
Abstract
Both lysosomal storage diseases and mitochondrial diseases are a group of genetic-inherited metabolic disorders. In an era, where “old fashioned methods” are apparently being replaced by evolving molecular techniques (i.e. exome and whole genome sequencing), the “old fashioned methods” might help to characterise and thus narrow down the potential differential diagnosis. Therefore, we retrospectively evaluated the relevance of electron microscopy of axillary skin for the diagnosis of lysosomal storage or mitochondrial diseases (=inherited metabolic disorders of energy metabolism). Methods and patients: We included 74 patients with developmental delay with regression or neurodegeneration who underwent an axillary skin biopsy for both fibroblast culture and electron microscopy. Because of insufficient skin biopsy quality, for 8 patients no electron microscopy result was obtained. The electron microscopy biopsies revealed abnormalities in 37/66 (56.1%) patients. 29/66 electron microscopy biopsies showed normal results. A definite diagnosis was established in 21/66 (31.8%) patients with a pathological results of axillary skin electron microscopy analysis. In total, in 25/66 (37.8%) of the patients who underwent an axillary skin electron microscopy analysis, a definite diagnosis was finally established. Taking an axillary skin biopsy during anaesthesia or with use of local intradermal lidocaine application is an inexpensive alternative and useful to establish a diagnosis in patients suspected to have a lysosomal storage disease (or inherited metabolic disorder of energy metabolism).
Similar content being viewed by others


Introduction
Lysosomal storage diseases and inherited metabolic disorders of energy metabolism (e.g. mitochondrial disorders) are rare diseases. Up to date, lysosomal storage diseases comprise a group of over 50 different inherited metabolic disorders with a combined incidence of estimated 1:7.000 births [1]. The phenotypical characteristics of each disorder show a broad spectrum. Lysosomal storage disease patients often present with neurological disorders, hepatosplenomegaly, cloudy corneas, skeletal abnormalities or a combination of the above-mentioned symptoms [1, 2]. Developmental delay with regression or neurodegeneration are non-specific symptoms, thus, a great variety of disorders must be considered in the differential diagnosis (e.g. mitochondrial disorders or other non-lysosomal-inherited metabolic disorders).
The majority of lysosomal storage diseases or inherited metabolic disorders of energy metabolism are caused by single enzyme deficiencies, defective enzyme targeting, defective lysosomal membrane transport, or deficiency of a protective enzyme [2,3,4]. The age of disease onset and the specific phenotype differ between diseases. Selective diagnostic screening of patients with unclear neurological metabolic disease can be challenging including determination of: plasma amino acids, urinary amino acids, acylcarnitine profile, chitotriosidase concentrations, urinary organic acids, urinary mucopolysaccharides and oligosaccharides, specific enzymes activities in plasma, leucocytes or cultured fibroblasts and genetic testing. However, sometimes even with a precise clinical and laboratory characterisation, the diagnostic work up does not reveal a diagnosis. Nowadays, in such cases molecular techniques like exome and whole genome sequencing can allow a diagnosis. The costs of these tests are expensive.
On the other hand, since the 1970s electron microscopy of skin biopsies has been used to narrow down the diagnosis of lysosomal storage diseases (e.g. mucopolysaccharidosis I–VII, Pompe disease, fucosidosis, sialidosis, neuronal ceroid lipofuscinosis, etc.) and heritable disorders of connective tissue (e.g. Ehlers–Danlos syndrome, osteogenesis imperfecta) [2, 3, 5,6,7,8,9]. For patients with lysosomal storage diseases, electron microscopy of skin biopsies was proven to be an inexpensive and sensitive screening tool [2, 3, 5,6,7,8]. Furthermore, skin biopsies can be taken from patients during anaesthesia for other examinations (e.g. magnetic resonance imaging (MRI)).
In this retrospective study, we evaluated whether electron microscopy of axillary skin is useful, and a rather inexpensive diagnostic screening tool, which should be considered as an alternative to exome and whole genome sequencing.
Material and methods
Sample collection
Since 2001, 74 axillary skin biopsies have been taken from patients with developmental delay with regression and from patients with neurological abnormalities and/or neurodegeneration at our department. Biopsies were most commonly performed due to developmental delay ± regression (22/74), epilepsia (23/74), ataxia or other neurological impairment (22/74), elevation of liver function parameters and icterus prolongatus (2/74), encephalopathy (1/74), positive family history (1/74), weak muscle control (1/74), dysmorphic signs (1/74) or a combination of the mentioned symptoms (12/74). Most of the samples were punch skin biopsies with a mean diameter of 3 mm and included epidermis and dermis from patient’s axilla, to include peripheral nerve endings. Skin was taken during anaesthesia conducted within the scope of other diagnostic procedures (e.g. magnet resonance imaging). Due to the heterogeneous study population included, patients age at the time of investigation varies from the first month of livelihood to adulthood (new borns–55 years of life). Samples were fixated in 3% glutaraldehyde–phosphate buffer solution and sent to the Institute of Clinical Pathology, University Hospital Zürich, Switzerland.
During the study period the charges for electron microscopy were kept approximately stable, whereas charges for genetic testing (e.g. whole exome sequencing) fluctuated with ongoing time.
The study was approved by local ethical committee. All parents gave written informed consent before biopsy and the study was conducted following the guidelines of the Declaration of Helsinki (2013).
Sample fixation and electron microscopy
The biopsies were fixed in 3% buffered glutaraldehyde. After post-fixation with buffered osmium tetroxide 1%, dehydration with ethanol and propylenoxide, they were embedded in epon. Semithin sections were stained with methyleneblue/azur II. Ultrathin sections were stained with uranyl acetate and lead citrate. The probes were examined using a Hitachi H-7650 TEM (Tokyo, Japan).
Results
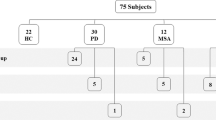
In our study, we included examinations of 74 axillary skin biopsies from patients with either developmental delay with regression, neurological abnormalities and/or neurodegeneration (see Flowchart). Punch skin biopsies were performed at the Department of Paediatrics, Medical University of Innsbruck, Austria. Because of insufficient skin biopsy quality, for 8 patients no electron microscopy result was obtained.
Of the 66 included biopsies, in 37 patients abnormalities were detected by electron microscopy (56.1%) (Fig. 1).
37/66 patients with abnormal electron microscopic findings
In 21/37 a diagnosis exists, in 16 the diagnosis remains unclear.
In the six patients with neuronal ceroid lipofuscinosis (6/21 (28.6%) electron microscopy provided a definitive diagnosis. In further three electron microscopy found storage pointing to a lysosomal storage diseases which was confirmed. In two later diagnosed mitochondrial disorders, electron microscopy found abnormal hypertrophic polytrophic mitochondria with normal lysosomes. In six patients neurological syndromes were identified, in these, electron microscopy found multiple unspecific ultrastructural changes. Especially, in the patient with later identified primary coenzyme Q10 deficiency mutation, the electron microscopy prompted further investigations (Table 1). 21, spondyloenchondrodysplasia,/21, spondyloenchondrodysplasia, DDHD2 mutation, phosphatidylinositol N-acetylglucosaminyltransferase subunit Q mutation and anhidrotic ectodermal dysplasia were identified (Table 2 and Fig. 1).
29/66 patients without abnormal electron microscopy findings
In 4/29 a diagnosis was found (13.8%), in 25/29 the diagnosis remains unclear.
Electron microscopy showed normal results in 29/66 (44%) patients. In four of these an underlying diagnosis was found: Gaucher’s disease type III, Di George syndrome, opsoclonus-myoclonus-syndrome (Kinsbourne syndrome), Canavan syndrome, respectively (Fig. 1). For the patient with Gaucher's disease type III, the material was rather autolytic (post mortem sample), thus difficult to evaluate. Even so, there are reports in literature with normal skin biopsy electron microscopy findings in confirmed Gaucher patients [2, 10].
Findings on electron microscopy
In six patients a neuronal ceroid lipofuscinosis was diagnosed through electron microscopy. The morphological diagnosis of neuronal ceroid lipofuscinosis on skin biopsy was established by the presence of a mixture of fingerprints and curvilinear structures in the electron microscopical examination (Fig. 2 and Table 1).

Flow Chart 1: electron microscopy results
In one case of aspartylglucosaminuria (Patient 7) the evidence of a lysosomal storage disease was established by the fibrillar material in fibroblasts and apocrine cells, as well as lamellated bodies in nerves and granules of glycogen in fibroblasts and apocrine cells (Fig. 3). In one patient (Patient 8), the morphological criteria of a Niemann–Pick disease were detected by electron microscopy (sphingomyelin and cholesterol stored as coarse, lamellated membrane structures) (Fig. 4).

Specific changes: Sweat gland cell with fingerprint, characteristic for neuronal ceroid lipofuscinosis; ×30,000

Non-specific changes: Fibroblast with enlarged secondary lysosome that contains varied amounts of fine fibrillary material and membrane structures; ×5000
In another patient (Patient 9), accumulated lamellated membrane structures were detected (Fig. 5). This patient was diagnosed having a ganglioside storage disease (GM1 gangliosidosis).

Specific changes: Fibrocyte with secondary lysosomes containing coarse lamellate membrane structures, characteristic for Niemann–Pick disease; ×6000
In two patients (Patient 10 and 11), a mitochondrial disease was suspected due to abnormal mitochondria on electron microscopy. Primary coenzyme Q10 deficiency (a mitochondrial disease) was diagnosed post mortem due to abnormal mitochondrial structure in various cell types (Fig. 6). In the second patient an ATP-synthetase deficiency was diagnosed. The electron microscopy of this patient showed increased, polymorphic mitochondria.

Changes suggestive for a certain abnormality: Sweat gland cell of a patient with GM1 gangliosidosis: secondary lysosomes containing lamellate membranes and fine fibrillary material; ×8000
An abnormal electron microscopical result was also detected in patients with other underlying diseases (4/66) (see Fig. 1 and Table 2). In Patient 12, the electron microscopic findings of lamellated membrane structures and fine fibrillary material was interpreted as possible lysosomal storage diseases, and later confirmed to be a glycosylphophaditylinositol anchor disease with compound heterozygote phosphatidylinositol N-acetylglucosaminyltransferase subunit Q mutation (Fig. 7). In Patient 12, the pathological electron microscopy finding prompted further genetic testing to pursue diagnosis. In Patient 13 with a DDHD2 mutation confirmed 2 months after skin biopsy, inclusions in mitochondria similar to alterations found in some lysosomal storage diseases were seen. In Patient 14 with spondyloenchondrodysplasia dense material (probably glycogen) was found. In Patient 15 with an anhidrotic ectodermal dysplasia alterations similar to mucolipidosis IV for example dense material (probably glycogen) in many organelles was detected.

Changes suggestive for a certain abnormality: Sweat gland cell of a patient with primary coenzyme Q10 deficiency (a mitochondrial disease): numerous, large abnormal mitochondria; ×7000

Changes suggestive for a certain abnormality: Sweat gland cell of a patient with glycosylphophaditylinositol anchor disease with compound heterozygote PIGQ mutation: secondary lysosomes containing fine fibrillary material and lamellate membranes; ×6000
Unspecific changes with diagnosis
In 6/20 cases with mainly neurological problems without developmental regression, microscopical alterations have been detected (Table 3). In patient 16 with hereditary spastic paraplegia, degenerative alterations within peripheral nerves were revealed by electron microscopy. In patient 17 with epileptical encephalopathy due to West Syndrome, storage of glycogen and oligosaccharides was found in eccrine ducts and Schwann cells.
In patient 18 with progressive myoclonic epilepsy, oligosaccharides, glycogen and fibrillary material was detected in Schwann cells and nerves. In patient 19 with early onset autism, degenerative alterations within the mitochondria were seen by electron microscopy.
In the skin biopsy from Patient 20 with febrile infection-related-epilepsy syndrome, degenerative alterations in myelinated axons of peripheral nerves were seen. In Patient 21 with hypomyelinating leucodystrophia Type 4 scattered lysosomal vacuoles have been found in fibrocytes of the dermis.
Unspecific changes without diagnosis
9/66 skin biopsies from patients with neurological symptoms revealed alterations, but no diagnosis was found so far (Table 4). In Patient 25 with dyskinetic cerebral palsy and epilepsy, fingerprints and curvilinear structures were detected, but enzymatic and genetic tests for lysosomal storage disease were normal, especially for neuronal ceroid lipofuscinosis. Skin biopsy of Patient 27 with developmental delay revealed lamellated material in several cell lines (e.g. oligosaccharides). No hint for a lysosomal storage disease was found.
In seven other cases there were morphological alterations of lysosomes and/or mitochondria on electron microscopy, but no definite diagnosis could be established so far (Table 4).
In an additional patient, where no diagnosis has yet been found, electron microscopy detected lysosomal storage, raising the suspicion of Pompe disease, deposition of oligosaccharides, and unspecific alterations, respectively. However, the further diagnostic work up did not confirm any of the suspected diagnoses.
Discussion
Electron microscopic examination of skin biopsy samples provides essential information about the number, size and content of various cell types (ectodermal, endodermal and mesodermal cell types including neuronal elements) [2, 3, 5]. Prior studies from the mid-1970s have shown that electron microscopy of skin biopsies is a proven inexpensive and sensitive screening tool for detection of certain lysosomal storage diseases [2, 3, 5,6,7,8,9].
In our study, in 25/66 (37.8%) patients who underwent an axillary skin biopsy, a definitive diagnosis was found due to findings of electron microscopic axillary skin biopsy. In patients with a neuronal ceroid lipofuscinosis electron microscopic examination is currently a proven method to diagnose the various forms of the disease [2, 10]. Six within our study population suffer from a neuronal ceroid lipofuscinosis, which was primarily diagnosed by electron microscopy. Neuronal ceroid lipofuscinosis comprises a group of nine different lysosomal storage diseases with various etiologies [2]. In some cases the ultrastructure of the storage material is unique for a defined type of neuronal ceroid lipofuscinosis. Via electron microscopy a mixture of fingerprints and curvilinear structures can be identified characterising the different types of neuronal ceroid lipofuscinosis [2, 10]. Accordingly, in six neuronal ceroid lipofuscinosis patients with curvilinear bodies and fingerprints electron microscopy gave a definitive diagnosis.
In the 1990s, in Niemann–Pick disease loosely arrayed dark lamellated structures within a clear matrix were seen in macrophages, axons, pericytes, Schwann cells, smooth muscle cells and fibroblasts [5]. In our patient with Niemann–Pick type C disease (Patient 8), similar alterations were present. Comparable to former results, we detected fine lamellated membrane structures, and coarsed vacuoles of ganglioside in a patient with GM1 (Patient 9) [2].
A histological examination of the skin provides an insight into various cell types present [8]. In our patients with primary coenzyme Q10 deficiency, aspartylglucosaminuria and ATP-synthase deficiency, ultrastructural study by electron microscopy revealed interesting findings. According to a mitochondrial disease, altered mitochondrial structures were detected in ATP-synthase deficiency (Patient 10) and coenzyme Q10 deficiency (Patient 11). Thus, also in mitochondrial disease the specific structure of different cell types in electron microscopical examinations might be a diagnostic screening tool. Up to date there is no specific classification on a phenotype/genotype correlation for electron microscopical examination for different mitochondrial diseases. Considering the wide range of mitochondrial disease and the often secondary involvement of mitochondria in multiple disorders, we would not expect that to be possible. However, if an altered mitochondrial structure is seen, it supports further follow-up. One limitation of our study is the small number of patients with mitochondrial disease examined by electron microscopy. However, mitochondrial disorders were not the main target, but storage disorders (i.e. lysosomal storage diseases).
In the skin biopsy samples of patients with primarily neurological disorders (Patients 16–20] specific alterations within nerves and neuronal structures of the skin were detected (Table 3). The definite diagnosis was genetically found and confirmed (Table 3). It is so far unclear, whether these findings could be disease specific. Former studies revealed that in some skin biopsies storage of certain lipids occurs which cannot be distinguished from alterations found in classical lysosomal storage disorders [2, 11]. Some systemic cationic amphophilic drugs (amiodarone, chloroquine, etc.) and certain locally administered topical anaesthetic creams (e.g. Emla Crème®), can lead to storage of certain substances within a patients’ skin [12, 13]. Our study cannot support this data. Biopsy was either taken under general anaesthesia or with the use of Emla Crème® in our patients. However, the use of Emla Crème was not a predictive marker for the lack of a diagnosis in spite of a pathological biopsy result. On the other hand, these data show that a detailed history including the used drugs and possible pre-treatment procedures is essential to interpret results.
The morphologic examination of skin biopsy samples by electron microscopy might reveal structural alterations specific for a defined disease, in these cases leading to a rapid confirmation of a definitive diagnosis. However, this is only valid for very few of the diseases. In one of our patients skin biopsy was negative despite later confirmation of Gaucher’s disease. This might be due to the fact of insufficient biopsy material.
Mostly, the findings of storage material on electron microscopy help to confirm the clinical suspicion of, i.e. lysosomal storage diseases and narrow down further work-up. This is even truer for patients with mitochondrial disease, as there ultrastructural alterations on skin biopsy point to a mitochondrial disturbance but rarely give direction to the exact underlying defect.
Although our data are a retrospective collection, we illustrated that many inherited disorders (lysosomal storage disease, mitochondrial diseases, and certain inherited neurological disorders) show ultrastructural alterations within skin biopsies. In the last century electron microscopic examination of skin biopsy was proven to have a high significance in the diagnostic work up for lysosomal storage diseases (e.g. neuronal ceroid lipofuscinosis). Our data emphasises that for narrowing down diagnosis of disorders when the direction of work-up is unclear, “old fashioned” electron microscopic examination of skin biopsies may give a hint for the direction of further diagnostic testing. Taken together, electron microscopic evaluation of skin biopsies is a quick, useful and particularly relatively inexpensive method to screen for certain inherited disorders which may help to narrow down potential further diagnostic procedures (e.g. exome and whole-genome sequencing).
References
Seregin SS, Amalfitano A. Gene therapy for lysosomal storage diseases: progress, challenges and future prospects. Curr Pharm Des. 2011;17:2558–74.
Alroy J, Ucci AA. Skin biopsy: a useful tool in the diagnosis of lysosomal storage diseases. Ultrastruct Pathol. 2006;30:489–503.
Prasad A, Kaye EM, Alroy J. Electron microscopic examination of skin biopsy as a cost-effective tool in the diagnosis of lysosomal storage diseases. J Child Neurol. 1996;11:301–8.
Neufeld EF. Lysosomal storage diseases. Annu Rev Biochem. 1991;60:257–80.
Boustany RN, Kaye E, Alroy J. Ultrastructural findings in skin from patients with Niemann–Pick disease, type C. Pediatr Neurol. 1990;6:177–83.
Burck U, Moser HW, Goebel HH, Grüttner R, Held KR. A case of lipogranulomatosis Farber: some clinical and ultrastructural aspects. Eur J Pediatr. 1985;143:203–8.
Goebel HH, Kimura S, Harzer K, Klein H. Ultrastructural pathology of eccrine sweat gland epithelial cells in globoid cell leukodystrophy. J Child Neurol. 1993;8:173–4.
Holbrook KA, Byers PH. Skin is a window on heritable disorders of connective tissue. Am J Med Genet. 1989;34:105–21.
Merin S, Livni N, Berman ER, Yatziv S, Mucolipidosis IV. Ocular, systemic, and ultrastructural findings. Invest Ophthalmol. 1975;14:437–48.
Nijssen PCG, Ceuteric C, van Diggelen OP, Elleder M, Martin JJ, Teepen JL, et al. Autosomal dominant adult neuronal ceroid lipofuscinosis: a novel form of NCL with granular osmiophilic deposits without palmitoyl protein thioesterase deficiency. Brain Pathol. 2003;13:574–81.
Elias PM, Williams ML. Neutral lipid storage with ichthyosis, defective lamellar body contents and intracellular dispersion. Arch Dermatol. 1985;121:1000–8.
Hruban Z. Pulmonary and generalized lysosomal storage induced by amphophilic drug. Environ Health Perspect. 1984;55:53–76.
Vallance H, Chaba T, Clarke L, Taylor G. Pseudo-lysosomal storage disease caused by EMLA cream. J Inherit Metab Dis. 2004;27:507–11.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Shared senior authorship: Daniela Mihic-Probst, Daniela Karall.
Rights and permissions
About this article

Cite this article
Zlamy, M., Hofstätter, J., Albrecht, U. et al. The value of axillary skin electron microscopic analysis in the diagnosis of lysosomal storage disorders. Mod Pathol 32, 755–763 (2019). https://doi.org/10.1038/s41379-019-0201-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-019-0201-4
This article is cited by
-
Auf der Suche nach der Nadel im Heuhaufen
Pädiatrie (2021)
-
Auf der Suche nach der Nadel im Heuhaufen
Pädiatrie & Pädologie (2021)
