
Abstract
Dedifferentiated liposarcoma is defined as progression of atypical lipomatous tumor/well-differentiated liposarcoma to a higher grade usually non-lipogenic sarcoma, with amplification of 12q13-15. This region contains several genes involved in liposarcoma pathogenesis, including MDM2, CDK4, and DDIT3. While the former two are thought of as the main drivers in dedifferentiated liposarcoma, DDIT3 is typically rearranged in myxoid liposarcoma. Overexpression of DDIT3, along with MDM2 and CDK4, may contribute to the pathogenesis of dedifferentiated liposarcoma by interfering with adipocytic differentiation. Dedifferentiated liposarcoma with DDIT3 amplification has not been well characterized. In this study we evaluate the presence of DDIT3 amplification in 48 cases of dedifferentiated liposarcoma by cytogenomic microarray analysis and its correlation with demographic, clinical, and morphologic characteristics. Data from The Cancer Genome Atlas were also evaluated to determine a relationship between DDIT3 amplification and prognostic outcomes. Of the 48 cases, 16 (33%) had amplification of DDIT3; these patients were on average 11 years younger than patients without DDIT3 amplification (P < 0.05). Myxoid liposarcoma-like morphologic features were identified in 12/16 (75%) cases with DDIT3 amplification and in 7/32 (22%) cases without amplification (P < 0.05). Homologous lipoblastic differentiation was seen in 6/16 (38%) cases with DDIT3 amplification and 2/32 (6%) cases without it (P < 0.05). There was no significant correlation between DDIT3 amplification and tumor location, disease-specific or recurrence-free survival, and distant metastasis. DDIT3 amplification appears to interfere with the adipogenic molecular program and plays a role in inducing or maintaining a lipogenic phenotype in dedifferentiated liposarcoma. From a diagnostic standpoint, it is important to consider DDIT3-amplified dedifferentiated liposarcoma in the differential diagnosis of myxoid liposarcoma, particularly in small biopsies. Further studies evaluating the significance of DDIT3 amplification in the pathogenesis of dedifferentiated liposarcoma, as well as a potential predictor of tumor behavior in well-differentiated liposarcoma, are needed.
Similar content being viewed by others
Introduction
Liposarcoma is the most common type of sarcoma in humans, accounting for approximately 20% of all cases, and is subdivided into three categories: atypical lipomatous tumor/well differentiated liposarcoma/dedifferentiated liposarcoma, myxoid liposarcoma, and pleomorphic liposarcoma. These neoplasms have a wide morphologic spectrum with different clinical behaviors and defining genetic alterations. Among these, well-differentiated liposarcoma/atypical lipomatous tumor/dedifferentiated liposarcoma is the most prevalent subtype [1]. Atypical lipomatous tumor/well-differentiated liposarcoma is a low-grade, slowly growing neoplasm with a high rate of local recurrence, which typically arises in the extremities, retroperitoneum, and, less commonly, in the inguinal/paratesticular region. It has a tendency to present in older adults of either sex. Atypical lipomatous tumor/well-differentiated liposarcoma has a wide morphologic spectrum, including tumors composed of predominantly mature adipose tissue to myxoid, inflammatory, and sclerosing morphologies with a less conspicuous adipocytic component. Dedifferentiated liposarcoma is generally defined as the development of higher grade sarcoma arising from atypical lipomatous tumor/well-differentiated liposarcoma in which lipogenic differentiation is most often lost; however, a subset of dedifferentiated liposarcoma may contain homologous lipoblastic differentiation and morphologically resemble pleomorphic liposarcoma. Dedifferentiated liposarcoma is characterized as having more aggressive behavior, including metastatic potential. Whether or not there is a low-grade variant of dedifferentiated liposarcoma remains an unresolved question in the literature.
At a genetic level, atypical lipomatous tumor/well-differentiated liposarcoma and dedifferentiated liposarcoma are characterized by the presence of supernumerary ring and giant marker chromosomes containing amplified sequences from the 12q13-15 region that include gene loci for MDM2 (murine double minute 2), CDK4 (cyclin-dependent kinase 4), and others [1,2,3,4]. Detection of MDM2 gene amplification by fluorescence in-situ hybridization (FISH) or MDM2 protein expression by immunohistochemistry are common methods currently used to help confirm the diagnosis of atypical lipomatous tumor/well-differentiated liposarcoma and dedifferentiated liposarcoma. Additionally, high copy numbers of MDM2 and CDK4 have been correlated with a lower disease-free survival in dedifferentiated liposarcoma [1], and direct inhibitors of CDK4 have been developed as a targeted therapeutic option for these entities [5].
In contrast to atypical lipomatous tumor/well-differentiated liposarcoma and dedifferentiated liposarcoma, myxoid liposarcoma typically arises in the extremities of young adults and has a tendency to metastasize to unusual locations, such as the contralateral extremity and bone. These lesions are morphologically characterized by having uniform spindle cells intermixed with univacuolated lipoblasts in an abundant myxoid background with a characteristic “chicken-wire” vascular pattern. A round cell component can also be observed, which is typically associated with more aggressive clinical behavior [6]. These neoplasms are genetically defined by having fusions of the DDIT3 (DNA damage inducible transcript 3) gene located on 12q13.2 with partners belonging to the TET family, most commonly FUS, followed by EWSR1 [2, 6, 7].
Given the geographic proximity of MDM2, CDK4, and DDIT3, it is possible for the latter to be amplified in atypical lipomatous tumor/well-differentiated liposarcoma and dedifferentiated liposarcoma. Amplification of DDIT3 has been previously described in a number of cases in which it was loosely associated with the presence of myxoid liposarcoma-like features [7, 8]; however, specific associations between DDIT3 gene amplification and clinical, morphologic, and prognostic features have not yet been fully characterized. In this study we evaluate the clinical, pathologic, and prognostic significance of DDIT3 gene amplification in 48 cases of dedifferentiated liposarcoma.
Methods
Patient selection
The institutional electronic pathology database was searched to retrospectively identify patients diagnosed with dedifferentiated liposarcoma and treated at our institution between January 1999 and August 2017. The study protocol was reviewed and approved by the institutional review board (University of Washington, Human Subjects Division). Cases that had sufficient material available for review were included and any material appropriate for cytogenomic microarray analysis was acquired. Of these 48 cases of dedifferentiated liposarcoma, 47 were selected as part of the cited study by Ricciotti et al. [1].
Clinical and pathologic review
All archived slides for cases with diagnosis of dedifferentiated liposarcoma were reviewed by 4 soft tissue pathologists in conjunction with information contained in the surgical pathology report. Clinical follow-up information was acquired from the institutional electronic medical records.
The diagnosis in all cases was confirmed by using morphologic findings as well as any pre-existing immunohistochemical, cytogenetic, and FISH testing results available for review. Dedifferentiated liposarcoma was defined as a region within a background of atypical lipomatous tumor/well-differentiated liposarcoma completely devoid of lipoblastic differentiation occupying at least 10% of the tumor or two continuous low-power (×4 objective) microscopic fields [9]. Homologous lipoblastic differentiation resembling pleomorphic liposarcoma was also considered part of the dedifferentiated component. The Federation Nationale des Centres de Lutte Contre le Cancer (FNCLCC) grading system was used to grade the dedifferentiated areas according to tumor differentiation (score 1–3), mitotic count per 10 high-power fields in the most mitotically active area (score 1–3), and tumor necrosis (0–2). This is a modification of the FNCLCC grading system, allowing for a differentiation score of 1 in contrast to the World Health Organization (WHO) recommendations, as used in a previous study by Jour et al. [9]. For the differentiation score, tumors with bland spindle cells with at most mild nuclear atypia and morphologic features resembling benign or low-grade fibroblastic/myofibroblastic neoplasms (fibromatosis-like) were given a score of 1. Tumors with moderate atypia and/or a morphologically recognizable line of differentiation were given a differentiation score of 2 (i.e., myxofibrosarcoma-like). A differentiation score of 3 was assigned to tumors having pleomorphic cells with severe atypia and/or no morphologic evidence of a particular line of differentiation (i.e., undifferentiated pleomorphic sarcoma-like morphology). The final grade was assigned based on the highest grade present in the tumor.
The well-differentiated and dedifferentiated regions of all cases were evaluated for additional histologic features, including the presence of myxoid liposarcoma-like features (prominent myxoid stroma with branching capillary network and associated uniform, oval neoplastic cells lacking pleomorphism), round cell component, and homologous lipoblastic differentiation resembling pleomorphic liposarcoma including pleomorphic lipoblasts with multivacuolated cytoplasm.
Cytogenomic microarray analysis
All specimens used in this study were archived formalin-fixed, paraffin-embedded tissue specimens. Whole genome microarray analysis was performed using formalin-fixed, paraffin-embedded tissues of 48 dedifferentiated liposarcomas. The methods for DNA isolation from formalin-fixed, paraffin-embedded specimens, cytogenomic microarray analysis, and copy number evaluation have been described previously [1, 10]. Cytogenomic microarray analysis was performed using the Agilent SurePrint G3 Cancer CGH+SNP 4×180K Array (Agilent Technologies, Santa Clara, CA, USA). Amplification was defined as 5 or more gene copies.
Evaluation of TCGA data
To evaluate the prognostic significance of DDIT3 amplification in dedifferentiated liposarcoma, an additional subset of 56 cases were extracted from The Cancer Genome Atlas (TCGA) through cBioPortal (http://www.cbioportal.org/), and cases with amplification of DDIT3 were identified. Prognostic data, including length of follow-up, local recurrence, disease specific mortality and metastasis were reviewed for both The Cancer Genome Atlas (TCGA) and University of Washington datasets.
Statistical analysis
To evaluate the correlation between specific morphologic features and the presence of DDIT3 amplification, as well as to compare clinical outcomes and categorical demographic characteristics, such as sex and anatomic location, data were compared using Fisher’s exact test and patient age was compared using independent T-test. In all variables, statistical significance was determined as P < 0.05. Analysis evaluating potential differences in recurrence-free and disease-related survival was undertaken using the Kaplan–Meier method on two groups (patients with DDIT3 amplification and patients without amplification) built from the combined cohorts of the University of Washington and the Cancer Genome Atlas, and the survival curves were compared using log-rank tests.
Results
Morphologic features and correlation with DDIT3 amplification status
The 48 cases of dedifferentiated liposarcoma were classified by the modified FNCLCC criteria as follows: 4 cases as grade 1, 29 as grade 2, and 15 as grade 3, and DDIT3 gene amplification was present in 3 of 4 (75%) grade 1, 11/29 (38%) grade 2, and 2/15 (13%) grade 3 tumors. While there appears to be a trend between histological grade, with grade 1 and 2 tumors more commonly having DDIT3 amplification, no statistically significant association between histologic grade and DDIT3 amplification was identified (P = 0.052).
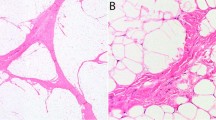
Morphologic features resembling myxoid liposarcoma were defined by the presence of myxoid stroma in combination with a branching, capillary vascular pattern and neoplastic cells lacking pleomorphism. These features were identified in variable proportion, ranging from less than 5% to up to 70% of the primary tumor, in a total of 19 cases (40%). There was a statistically significant association between amplification of DDIT3 and myxoid liposarcoma-like features, which were identified in 12/16 (75%) cases of dedifferentiated liposarcoma with DDIT3 amplification, and 7/32 cases (22%) without amplification (P = 0.0006). These areas were most commonly observed in areas transitioning from the well-differentiated component to areas of higher grade dedifferentiation (Table 1 and Fig. 1). One case with extensive myxoid liposarcoma-like areas, initially diagnosed as well-differentiated liposarcoma, developed bone metastases (without lung metastases) that were entirely composed of myxoid liposarcoma-like areas.

Morphologic features resembling myxoid liposarcoma in cases of dedifferentiated liposarcoma, including the presence of cytologically bland spindle cells, loose myxoid stroma, and a characteristic thin, branching vascular pattern in variable proportions (a–c). These features were significantly associated with DDIT3 amplification. Areas resembling round cell liposarcoma (d) were noticed in two cases of dedifferentiated liposarcoma with DDIT3 amplification (hematoxylin and eosin (H&E), ×200)
Homologous lipoblastic differentiation, defined as the presence of pleomorphic lipoblasts with features resembling pleomorphic liposarcoma, was identified in 8 cases of dedifferentiated liposarcoma, including 6/16 with DDIT3 amplification (38%), and 2/32 cases (6%) without amplification. This finding was also significantly associated with DDIT3 gene amplification (P = 0.0115) (Fig. 2). These areas were always seen as part of the dedifferentiated component. Of the 8 cases with homologous lipoblastic differentiation, 6 (75%) also had myxoid liposarcoma-like morphology. Additionally, a distinct component closely resembling round cell liposarcoma was identified in 2 cases, both of which had DDIT3 amplification. Both of these cases also contained areas of myxoid liposarcoma and homologous lipoblastic differentiation.

Homologous lipoblastic differentiation consisting of pleomorphic lipoblasts within regions resembling pleomorphic liposarcoma (a–d) was significantly associated with amplification of DDIT3 in dedifferentiated liposarcoma (H&E, ×200)
Demographic characteristics and clinical outcomes
Patients with DDIT3-amplified liposarcomas were significantly younger, with a mean age of 54 years, compared to 65.5 years in patients without DDIT3 amplification (P = 0.022). No significant differences in male to female ratio (P = 0.76) or anatomic location were observed between patients with and without DDIT3 gene amplification in our cohort. These findings are summarized in Table 2.
Comparison of clinical outcomes in the combined University of Washington and the Cancer Genome Atlas cohorts demonstrated no statistically significant difference in recurrence-free survival or disease specific survival for both groups between cases with and without DDIT3 amplification (Fig. 3). No difference in the rate of distant metastasis was observed between both groups (P = 1) (Table 3); however, the number of metastatic events was low.

Survival curves comparing cases of dedifferentiated liposarcoma with and without amplification of DDIT3. No statistically significant difference was observed between both groups in terms of disease-related survival and recurrence-free survival
Amplification status of DDIT3, MDM2 and CDK4
Of the 48 cases evaluated by cytogenomic microarray analysis, DDIT3 gene amplification was present in 16 (33%). While the entire gene was amplified in 15 cases, we found amplification of a truncated DDIT3 gene in one case, in which exon 1 was not amplified. The mean copy number in cases with DDIT3 amplification was 18 (range 6–45). The presence or absence of DDIT3 amplification showed no significant correlation with the number of copies of MDM2 and/or CDK4 (Table 4).
Discussion
Atypical lipomatous tumor/well-differentiated liposarcoma and dedifferentiated liposarcoma combined represent the most common category of liposarcoma and are characterized by amplification of genes from the 12q13‒15 region including MDM2, CDK4, DDIT3, HMGA2, FRS2, and others [1]. The pathogenesis of atypical lipomatous tumor/well-differentiated liposarcoma/dedifferentiated liposarcoma is not completely understood. While amplification of genes at 12q13-15 has been known for quite some time, the precise molecular mechanisms resulting in initiation and propagation of adipocytic neoplastic cells remains to be fully elucidated. The amplicons in dedifferentiated liposarcoma can be complex and contain a variety of genes with functions ranging from cell cycle regulation (e.g., MDM2 and CDK4) to proliferation and growth factor receptor signaling (e.g., FRS2). These are amplified to varying degrees and some genes may even be truncated, as seen in our series. HMGA2, MDM2, CDK4, and JUN have been implicated as playing major roles in the pathogenesis of atypical lipomatous tumor/well-differentiated liposarcoma and dedifferentiated liposarcoma and have shown clinical prognostic significance [11]. HMGA2 may be the genetic cornerstone for lipomatous neoplasms by maintaining a degree of adipocytic differentiation [11]. These genes have oncogenic effects but do not necessarily inhibit the adipocytic differentiation program. More recent studies have demonstrated amplification of receptor tyrosine kinase genes in approximately one-third of atypical lipomatous tumor/dedifferentiated liposarcoma [12], and have implicated a role for micro-RNA in liposarcoma. A recent study demonstrated miR-193b tumor suppressor activity and oncogenic roles of FAK–SRC–CRKL (focal adhesion kinase–steroid receptor coactivator–CRK-like proto-oncogene) signaling and MRSA (methicillin-resistant Staphylococcus aureus)-regulated reactive oxygen species signaling in atypical lipomatous tumor/dedifferentiated liposarcoma [13].
The presence of myxoid liposarcoma-like features in MDM2-amplified liposarcoma was described by Sioletic et al. [14], who noticed the presence of myxoid stroma in 56 cases, including 22 well-differentiated and 34 dedifferentiated liposarcoma, and a myxoid liposarcoma-like vascular pattern in 11/22 well-differentiated liposarcomas. These features have now been well characterized in a subset of dedifferentiated liposarcomas [7, 8, 15]. Homologous lipoblastic differentiation, defined as the presence of pleomorphic lipoblasts, either admixed within spindle cell areas or in solid sheets closely resembling pleomorphic liposarcoma [16], has been previously described in a subset of dedifferentiated liposarcomas [16,17,18]. However, these studies did not assess the association of myxoid liposarcoma-like features or homologous lipoblastic differentiation with a specific genetic alteration or prognostic implications.
In our study, we observe that DDIT3 is amplified in 33% of the cases of dedifferentiated liposarcoma and significantly associated with the presence of myxoid liposarcoma-like features compared to cases without amplification. The proportion of the myxoid liposarcoma-like component was variable throughout the cases in which these features were identified. In addition, a significant percentage of dedifferentiated liposarcomas with DDIT3 amplification contained areas of homologous lipoblastic differentiation in the dedifferentiated component, supporting an association between both of these morphologic features and amplification of DDIT3. Rarely, dedifferentiated liposarcoma with DDIT3 amplification contained round cell liposarcoma-like regions, the significance of which is unknown given the limited number of cases.
DDIT3 (formerly CHOP) encodes for a nuclear protein in the CCAAT/enhancer-binding protein (C/EBP) family, which functions primarily as a transcriptional regulator, and has a wide spectrum of biological functions [19], including cell survival and apoptosis in association with endoplasmic reticulum stress [20], odontogenesis [21], and adipogenesis [22, 23]. With respect to adipogenesis, DDIT3 is normally expressed late in adipocytic differentiation and the timing of expression is critical for normal adipogenesis [24]. It has been postulated that over-expression of DDIT3 can block terminal differentiation of pre-adipocytic cells [25]. More so, in the context of liposarcoma pathogenesis, aberrant DDIT3 expression in neoplastic cells may induce an adipocytic differentiation program while simultaneously blocking later stages of adipogenesis [26].
In prior studies, transfection of HT1080 fibrosarcoma cells with DDIT3 has been associated with markedly increased adipogenesis in vitro, including the presence of abundant univacuolated signet ring cell-like lipoblasts, when compared to control cells [25]. The same study also demonstrated that inoculation with DDIT3 and FUS-DDIT3-expressing human xenografts established in severe combined immunodeficient (SCID) mice led to the development of multiple tumors with liposarcoma-like morphology, including the presence of branching capillary vessels similar to those seen in myxoid liposarcoma; these features were not identified in mice inoculated with non-transfected cells of the same line. Similarly, transgenic mice overexpressing FUS-DDIT3 have been shown to develop tumors that are morphologically similar to human myxoid liposarcoma [27]. Based on these findings, over-expression of DDIT3 plays a major role in inducing morphologic features resembling myxoid liposarcoma. However, current evidence does not support the normal DDIT3 protein having a role as a driving oncoprotein in the development of dedifferentiated liposarcoma as it seems to have in myxoid liposarcoma [26].
Homologous lipoblastic differentiation in dedifferentiated liposarcoma manifested as a pleomorphic liposarcoma-like dedifferentiated component is a relatively recently recognized variant of dedifferentiated liposarcoma. Our study is the first to demonstrate a significant association between DDIT3 amplification and pleomorphic liposarcoma-like morphology in dedifferentiated liposarcoma. As previously described, DDIT amplification usually produces myxoid liposarcoma-like morphology. How DDIT3 amplification might drive a pleomorphic liposarcoma-like phenotype in addition to myxoid liposarcoma-like morphology remains to be elucidated.
Based on prior evidence supporting the role of DDIT3 in adipogenic differentiation, our findings suggest that its amplification contributes to adipocytic morphological phenotype in a subset of dedifferentiated liposarcoma, characterized by variable amounts of myxoid liposarcoma-like differentiation and/or homologous lipoblastic differentiation. DDIT3 may lead to this phenotype by interfering with normal molecular programs of adipogenesis, resulting in molecular dynamics that favor lipogenic differentiation. In at least one case, the primary tumor had extensive myxoid liposarcoma-like areas and bone metastases composed entirely of myxoid liposarcoma-like morphological phenotype; this exemplifies how DDIT3 amplification can produce morphological changes that can obscure the dedifferentiated nature of a given case. Similar to HMGA2, DDIT3 may contribute to the maintenance of an adipocytic phenotype, despite the underlying mechanisms that are driving biological behavior. Cases with more extensive myxoid liposarcoma-like features raise the question of the existence of a separate myxoid liposarcoma-like variant of dedifferentiated liposarcoma. However, large numbers of cases would need to be analyzed to answer such a question.
Our study only analyzed the presence of DDIT3 amplification and did not look at the relevance of the levels of amplification. Furthermore, we did not correlate gene amplification with RNA or protein expression. Understanding the full significance of DDIT3 amplification and the mechanisms by which it induces adipocytic phenotypic changes in dedifferentiated liposarcoma requires further analysis including at the protein, RNA, and metabolic levels.
From a prognostic standpoint, no statistically significant difference in disease-related mortality, recurrence, or the incidence of metastatic disease was identified between dedifferentiated liposarcoma cases with DDIT3 amplification and those without it. This seems to be in keeping with the current understanding of normal DDIT3 not having a major role as a driving oncogene in the development of liposarcoma. However, our study did demonstrate a trend toward DDIT3 amplification correlating with low histologic grade, which has been shown to have prognostic significance [10]. Of note, one case in which DDIT3 was truncated is interesting in that it raises the question of whether or not abnormal DDIT3 has an oncogenic role in the pathogenesis of liposarcoma, or as a prognostic factor, given the presence of metastatic disease in one of these cases. Additional study of a larger series of cases with truncated DDIT3 is required to better understand the significance of abnormal forms of DDIT3 in dedifferentiated liposarcoma.
From a diagnostic standpoint, DDIT3-amplified dedifferentiated liposarcoma with areas resembling myxoid liposarcoma and/or with homologous lipoblastic differentiation may be inaccurately classified as myxoid liposarcoma or pleomorphic liposarcoma, respectively, particularly in small biopsy specimens or with lesions located in anatomic areas where these subtypes are more likely to arise, such as in the extremities. Given the differences in surgical and medical treatment between these entities, it is important to be aware of this potential diagnostic pitfall. Notably, dedifferentiated liposarcoma with DDIT3 amplification likely accounts for a subset, if not the majority, of cases historically diagnosed as retroperitoneal myxoid liposarcoma.
Finally, dedifferentiated liposarcoma with DDIT3 amplification significantly correlated with younger patient age. Based on available information, it is unclear why cases with DDIT3 amplification would occur in younger patients. Analysis of a larger series of cases would be needed to confirm the correlation between younger age and DDIT3 amplification.
In summary, amplification of DDIT3 was identified in 33% of all cases of dedifferentiated liposarcoma evaluated and was significantly associated with myxoid liposarcoma-like morphologic features and homologous lipoblastic differentiation. This is the first study to demonstrate such a relationship. Based on current evidence, DDIT3 in its normal state does not appear to have a driving oncogenic role and is not associated with a different prognosis in dedifferentiated liposarcoma. Importantly, DDIT3 amplification can maintain a differentiated adipocytic phenotype even in cases with aggressive clinical behavior. Future studies to more fully elucidate the role of DDIT3 and its relationship to other pathogenic molecular mechanisms in dedifferentiated liposarcoma, and to evaluate the clinicopathologic significance of these factors in atypical lipomatous tumor/well-differentiated liposarcoma, are necessary.
References
Ricciotti RW, Baraff AJ, Jour G, Kyriss M, Wu Y, Liu Y, et al. High amplification levels of MDM2 and CDK4 correlate with poor outcome in patients with dedifferentiated liposarcoma: A cytogenomic microarray analysis of 47 cases. Cancer Genet. 2017;218-219:69–80.
Sugita S, Hasegawa T. Practical use and utility of fluorescence in situ hybridization in the pathological diagnosis of soft tissue and bone tumors. J Orthop Sci. 2017;22:601–12.
Weaver J, Downs-Kelly E, Goldblum JR, Turner S, Kulkarni S, Tubbs RR, et al. Fluorescence in situ hybridization for MDM2 gene amplification as a diagnostic tool in lipomatous neoplasms. Mod Pathol. 2008;21:943–9.
Kashima T, Halai D, Ye H, Hing SN, Delaney D, Pollock R, et al. Sensitivity of MDM2 amplification and unexpected multiple faint alphoid 12 (alpha 12 satellite sequences) signals in atypical lipomatous tumor. Mod Pathol. 2012;25:1384–96.
Laroche-Clary A, Chaire V, Algeo MP, Derieppe MA, Loarer FL, Italiano A. Combined targeting of MDM2 and CDK4 is synergistic in dedifferentiated liposarcomas. J Hematol Oncol. 2017;10:123.
Smith TA, Easley KA, Goldblum JR. Myxoid/round cell liposarcoma of the extremities. A clinicopathologic study of 29 cases with particular attention to extent of round cell liposarcoma. Am J Surg Pathol. 1996;20:171–80.
Rao UN, Cieply K, Sherer C, Surti U, Gollin SM. Correlation of classic and molecular cytogenetic alterations in soft-tissue sarcomas: analysis of 46 tumors with emphasis on adipocytic tumors and synovial sarcoma. Appl Immunohistochem Mol Morphol. 2017;25:168–77.
Ma Y, Wei S, Peker D. An extremely rare primary gallbladder myxoid liposarcoma associated with amplification of DDIT3 gene. J Gastrointestin Liver Dis. 2014;23:460–1.
Jour G, Gullet A, Liu M, Hoch BL. Prognostic relevance of Fédération Nationale des Centres de Lutte Contre le Cancer grade and MDM2 amplification levels in dedifferentiated liposarcoma: a study of 50 cases. Mod Pathol. 2015;28:37–47.
Liu YJ, Zhou Y, Yeh MM. Recurrent genetic alterations in hepatitis C-associated hepatocellular carcinoma detected by genomic microarray: a genetic, clinical and pathological correlation study. Mol Cytogenet. 2014;7:81.
Saada-Bouzid E, Burel-Vandenbos F, Ranchere-Vince D, Birtwisle-Peyrottes I, Chetaille C, Bouvier C, et al. Prognostic significance of HMGA2, CDK4, JUN amplification in well-differentiated and dedifferentiated liposarcoma. Mod Pathol. 2015;28:1404–14.
Asano N, Akihiko Y, Mitani S, Kobayashi E, Shiotani B, Komiyama M, et al. Frequent amplification of receptor tyrosine kinases in well-differentiated/dedifferentiated liposarcoma. Oncotarget. 2017;8:12941–52.
Mazzu YZ, Hu Y, Soni RK, Mojica KM, Qin L-X, Agius P, et al. miR-193b-regulated signaling networks serve as tumor suppressors in liposarcoma and promote adipogenesis in adipose-derived stem cells. Cancer Res. 2017;77:5728–40.
Sioletic S, Dal Cin P, Fletcher CD, Hornick JL. Well-differentiated and dedifferentiated liposarcomas with prominent myxoid stroma: analysis of 56 cases. Histopathology. 2013;62:287–93.
Thway K, Jones RL, Noujaim J, Zaidi S, Miah AB, Fisher C. Dedifferentiated liposarcoma: updates on morphology, genetics, and therapeutic strategies. Adv Anat Pathol. 2016;23:30–40.
Mariño-Enríquez A, Fletcher CD, Dal Cin P, Hornick JL. Dedifferentiated liposarcoma with “homologous” lipoblastic (pleomorphic liposarcoma-like) differentiation: clinicopathologic and molecular analysis of a series suggesting revised diagnostic criteria. Am J Surg Pathol. 2010;34:1122–31.
Liau JY, Lee JC, Wu CT, Kuo KT, Huang HY, Liang CW. Dedifferentiated liposarcoma with homologous lipoblastic differentiation: expanding the spectrum to include low-grade tumours. Histopathology. 2013;62:702–10.
Saeed-Chesterman D, Thway K. Homologous lipoblastic differentiation in dedifferentiated liposarcoma. Int J Surg Pathol. 2016;24:237–9.
Crozat A, Aman P, Mandahl N, Ron D. Fusion of CHOP to a novel RNA-binding protein in human myxoid liposarcoma. Nature. 1993;363:640–4.
Ron D, Habener JF. CHOP, a novel developmentally regulated nuclear protein that dimerizes with transcription factors C/EBP and LAP and functions as a dominant-negative inhibitor of gene transcription. Genes Dev. 1992;6:439–53.
Li Y, Guo Y, Tang J, Jiang J, Chen Z. New insights into the roles of CHOP-induced apoptosis in ER stress. Acta Biochim Biophys Sin (Shanghai). 2014;46:629–40.
Wu Y, Sun H, Song F, Fu D, Wang J. DDIT3 overexpression increases odontoblastic potential of human dental pulp cells. Cell Prolif. 2014;47:249–57.
Engström K, Willén H, Kåbjörn-Gustafsson C, Andersson C, Olsson M, Göransson M, et al. The myxoid/round cell liposarcoma fusion oncogene FUS-DDIT3 and the normal DDIT3 induce a liposarcoma phenotype in transfected human fibrosarcoma cells. Am J Pathol. 2006;168:1642–53.
Darlington GJ, Ross SE, MacDougald OA. The role of C/EBP genes in adipocyte differentiation. J Biol Chem. 1998;273:30057–60.
Adelmant G, Gilbert JD, Freytag SO. Human translocation liposarcoma-CCAAT/enhancer binding protein (C/EBP) homologous protein (TLS-CHOP) oncoprotein prevents adipocyte differentiation by directly interfering with C/EBPβ function. J Biol Chem. 1998;273:15574–81.
Kåbjörn Gustafsson C, Engström K, Aman P. DDIT3 expression in liposarcoma development. Sarcoma. 2014;2014:954671.
Pérez-Losada J, Pintado B, Gutiérrez-Adán A, Flores T, Bañares-González B, del Campo JC, et al. The chimeric FUS/TLS-CHOP fusion protein specifically induces liposarcomas in transgenic mice. Oncogene. 2000;19:2413–22.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Mantilla, J.G., Ricciotti, R.W., Chen, E.Y. et al. Amplification of DNA damage-inducible transcript 3 (DDIT3) is associated with myxoid liposarcoma-like morphology and homologous lipoblastic differentiation in dedifferentiated liposarcoma. Mod Pathol 32, 585–592 (2019). https://doi.org/10.1038/s41379-018-0171-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-018-0171-y
