology. 2010;138:2073–87 e2073.
Lynch HT, Snyder CL, Shaw TG, Heinen CD, Hitchins MP. Milestones of Lynch syndrome: 1895-2015. Nat Rev Cancer. 2015;15:181–94.
Rodriguez-Soler M, Perez-Carbonell L, Guarinos C, Zapater P, Castillejo A, Barbera VM, et al. Risk of cancer in cases of suspected lynch syndrome without germline mutation. Gastroenterology. 2013;144:926–932e921. quize913–924
Buchanan DD, Rosty C, Clendenning M, Spurdle AB, Win AK. Clinical problems of colorectal cancer and endometrial cancer cases with unknown cause of tumor mismatch repair deficiency (suspected Lynch syndrome). Appl Clin Genet. 2014;7:183–93.
Cunningham JM, Kim CY, Christensen ER, Tester DJ, Parc Y, Burgart LJ, et al. The frequency of hereditary defective mismatch repair in a prospective series of unselected colorectal carcinomas. Am J Hum Genet. 2001;69:780–90.
Hartman DJ, Brand RE, Hu H, Bahary N, Dudley B, Chiosea SI, et al. Lynch syndrome-associated colorectal carcinoma: frequent involvement of the left colon and rectum and late-onset presentation supports a universal screening approach. Hum Pathol. 2013;44:2518–28.
Poynter JN, Siegmund KD, Weisenberger DJ, Long TI, Thibodeau SN, Lindor N, et al. Molecular characterization of MSI-H colorectal cancer by MLHI promoter methylation, immunohistochemistry, and mismatch repair germline mutation screening. Cancer Epidemiol Biomark Prev. 2008;17:3208–15.
Jass JR. Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology. 2007;50:113–30.
Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38:787–93.
Parsons MT, Buchanan DD, Thompson B, Young JP, Spurdle AB. Correlation of tumour BRAF mutations and MLH1 methylation with germline mismatch repair (MMR) gene mutation status: a literature review assessing utility of tumour features for MMR variant classification. J Med Genet. 2012;49:151–7.
Farchoukh L, Kuan SF, Dudley B, Brand R, Nikiforova M, Pai RK. MLH1-deficient colorectal carcinoma with wild-type BRAF and MLH1 promoter hypermethylation harbor KRAS mutations and arise from conventional adenomas. Am J Surg Pathol. 2016;40:1390–9.
Lv X, Zhao M, Yi Y, Zhang L, Guan Y, Liu T, et al. Detection of rare mutations in CtDNA using next generation sequencing. J Vis Exp. 2017;126:e56342.
Yang X, Chu Y, Zhang R, Han Y, Zhang L, Fu Y, et al. Technical validation of a next-generation sequencing assay for detecting clinically relevant levels of breast cancer-related single-nucleotide variants and copy number variants using simulated cell-free DNA. J Mol Diagn. 2017;19:525–36.
Chen W, Swanson BJ, Frankel WL. Molecular genetics of microsatellite-unstable colorectal cancer for pathologists. Diagn Pathol. 2017;12:24.
Stransky N, Cerami E, Schalm S, Kim JL, Lengauer C. The landscape of kinase fusions in cancer. Nat Commun. 2014;5:4846.
Yakirevich E, Resnick MB, Mangray S, Wheeler M, Jackson CL, Lombardo KA, et al. Oncogenic ALK fusion in rare and aggressive subtype of colorectal adenocarcinoma as a potential therapeutic target. Clin Cancer Res. 2016;22:3831–40.
Houang M, Toon CW, Clarkson A, Siosen L, de Silva K, Watson N, et al. ALK and ROS1 overexpression is very rare in colorectal adenocarcinoma. Appl Immunohistochem Mol Morphol. 2015;23:134–8.
Lipson D, Capelletti M, Yelensky R, Otto G, Parker A, Jarosz M, et al. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat Med. 2012;18:382–4.
Ardini E, Bosotti R, Borgia AL, De Ponti C, Somaschini A, Cammarota R, et al. The TPM3-NTRK1 rearrangement is a recurring event in colorectal carcinoma and is associated with tumor sensitivity to TRKA kinase inhibition. Mol Oncol. 2014;8:1495–507.
Creancier L, Vandenberghe I, Gomes B, Dejean C, Blanchet JC, Meilleroux J, et al.Chromosomal rearrangements involving the NTRK1 gene in colorectal carcinoma. Cancer Lett. 2015;365:107–11.
Le Rolle AF, Klempner SJ, Garrett CR, Seery T, Sanford EM, Balasubramanian S, et al. Identification and characterization of RET fusions in advanced colorectal cancer. Oncotarget. 2015;6:28929–37.
Kloosterman WP, Coebergh van den Braak RRJ, Pieterse M, van Roosmalen MJ, Sieuwerts AM, Stangl C, et al. A Systematic analysis of oncogenic gene fusions in primary colon cancer. Cancer Res. 2017;77:3814–22.
Pietrantonio F, Di Nicolantonio F, Schrock AB, Lee J, Morano F, Fuca G, et al. RET fusions in a small subset of advanced colorectal cancers at risk of being neglected. Ann Oncol. 2018;29:1394–401.
Pietrantonio F, Di Nicolantonio F, Schrock AB, Lee J, Tejpar S, Sartore-Bianchi A, et al. ALK, ROS1, and NTRK rearrangements in metastatic colorectal cancer. J Natl Cancer Inst. 2017;109:1.
Amatu A, Somaschini A, Cerea G, Bosotti R, Valtorta E, Buonandi P, et al. Novel CAD-ALK gene rearrangement is drugable by entrectinib in colorectal cancer. Br J Cancer. 2015;113:1730–4.
Sartore-Bianchi A, Ardini E, Bosotti R, Amatu A, Valtorta E, Somaschini A, et al. Sensitivity to entrectinib associated with a novel LMNA-NTRK1 gene fusion in metastatic colorectal cancer. J Natl Cancer Inst. 2016;108:1.
Drilon A, Laetsch TW, Kummar S, DuBois SG, Lassen UN, Demetri GD, et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N Engl J Med. 2018;378:731–9.
Ross JS, Wang K, Chmielecki J, Gay L, Johnson A, Chudnovsky J, et al. The distribution of BRAF gene fusions in solid tumors and response to targeted therapy. Int J Cancer. 2016;138:881–90.
Wu YM, Su F, Kalyana-Sundaram S, Khazanov N, Ateeq B, Cao X, et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov. 2013;3:636–47.
Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626–34.
Lievre A, Bachet JB, Boige V, Cayre A, Le Corre D, Buc E, et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol. 2008;26:374–9.
Medico E, Russo M, Picco G, Cancelliere C, Valtorta E, Corti G, et al. The molecular landscape of colorectal cancer cell lines unveils clinically actionable kinase targets. Nat Commun. 2015;6:7002.
Cremolini C, Morano F, Moretto R, Berenato R, Tamborini E, Perrone F, et al. Negative hyper-selection of metastatic colorectal cancer patients for anti-EGFR monoclonal antibodies: the PRESSING case-control study. Ann Oncol. 2017;28:3009–14.
National Comprehensive Cancer Network. Bone Cancer (Version 3.2018). https://www.nccn.org/professionals/physician_gls/pdf/colon.pdf. Accessed October 6, 2018.
National Comprehensive Cancer Network. Rectal Cancer (Version 3.2018). https://www.nccn.org/professionals/physician_gls/pdf/rectal.pdf. Accessed October 6, 2018.
Adar T, Rodgers LH, Shannon KM, Yoshida M, Ma T, Mattia A, et al. A tailored approach to BRAF and MLH1 methylation testing in a universal screening program for Lynch syndrome. Mod Pathol. 2017;30:440–7.
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–20.
Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014;515:558–62.
Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–71.
Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371:2189–99.
Rosenberg JE, Hoffman-Censits J, Powles T, van der Heijden MS, Balar AV, Necchi A, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. 2016;387:1909–20.
Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–8.
Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350:207–11.
Gibney GT, Weiner LM, Atkins MB. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016;17:e542–51.
Giannakis M, Mu XJ, Shukla SA, Qian ZR, Cohen O, Nishihara R, et al. Genomic correlates of immune-cell infiltrates in colorectal carcinoma. Cell Rep. 2016;15:857–65.
Acknowledgements
This work was supported by Chinese Academy of Medical Sciences (CAMS) Initiative for Innovative Medicine (CAMS-I2M) 2016-I2M-1-002, Chinese Academy of Medical Sciences (CAMS) Initiative for Innovative Medicine (CAMS-I2M) 2017-I2M-1-001, Center for molecular pathology, Chinese Academy of Medical Sciences & Peking Union Medical College (2017PT31008).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Wang, J., Yi, Y., Xiao, Y. et al. Prevalence of recurrent oncogenic fusion in mismatch repair-deficient colorectal carcinoma with hypermethylated MLH1 and wild-type BRAF and KRAS. Mod Pathol 32, 1053–1064 (2019). https://doi.org/10.1038/s41379-019-0212-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-019-0212-1
This article is cited by
-
Systematic review of NTRK 1/2/3 fusion prevalence pan-cancer and across solid tumours
Scientific Reports (2023)
-
SWI/SNF complex gene variations are associated with a higher tumor mutational burden and a better response to immune checkpoint inhibitor treatment: a pan-cancer analysis of next-generation sequencing data corresponding to 4591 cases
Cancer Cell International (2022)
-
Liquid biopsy using ascitic fluid and pleural effusion supernatants for genomic profiling in gastrointestinal and lung cancers
BMC Cancer (2022)
-
A lung adenocarcinoma patient with co-mutations of MET and EGFR exon20 insertion responded to crizotinib
BMC Medical Genomics (2022)
-
Gene fusions and oncogenic mutations in MLH1 deficient and BRAFV600E wild-type colorectal cancers
Virchows Archiv (2022)
