
Abstract
Information on the heterogeneity of phosphaturic mesenchymal tumor, a rare entity associated with tumor-induced osteomalacia, is limited. In this retrospective analysis of 222 phosphaturic mesenchymal tumors, 22 cases exhibited mixed mesenchymal and epithelial elements, which we propose to term “phosphaturic mesenchymal tumor, mixed epithelial, and connective tissue type.” Phosphaturic mesenchymal tumor of the mixed epithelial and connective tissue type showed a distinctive and significant male predominance (male:female = 2.67:1), with most patients diagnosed at <40 years old. Moreover, all tumors were mainly located in the alveolar bone with focal invasion into surrounding soft tissue and oral mucosa, which could be detected preoperatively by oral examination. The mesenchymal component, composed of spindled cells resembling fibroblasts or myofibroblasts arranged in a storiform or fascicular pattern, exhibited a less prominent vasculature and lower cellularity than the typical phosphaturic mesenchymal tumor (mixed connective tissue type). The epithelial component was typically haphazardly and diffusely distributed throughout the tumor, forming small, irregular nests resembling odontogenic epithelial nests. All cases were immunoreactive for fibroblast growth factor-23, somatostatin receptor 2A, and NSE in both components. Mostly also demonstrated positive staining for CD99 (21/22, 96%), CD56 (16/22, 73%), Bcl-2 (21/22, 96%), and D2-40 (19/22, 86%) in one or both components. S100 was positive in both components in one of seven cases. Interestingly, immunoreactivity was typically stronger and more diffuse in the epithelial than in the paired mesenchymal components. The mesenchymal component was also diffusely positive for CD68 (17/17, 100%) and showed variable focal staining for SMA (15/22, 68%) and CD34 (9/19, 47 %). These results indicate that phosphaturic mesenchymal tumor of the mixed epithelial and connective tissue type has distinctive clinicopathological characteristics and a polyimmunophenotypic profile.
Similar content being viewed by others
Introduction
Tumor-induced osteomalacia, also known as oncogenic osteomalacia, is a rare paraneoplastic syndrome characterized by abnormal phosphate and vitamin D metabolism (abnormal phosphate homeostasis) due to renal phosphate wasting. McCance [1] reported the first case of tumor-induced osteomalacia in 1947, although unaware of the causative relation between his patient’s femur tumor and osteomalacia. It was not until 1959 that Prader et al. [2] first recognized a neoplasm as the cause of osteomalacia. Tumor-induced osteomalacia is typically caused by small endocrine tumors that secrete the phosphaturic hormone fibroblast growth factor-23 (FGF23), inhibiting renal phosphate reabsorption and renal 1,25-dihydroxy vitamin D production [3]. Patients frequently present with bone pain, muscle weakness, and multiple fractures due to systemic bone demineralization. Elevated or inappropriately normal plasma FGF23, inappropriately normal or low 1,25-dihydroxy vitamin D, hypophosphatemia, and resistance to vitamin D supplementation are the biochemical hallmarks of the disorder. Osteomalacia-associated tumors are often small and difficult to locate, with an average period of 5 years between time of suspecting tumor-induced osteomalacia to identification of the tumor [4, 5]. Most patients can be cured with complete tumor resection.
It is widely accepted that tumor-induced osteomalacia-associated tumors are generally of mesenchymal origin [6, 7]. These mesenchymal tumors are histologically polymorphous and have been diagnosed as giant cell tumors, hemangiopericytomas, non-ossifying fibromas, fibrosarcomas, osteosarcomas, osteoblastomas, chondroblastomas, chondrosarcomas, sclerosing hemangiomas, angiofibromas, angiolipomas, or other mesenchymal tumors [3, 7, 8]. There is limited information of this rare entity in the literature regarding its morphological diversity. In 1987, Weidner and Santa Cruz [7] coined the term “phosphaturic mesenchymal tumor” and categorized these mesenchymal tumors into four distinct morphological subtypes: (1) phosphaturic mesenchymal tumor, mixed connective tissue type; (2) phosphaturic mesenchymal tumor, osteoblastoma-like; (3) phosphaturic mesenchymal tumor, non-ossifying fibroma-like; and (4) phosphaturic mesenchymal tumor, ossifying fibroma-like. Another landmark study in 2004 analyzed 32 cases of tumor-induced osteomalacia-associated mesenchymal tumors with a comprehensive review of 106 cases in the literature and concluded that most tumors, both in their series and in the literature, were a single entity (phosphaturic mesenchymal tumor, mixed connective tissue type) with a wide histological spectrum. The remaining 20% of cases consisted of other mesenchymal tumors such as hemangiopericytomas, giant cell tumors of the bone, osteosarcomas, sclerosing hemangiomas, and angiolipomas [3]. However, no other well-clarified variants other than the mixed connective tissue type have been revealed so far.
In the present study, we retrieved phosphaturic mesenchymal tumor cases diagnosed at our hospital (the largest series so far) and found a distinct clinicopathological variant of phosphaturic mesenchymal tumor exclusively involving alveolar bone. This variant, which we have proposed to term “phosphaturic mesenchymal tumor, mixed epithelial, and connective tissue type,” histologically consisted of both epithelial and mesenchymal elements. We further investigated its clinicopathological characteristics and immunohistochemical profiles and conducted a review of the literature.
Materials and methods
Case retrieval
We retrospectively identified 222 cases of mesenchymal tumors with clinical and laboratory evidence of phosphaturic mesenchymal tumor from the surgical pathology files of Peking Union Medical College Hospital. Patients had been seen between January 2004 and August 2017. A histological review was performed independently by two experienced pathologists (HW and DZ). Finally, 22 cases were histologically subclassified as “phosphaturic mesenchymal tumor, mixed epithelial, and connective tissue type,” which exhibited mixed epithelial and mesenchymal elements. The histopathological findings for all 22 cases (including epithelium distribution, matrix type and quality, perivascular myxoid change, “grungy” calcifications, slate-gray crystals, multinucleated giant cells, hemangiopericytoma-like vessels, cellularity, nuclear grade, mitotic figures/10 high-powered fields) were thoroughly reviewed, and the two pathologists reached consensus. The study was approved by Peking Union Medical College Hospital’s institutional review board, and was exempt from patient written informed consent due to its retrospective nature.
Clinical information review
We collected age, sex, tumor location, and clinical and biochemical features of the 222 phosphaturic mesenchymal tumors from patient medical records. Data on pre-biopsy duration of osteomalacia, radiological findings, tumor size, date of surgery, surgical procedures, time until return to normal phosphatemia after tumor resection, primary diagnosis, and follow-up information for the 22 patients with phosphaturic mesenchymal tumor of the mixed epithelial and connective tissue type were also obtained.
Immunohistochemistry
Archival formalin-fixed, paraffin-embedded tumor tissues were available for all 22 phosphaturic mesenchymal tumors of the mixed epithelial and connective tissue type. Immunohistochemical staining was performed using the EnVision system (DAKO, Glostrup, Denmark). Briefly, serial 5-μm-thick sections were cut from formalin-fixed, paraffin-embedded tumor blocks, dewaxed in xylene, rehydrated through sequential changes of alcohol, and then antigen retrieved. After the tissue sections were washed with phosphate-buffered saline, they were incubated with fresh 3% hydrogen peroxide for 20 min at room temperature. The sections were then blocked with 20% goat serum for 30 min and incubated with primary antibodies, including somatostatin receptor 2A (SSTR2A) (UMB1, 1:50 dilution; Abcam, Cambridge, UK), FGF23 (polyclonal, 1:1250 dilution; Abcam), CD99 (12E7, predilution; Dako), AE1/AE3 (AE1/AE3, predilution; Dako), vimentin (V9, 1:50 dilution; Dako), CD34 (QBEnd/10, 1:50 dilution; Dako), D2-40 (1:50 dilution; Dako), SMA (1A4,1:50 dilution; Dako), Bcl-2 (100/D5, 1:50 dilution; Dako), CD56 (1B6, predilution; Leica Biosystems, New Castle, UK), S100 (polyclonal, predilution; Leica), synaptophysin (27G12, predilution; Leica) and Ki-67 (UMAB107, predilution; ZsBio, Beijing, China) for 2 h. The sections were then incubated with a polymer horseradish peroxidase secondary antibody (Dako). Immunostaining was then developed with 3,3′-diaminobenzidine. Positive immunoreactivity was nuclear and cytoplasmic for S100, nuclear for Ki-67, and cytoplasmic for other antigens. The immunostaining was scored as negative (<5% tumor cells positive), focally positive (5–59% of tumor cells positive), or diffusely positive (≥50% of tumor cells positive); however, Ki-67 proliferation index was recorded as the percentage of tumor cells with Ki-67-positive nuclear immunostaining. The sections were assessed independently by two experienced pathologists (HW and DZ). The scoring for each section was determined by consensus.
Literature review
A comprehensive, retrospective review of the English language literature from 1972 to 2016 was carried out using PubMed. The following keywords were used: oncogenic osteomalacia, tumor-induced osteomalacia, phosphaturic mesenchymal tumor, head and neck, mandible, maxilla, paranasal sinus, sinonasal, nasopharyngeal, oral, and intracranial. Cases outside the head and neck or with no clinical or laboratory evidence of tumor-induced osteomalacia were excluded.
Results
Clinical findings
The anatomical site distribution of the 222 phosphaturic mesenchymal tumors is summarized in Table 1. More than half (52%) of the 222 phosphaturic mesenchymal tumors had involvement of extremities, with the femur (19%) most often involved, followed by foot and thigh soft tissue. Besides extremities, head and neck was the second most commonly involved site, accounting for 32% of all phosphaturic mesenchymal tumors. Among head and neck phosphaturic mesenchymal tumors, the tumors were most often located in the sinonasal area (13%), followed by the mandible (9%) and the maxilla (5%). Rarer sites included trunk, pelvis, and spine. Retroperitoneum phosphaturic mesenchymal tumors were extremely rare. Most interestingly, after careful radiological and pathological review, we identified 22 phosphaturic mesenchymal tumors of the mixed epithelial and connective tissue type, with all involving the maxilla (10 cases) and the mandible (12 cases) (Table 2). More specifically, all were located mainly at the alveolar bone (Fig. 1).

68Ga-DOTA TATE-PET/CT was performed to locate possible tumor-induced osteomalacia in a 22-year-old man who presented with bone pain for 3 years. The maximum intensity projection (MIP) image (right lower) revealed a small focus of elevated activity with standardized uptake value (SUVmax) of 8.24 in the right mandible. On axial view of the PET (right lower), CT (right lower), and fusion images (right lower), this activity was located in the alveolar bone. The images were suggestive of a causative tumor, which was subsequently histologically confirmed as phosphaturic mesenchymal tumor of the mixed epithelial and connective tissue type after tumor resection
For purposes of comparing analysis, phosphaturic mesenchymal tumors were further subclassified into three anatomic and morphologic subgroups: non-head and neck phosphaturic mesenchymal tumors, head and neck phosphaturic mesenchymal tumors (mixed epithelial and connective tissue type), and head and neck phosphaturic mesenchymal tumors (others). Table 3 summarizes the age and sex of all phosphaturic mesenchymal tumors and those within the subgroups. Compared with that shown in the other two subgroups, phosphaturic mesenchymal tumor of the mixed epithelial and connective tissue type showed a significant male predominance (male: female = 2.67:1). Moreover, in contrast to the other two groups, most patients with phosphaturic mesenchymal tumor of the mixed epithelial and connective tissue type were diagnosed at age <40 years.
All 22 patients with phosphaturic mesenchymal tumor of the mixed epithelial and connective tissue type presented with bone pain and muscle weakness with hypophosphatemia, high alkaline phosphatase levels, and inappropriately normal or low 1,25-dihydroxy vitamin D. A pre-biopsy presence of osteomalacia was shown in all cases, with duration ranging from 1 to 18 years (Table 2). It is worth noting that all tumors could be detected by oral physical examination preoperatively due to gingival and alveolar bone swelling and tooth loosening. In some cases, the mass could also be identified by patients themselves before or simultaneously with the presence of osteomalacia symptoms. Nuclear imaging was performed in 20/22 cases. The tumors were successfully located by octreotide scanning in 9 cases, whereas 68Ga-DOTA-TATE-PET/CT revealed tumors in the other 11 cases with negative or false-positive octreotide scans. The tumors measured between 1.4 and 6.0 cm at greatest dimension (median 2.05 cm). Nineteen patients underwent (wide) local tumor resection and tooth extraction, and two patients received subtotal hemi-maxillectomy. Patient 2 (Table 2) underwent partial tumor resection seven times from 1997 to 2017. The tumors were primarily diagnosed as odontogenic fibroma, giant cell reparative granuloma, myofibroblastic tumor, or phosphaturic mesenchymal tumor of the mixed connective tissue type/phosphaturic mesenchymal tumor with odontogenic epithelial component. Phosphatemia returned to normal within 1 week after tumor resection in all cases except for patient 2, who underwent partial resection.
Follow-up information
Among 21 patients with complete resection, 17 had follow-up information of more than 2 months (range 4 months to 9 years; median 24 months). All 17 patients were alive with no evidence of disease and normal phosphatemia at last follow-up (Table 2). Patient 2, who received repeated partial tumor resection since 1997, was still alive with disease at the last follow-up (June 2017), and nuclear imaging suggested multiple sites of involvement.
Histopathological characteristics
At low-power magnification, the tumors infiltrated and destroyed the trabecular meshwork with focal invasion into surrounding soft tissue and oral mucosa in all phosphaturic mesenchymal tumors of the mixed epithelial and connective tissue type, although most cases appeared relatively well circumscribed (Fig. 2a).

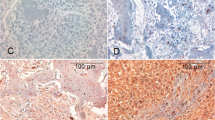
Phosphaturic mesenchymal tumor of the mixed epithelial and connective tissue type was histologically characterized by an admixture of neoplastic epithelial and mesenchymal elements with less prominent vasculature and lower cellularity than typical phosphaturic mesenchymal tumor (mixed connective tissue type). a At low-power magnification, the tumor infiltrated and destroyed the trabecular meshwork with focal invasion into surrounding mucosa. b The mesenchymal component was composed of spindle cells arranged in a fascicular or whorled, storiform pattern. c The neoplastic mesenchymal cells demonstrated an elongated and spindled morphology, resembling fibroblasts or myofibroblasts. The epithelial component was typically haphazardly and diffusely distributed throughout the tumor, forming small, irregular nests (b, c). The neoplastic epithelial cells had moderate eosinophilic (d) or clear cytoplasm (e) with uniform, small, round to oval nuclei and inconspicuous nucleoli, resembling odontogenic epithelium. The small epithelial nests could be overlooked or misinterpreted as multinucleated giant cells (f). d–f The epithelial nuclei were centrally located, evenly distributed, and not polarized. The black arrows indicate the epithelial nests
Phosphaturic mesenchymal tumor of the mixed epithelial and connective tissue type was histologically characterized by an admixture of neoplastic epithelial and mesenchymal elements and typically showed lower cellularity than phosphaturic mesenchymal tumor of the mixed connective tissue type. The mesenchymal component was composed of spindle cells that usually arranged in a fascicular or whorled, storiform pattern (Fig. 2b). When we compared the round, stellate to spindle primitive mesenchymal cells with the small round to oval nuclei in typical phosphaturic mesenchymal tumor (mixed connective tissue type), the neoplastic mesenchymal cells and nuclei in phosphaturic mesenchymal tumor mixed of the epithelial and connective tissue type usually demonstrated a more elongated and spindled morphology, resembling fibroblasts or myofibroblasts (Fig. 2c). The epithelial component was typically haphazardly and diffusely distributed throughout the tumor, forming small, irregular nests that resembled odontogenic epithelial nests, whereas a localized distribution of epithelial nests was found in four cases. The neoplastic epithelial cells had eosinophilic and/or clear cytoplasm, the epithelial nuclei were evenly distributed and not polarized, and stellate reticulum was not present in epithelial nests (Fig. 2d–f). Due to the somewhat indistinct epithelial–mesenchymal boundary and relatively ill-defined cell borders, the small epithelial nests might be overlooked or misinterpreted as multinucleated giant cells (Fig. 2d–f).
Tumor cells of both components were cytologically bland with normochromatic, small nuclei and inconspicuous nucleoli (Fig. 2d–f). Generally, we observed a low nuclear grade, and nuclear pleomorphism was absent or minimal. Mitotic figures were absent to rare (0 or 1/10 high-powered fields) in 16 cases, with 4 or 5/10 high-powered fields in the 6 other cases. Necrosis was absent in all cases (Table 4).
Focal osteoid matrix was observed in the great majority of cases (18/22) (Fig. 3a), whereas myxoid matrix and perivascular myxoid change was only detected in 3/22 and 2/22 cases, respectively. Eight of 22 showed “grungy” calcification (Fig. 3b). Slate-gray crystals were found in one case (Fig. 3c). Osteoclast-like giant cells were detected focally in areas of hemorrhages in 6/22 cases (Fig. 3d).

a Focal osteoid matrix was commonly appreciated histologically. “Grungy” calcification (b) and slate-gray crystals (c) could also be observed. d Osteoclast-like giant cells were detected focally in areas of hemorrhages. Focal hemangiopericytoma-like “staghorn” branching pattern of blood vessels were observed in approximately half cases (e), whereas one case demonstrated schwannoma-like perivascular hyalinization (f). g dilated thin-walled vessels were commonly found in the space between trabecular bones adjacent to the lesion. h In cases with focal epithelia, the neoplastic mesenchymal component showed a higher cellularity, a somewhat “patternless” pattern, and an elaborate intrinsic microvasculature, resembling those seen in typical phosphaturic mesenchymal tumor (mixed connective tissue type). i Focal areas with neoplastic cells showing enlarged hyperchromatic nuclei, obvious nucleoli, significant nuclear atypia and frequent mitotic figures (>20/10 high-powered fields) were observed in the latest specimen from the cases who underwent repeated resection
Phosphaturic mesenchymal tumors of the mixed epithelial and connective tissue type typically had a less prominent vasculature than those of the mixed connective tissue type. Focal hemangiopericytoma-like “staghorn” branching patterns of blood vessels were observed in 10/22 cases, whereas another case demonstrated schwannoma-like perivascular hyalinization (Fig. 3e, f). Dilated thin-walled vessels were found at the lesion periphery and in the space between trabecular bone adjacent to the lesion in 11/22 cases (Fig. 3g).
In the four cases with focal epithelia, the neoplastic mesenchymal component showed a higher cellularity and somewhat “patternless” pattern with elaborate intrinsic microvasculature, and the tumor cells became less spindle-shaped, more closely resembling those seen in typical phosphaturic mesenchymal tumor (mixed connective tissue type) (Fig. 3h). One of these four cases was from a patient who received repeated tumor resection from 1997 to 2017 (patient 2 in Table 2). There was a diminution in the quantity of the epithelial component in the series of specimens obtained from this patient, and the epithelial component finally disappeared since 2011. Moreover, focal areas with significant nuclear atypia and high mitotic activity (>20/10 high-powered fields) were observed in the latest specimen (Fig. 3i), suggestive of malignant transformation.
Immunohistochemical findings
Immunohistochemical results of 22 phosphaturic mesenchymal tumors of the mixed epithelial and connective tissue type are summarized in Table 5. The epithelial and mesenchymal components showed strong diffuse AE1/AE3 and vimentin positivity, respectively (Fig. 4a, b). Both components in all cases were immunoreactive for FGF23, SSTR2A, and NSE (Fig. 4c–e). Most of our cases also demonstrated positive staining for CD99 (21/22, 96%), CD56 (16/22, 73%), Bcl-2 (21/22, 96%), and D2-40 (19/22, 86%) in either one or both components (Fig. 4f–i). S100 was positive in both components in one of seven cases. Interestingly, immunoreactivity (shown with markers FGF23, NSE, CD99, CD56, Bcl-2, D2-40, and S100) was typically stronger and more diffuse in the epithelial components than in the paired connective tissue components. CD68 was diffusely positive with variable intensity in the connective tissue components in all cases (Fig. 4j). A variable focal positive staining for SMA (15/22, 68%) and CD34 (9/19, 47%) was also observed in the mesenchymal components (Fig. 4k, l). All cases were negative for desmin (0/22) and synaptophysin (0/6).

The epithelial and mesenchymal components showed strong and diffuse positivity for AE1/AE3 (a) and vimentin (b), respectively. Both components in all cases were immunoreactive for FGF23 (c), SSTR2A (d), and NSE (e). A dot-like perinuclear FGF23 staining as previously reported was only seen in the neoplastic mesenchymal cells. Most cases also demonstrated positive staining for CD99 (f), CD56 (g), Bcl-2 (h), and D2-40 (i) in both components, and immunoreactivity with the markers (FGF23, NSE, CD99, CD56, Bcl-2, and D2-40) was typically stronger and more diffuse in the epithelial component than in paired connective tissue components. All cases showed diffuse positivity for CD68 (j). Focal positive staining for SMA (k) and CD34 (l) was also observed in the mesenchymal components in some cases
The Ki-67 proliferation index of primary tumor ranged from <1 to 10%. The case from the patient who required repeated resection (patient 2 in Table 2), the Ki-67 labeling index increased from ≤1 to 25% during tumor progression.
Literature review
Our literature review focused on head and neck phosphaturic mesenchymal tumor. We identified 80 tumor-induced osteomalacia-associated mesenchymal tumors of the head and neck reported between 1972 and 2016 (Supplementary Table 1) [3, 4, 4, 8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70]. As expected, the sinonasal area was most often involved (44 cases, 55%). Other locations included the mandible (10 cases; 13%) and maxilla (4 cases; 5%).
Among the cases involving the mandible or maxilla, six cases seemed to fit our proposed definition of “phosphaturic mesenchymal tumor, mixed epithelial, and connective tissue type” (Table 6) [5, 28, 70,71,72,73]. The alveolus was involved in six of these cases, with a significant male predominance (5:1) and a median age of 42 years. Although an odontogenic epithelial component with a similar histology to our cases was identified by the authors in 3/6 cases (one case with focally distributed epithelium), these were considered as non-tumorous odontogenic epithelium or the epithelial component of an ameloblastic tumor, and the patients primarily received a diagnosis of phosphaturic mesenchymal tumor (mixed connective tissue type) or ameloblastic fibrosarcoma [5, 71, 73]. In the other three cases, the epithelial nests had probably been misinterpreted as giant cells according to their original figures and descriptions; therefore, the patients primarily received diagnoses of giant cell tumor, giant cell granuloma, and phosphaturic mesenchymal tumor (mixed connective tissue type), respectively [28, 70, 72]. We noticed that most cases showed a proliferation of spindle cells arranged in fascicles, which was similar to our cases. Interestingly, in the relapsed case, although the recurrent tumors were morphologically similar to the original tumors, there was a diminution in the quantity of the epithelial component and finally the recurrent and lung-metastasized tumors were composed solely of the neoplastic spindle cells [73], indicative of a malignant transformation.
There were also six other cases involving the mandible or maxilla (patients 8, 17, 21, 22, 70, and 73 in Table 6) that may have also fit the proposed criteria of “phosphaturic mesenchymal tumor, mixed epithelial, and connective tissue type” [16, 23, 27, 28, 66, 68]. All six of these cases had or were highly suspicious for alveolus involvement with a significant male predominance (5:1) and a median age of 33 years. Fascicular or storiform spindle cell proliferation with osteoid production was observed in three cases that had been diagnosed as ossifying fibroma-like phosphaturic mesenchymal tumor or ossifying fibroma [16, 28, 68]. Epithelial rests had been revealed in another case. Two cases, which had insufficient data for analysis, were primarily diagnosed as fibrosarcoma and central giant cell granuloma, respectively [23, 66]. The other one case was originally described as “chronic inflammatory tissue with fibrosis and epithelial rests.” [27]
The remaining 68 head and neck cases showed a significant female predominance (male:female = 0.48:1), with 69% (47/68) of patients diagnosed at age ≥40 years (median age of 48.5 years).
Discussion
According to previous reports, phosphaturic mesenchymal tumors have been reported in a wide variety of locations, most commonly involve the extremities and head and neck region, and are commonly diagnosed in the fourth to fifth decades of life with no age or sex predilection [51, 74]. Our single institution series also confirmed that the most commonly involved site of phosphaturic mesenchymal tumors is the lower extremities (particularly the femur), followed by the head and neck. Most cases occur in adults over 40 years, with a median age of 44 years and a slight predominance in men (male:female ratio = 1.22:1). Consistent with previous studies [32, 68, 75], our study also revealed that, for head and neck phosphaturic mesenchymal tumors, the mandible and maxilla are the second most commonly affected sites after the sinonasal cavity. Most importantly, we found that most phosphaturic mesenchymal tumors (22/30) in the mandible and maxilla had mixed epithelial and mesenchymal elements and exclusively involved the alveolar bone, which prompted our proposed designation of a new variant of phosphaturic mesenchymal tumor, namely “phosphaturic mesenchymal tumor, mixed epithelial, and connective tissue type.” Our literatures review unveiled six cases that fit nicely with the proposed new variant, with all involving the alveolar bone. We also found that the other six of eight remaining phosphaturic mesenchymal tumors in the mandible and maxilla from the literature could not be excluded from the possibility of the new variant. Compared with phosphaturic mesenchymal tumors outside the head and neck and other head and neck phosphaturic mesenchymal tumors, the proposed new variant demonstrated a remarkably different age and sex distribution. Most patients who fit the criteria for the proposed new variant are diagnosed at an age of <40 years and are predominantly males. In contrast, other head and neck phosphaturic mesenchymal tumors are predominantly female, with most patients diagnosed at an age of ≥40 years both in our series and in the literature. Phosphaturic mesenchymal tumors are often small, slow growing, and deeply located; therefore, these tumors may be missed by routine clinical examination [32]. However, all cases of the new variant in our study could be detected by clinical intraoral examination or even first discovered by patients themselves due to their relatively superficial location. Therefore, when tumor-induced osteomalacia are suspected, it is critical for clinicians to have a high index of suspicion for this variant of phosphaturic mesenchymal tumor. Careful review of previous related medical history and including periodontal and oral examinations into clinical routine workup, especially in younger male patients, could lead to earlier detection, prompt treatment by complete resection, and ultimately disease eradication.
The histological heterogeneity, the great histological diversity, and the rarity of tumor-induced osteomalacia-associated connective tissue tumors have often led to their misdiagnoses or the patient given a purely descriptive diagnoses [3, 51]. In addition to Weidner and Santa Cruz [7] first using the term phosphaturic mesenchymal tumor to describe these tumors, they also coined the term “phosphaturic mesenchymal tumor, mixed connective tissue type” to refer to a morphologically distinct entity of phosphaturic mesenchymal tumor. With an improved recognition of the histological spectrum, Folpe et al. [3] further confirmed in 2004 that most phosphaturic mesenchymal tumors are of a single histopathological entity (mixed connective tissue type), which is most fundamentally characterized by highly vascular proliferation of spindled to stellate neoplastic cells with a distinctive “smudgy” matrix. Prominent hemangiopericytoma-like vasculature and numerous osteoclast-like giant cells are also common findings in the mixed connective tissue type.
Besides the mixed connective tissue type, some other variants of phosphaturic mesenchymal tumors have also been reported. Three other variants (osteoblastoma-like, non-ossifying fibroma-like, and ossifying fibroma-like) have been proposed by Weidner and Santa Cruz [7]. Folpe et al. [3] also revealed that four cases did not resemble phosphaturic mesenchymal tumor of the mixed connective tissue type but were reminiscent of sinonasal hemangiopericytoma (two cases), conventional hemangiopericytoma (one cases), and sclerosing osteosarcoma (one cases). Sinonasal hemangiopericytoma-like tumors and osteosarcomas have also been reported to be associated with osteomalacia by other studies [16, 56, 62, 76, 77]. Recently, Agaimy et al. [78] revealed that phosphaturic mesenchymal tumors could highly resemble chondromyxoid fibromas, hemangio-/glomangiopericytomas with giant cells, angiomyolipomas, or reparative giant cell granulomas. However, the clinicopathological and immunohistochemical features of these variants except the mixed connective tissue type were not well clarified and accepted, mainly because of their rarity, the lack of distinct clinicopathological features, the polymorphous histological appearance, and the interobserver variability. Interestingly, it has been proposed that sinonasal phosphaturic mesenchymal tumor is generally similar in appearance and most reminiscent of sinonasal hemangiopericytoma and may be a rare variant of phosphaturic mesenchymal tumor with peculiar histological features differing from phosphaturic mesenchymal tumor of the mixed connective tissue type arising in soft tissue or bone [3, 56]. The relation between histology and tumor location in phosphaturic mesenchymal tumor needs to be further explored.
Herein, we proposed a new variant of phosphaturic mesenchymal tumor arising from alveolar bone, termed “phosphaturic mesenchymal tumor, mixed epithelial, and connective tissue type.” It is histologically characterized by a mixed proliferation of epithelial nests resembling odontogenic epithelium and spindled mesenchymal cells arranged in a fascicular or storiform pattern with or without osteoid matrix and could be distinguished from typical phosphaturic mesenchymal tumor (mixed connective tissue type). Besides the presence of an epithelial component, mesenchymal cells of the new variant resemble fibroblasts, or myofibroblasts, with tissue arranged in a fascicular or storiform pattern having less prominent vasculature and lower cellularity, whereas phosphaturic mesenchymal tumors of the mixed connective tissue type contain large numbers of small capillaries and typically consist of primitive mesenchymal cells with a higher cellularity and a more patternless growth pattern. Moreover, although focal osteoid matrix are found in most cases of the new variant, both “grungy” calcification and osteoclast-like giant cells are not common findings, and the latter have been usually only found in areas of hemorrhage. Myxoid matrix and perivascular myxoid changes are rare. Other features typical of phosphaturic mesenchymal tumors of the mixed connective tissue type including intralesional fat and microcystic change have not been observed.
Immunohistochemically, both components in all cases presented here demonstrated diffusely positive staining for FGF23 and SSTR2A. However, as previously reported, both FGF23 and SSTR2A are highly sensitive but not specific for the diagnosis of phosphaturic mesenchymal tumor; therefore, negative staining for the two markers could be used to rule out phosphaturic mesenchymal tumor [79,80,81]. The immunohistochemical application of the two markers might be further limited by antibody availability [80, 82]. Other methods have also been developed to detect FGF23 to confirm the diagnosis of phosphaturic mesenchymal tumor, including molecular tests for serum FGF23 by enzyme-linked immunosorbent assay or tumor FGF23 expression by reverse transcription-polymerase chain reaction, Western blot, or in situ hybridization [42, 83,84,85]. It has been reported that targeted venous sampling for FGF23 might be helpful in localizing occult phosphaturic mesenchymal tumor [83, 86]. The presence of somatostatin receptors on phosphaturic mesenchymal tumor has made nuclear imaging using radiolabeled analogs of somatostatin particularly useful in the detection of occult phosphaturic mesenchymal tumor that are not readily seen on conventional computed tomography or magnetic resonance imaging [86]. To support this, immunohistochemistry for SSTR2A has been shown to correlate with positivity by 68Ga-DOTA-TATE-PET/CT [87, 88]. According to our new variant cases, both 68Ga-DOTA-TATE-PET/CT and octreotide scanning were helpful in identifying occult tumors, but 68Ga-DOTA-TATE-PET/CT appeared more sensitive and specific than the latter.
Not surprisingly, the epithelial nests that are easily misinterpreted or ignored on hematoxylin and eosin-stained slides can be revealed by AE1/AE3 staining, whereas the mesenchymal cells are positive for vimentin staining. We also found that NSE, CD99, CD56, Bcl-2, and D2-40 were positive in all or almost all phosphaturic mesenchymal tumor of the mixed epithelial and connective tissue type. More interestingly, the epithelial component usually demonstrated a stronger and/or more diffuse staining for FGF23, NSE, CD99, CD56, Bcl-2, and D2-40 than the paired mesenchymal component. Our results indicated that the mesenchymal and epithelial components of the new variant shared a similar immunohistochemical profile, further supporting the neoplastic nature of the epithelial component and suggesting that the two components are of the same origin. Although none of these markers is both highly sensitive and highly specific, the typical polyimmunophenotypic pattern (AE1/AE3, vimentin, SSTR2A, FGF23, NSE, CD99, CD56, Bcl-2, and D2-40) is distinctive and valuable for the differential diagnosis of phosphaturic mesenchymal tumor of the mixed epithelial and connective tissue type from its histological mimickers. Consistent with this, the use of CD56 has been recommended when phosphaturic mesenchymal tumors are suspected, and a recent study has revealed consistent coexpression of SATB2, SSTR2A, ERG, and CD56 in phosphaturic mesenchymal tumors [78, 82]. As far as we know, the value of NSE, CD99, Bcl-2, and D2-40 has not been explored in phosphaturic mesenchymal tumors before. Our study further expanded the immunophenotypic spectrum of phosphaturic mesenchymal tumors with AE1/AE3, NSE, CD99, Bcl-2, and D2-40.
Although phosphaturic mesenchymal tumors have been reported to be occasionally positive for SMA and negative for S100, CD68, desmin, and CD34 [63, 67, 68], mesenchymal markers has not been well clarified in phosphaturic mesenchymal tumors. The mesenchymal components in phosphaturic mesenchymal tumor of the mixed epithelial and connective tissue type showed consistent diffuse staining for CD68 and variable focal staining for SMA, CD34, and S100, which should be kept in mind to avoid misdiagnosis such as fibrohistiocytic tumor, myofibroblastic tumor, solitary fibrous tumor, and tumor of neural origin. Based on our own data, other variants of phosphaturic mesenchymal tumors showed a similar immunohistochemical pattern to the mesenchymal components of our proposed new variant (data not shown). We speculate that the polyimmunophenotypic pattern in phosphaturic mesenchymal tumors might be attributed to the multidirectional differentiation potential of tumor cells, which might also underlay the broad histological heterogeneity of phosphaturic mesenchymal tumors. The presence of odontogenic epithelium in the new variant further supported the multidirectional differentiation potential of phosphaturic mesenchymal tumor, and suggested that the multipotent differentiation might be site-specific under certain circumstances. In consistent with this, sinonasal phosphaturic mesenchymal tumor had also been reported to show peculiar histological features differing from phosphaturic mesenchymal tumor of the mixed connective tissue type of soft tissue or bone [3, 56].
It is important to differentiate phosphaturic mesenchymal tumor of the mixed epithelial and connective tissue type from other histological mimickers due to differences in treatment and prognosis. Osteoid matrix production in a background of a spindle cell proliferation in phosphaturic mesenchymal tumor of the mixed epithelial and connective tissue type is reminiscent of ossifying fibromas and osteosarcoma. However, the characteristic location and the presence of epithelial components support the diagnosis of this new variant. Moreover, tumor cells in phosphaturic mesenchymal tumor of the mixed epithelial and connective tissue type typically lack cytological atypia and have few mitotic figures compared with osteosarcoma. Actually, all three phosphaturic mesenchymal tumor cases that had been diagnosed as ossifying fibroma-like phosphaturic mesenchymal tumor or ossifying fibroma in the literature [16, 28, 68] were highly suspicious for alveolus involvement and could not be excluded from the possibility of phosphaturic mesenchymal tumor of the mixed epithelial and connective tissue type. The relation between phosphaturic mesenchymal tumor of the mixed epithelial and connective tissue type and the so-called “ossifying fibroma-like variant of phosphaturic mesenchymal tumor” proposed by Weidner and Santa Cruz needs further clarification. Giant cell lesions (including giant cell tumors and giant cell granulomas) are also important consideration in differential diagnosis. The epithelial nests in phosphaturic mesenchymal tumor of the mixed epithelial and connective tissue type may have been misinterpreted as giant cells in the literature and thus originally diagnosed as giant cell tumors or giant cell granulomas [28, 70, 72]. Similarly, one of our new variant cases was primarily diagnosed as giant cell reparative granuloma. The most useful histological features in making this distinction include the uniformity of nuclear distribution of the epithelia and the presence of epithelial cells with clear cytoplasm in phosphaturic mesenchymal tumor of the mixed epithelial and connective tissue type. Tumors with both mesenchymal and odontogenic epithelial components including odontogenic fibromas/fibrosarcomas and ameloblastic fibromas/fibrosarcomas are also considerations in the differential diagnosis. Odontogenic fibroma histologically consists of a collagenous stroma with a uniform distribution of fibroblasts and a variable odontogenic epithelial component. However, these epithelial nests are often localized and seldom numerous. Moreover, odontogenic fibroma had no capability of invasion, whereas focal invasion into surrounding soft tissue and oral mucosa is always seen in phosphaturic mesenchymal tumor of the mixed epithelial and connective tissue type. In ameloblastic fibroma, odontogenic epithelial nests are larger, exhibit peripheral cell palisading and reverse nuclear polarization, and may show a central area of stellate reticulum, which are not present in phosphaturic mesenchymal tumor of the mixed epithelial and connective tissue type. Synovial sarcoma might also show biphasic differentiation. However, the epithelial component in biphasic synovial sarcoma is quite distinct from that in phosphaturic mesenchymal tumor of the mixed epithelial and connective tissue type and may form glands, tubules, or papillae. Moreover, synovial sarcoma has more pronounced cytological atypia and mitotic activity and higher nucleus-to-cytoplasm ratio. Other differential diagnoses that should be considered include sinonasal-type hemangiopericytoma, fibrohistiocytic tumor, myofibroblastic tumor, and solitary fibrous tumor, due to fascicular or storiform spindle cell proliferation, staghorn vessels, and CD68/SMA/CD34 expression. In addition to use of histology, characteristic clinical features, laboratory chemistry results, and immunohistochemistry profiles are equally critical to confirming the final diagnosis of phosphaturic mesenchymal tumor of the mixed epithelial and connective tissue type.
As with other variants of phosphaturic mesenchymal tumor, despite local invasion of surrounding tissues, phosphaturic mesenchymal tumor of the mixed epithelial and connective tissue type is usually benign, as shown in our series and in the literature. Most patients are cured with complete resection, and there is an immediate and dramatic clinical and biochemical improvement after tumor resection. However, multiple local recurrence and metastasis have also been reported [73]. It is worth noting that phosphaturic mesenchymal tumor of the mixed epithelial and connective tissue type may demonstrate a diminution or even disappearance of the epithelial component during tumor progression, recurrence, and metastasis, and malignant transformations with significant nuclear atypia and high mitotic activity may occur, which would likely lead to a diagnosis of fibrosarcoma or osteosarcoma without knowing the medical history. Given that the correlation between histology and clinical behavior of phosphaturic mesenchymal tumor including the mixed epithelial and connective tissue type has not been well established, postoperative clinical, and laboratory follow-up is mandatory.
In summary, our proposed new variant, phosphaturic mesenchymal tumor arising from alveolar bone, termed “phosphaturic mesenchymal tumor, mixed epithelial, and connective tissue type,” is histologically characterized by an admixture of an epithelial component resembling odontogenic epithelium and mesenchymal spindle cells arranged in a fascicular or storiform pattern that show less prominent vasculature and lower cellularity than typical phosphaturic mesenchymal tumor (mixed connective tissue type). We found that phosphaturic mesenchymal tumor of the mixed epithelial and connective tissue type shows a polyimmunophenotypic profile. When tumor-induced osteomalacia is suspected, clinicians should consider this rare variant of phosphaturic mesenchymal tumor, and periodontal examinations should be included into clinical routine workup, especially in younger male patients. Early detection and complete resection of the tumor will cure the disease.
References
McCance RA. Osteomalacia with Looser’s nodes (Milkman’s syndrome) due to a raised resistance to vitamin D acquired about the age of 15 years. Q J Med. 1947;16:33–46.
Prader A, Illig R, Uehlinger E, Stalder G. Rachitis infolge knochentumors [Rickets caused by bone tumors]. Helv Pediatr Acta. 1959;14:554–65.
Folpe AL, Fanburg-Smith JC, Billings SD, Bisceglia M, Bertoni F, Cho JY, et al. Most osteomalacia-associated mesenchymal tumors are a single histopathologic entity: an analysis of 32 cases and a comprehensive review of the literature. Am J Surg Pathol. 2004;28:1–30.
Ungari C, Rocchi G, Rinna C, Agrillo A, Lattanzi A, Pagnoni M. Hypophosphaturic mesenchymal tumor of the ethmoid associated with oncogenic osteomalacia. J Craniofac Surg. 2004;15:523–7.
Mori Y, Ogasawara T, Motoi T, Shimizu Y, Chikazu D, Tamura K, et al. Tumor-induced osteomalacia-associated with a maxillofacial tumor producing fibroblast growth factor-23: report of a case and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2010;109:e57–63.
Weidner N, Bar RS, Weiss D, Strottmann MP. Neoplastic pathology of oncogenic osteomalacia/rickets. Cancer. 1985;55:1691–705.
Weidner N, Santa Cruz D. Phosphaturic mesenchymal tumors. A polymorphous group causing osteomalacia or rickets. Cancer. 1987;59:1442–54.
Fatani HA, Sunbuli M, Lai SY, Bell D. Phosphaturic mesenchymal tumor: a report of 6 patients treated at a single institution and comparison with reported series. Ann Diagn Pathol. 2013;17:319–21.
Olefsky J, Kempson R, Jones H, Reaven G. Tertiary hyperparathyroidism and apparent “cure” of vitamin-D-resistant rickets after removal of an ossifying mesenchymal tumor of the pharynx. N Engl J Med. 1972;286:740–5.
Renton P, Shaw DG. Hypophosphatemic osteomalacia secondary to vascular tumors of bone and soft tissue. Skelet Radiol. 1976;1:21–4.
Lejeune E, Bouvier M, Meunier P, Vauzelle JL, Deplante JP, David L, et al. Osteomalacia of mesenchymatous tumors. Apropos of a new case. Rev Rhum Mal Osteoartic. 1979;46:187–93.
Camus JP, Crouzet J, Prier A, Bergevin H. Osteomalacies hypophosphoremiques gueries par l’ablation de tumeurs benignes du tissu conjonctif. Etude de trois observations avec dosages pre- et post-operatoires des metabolites de la vitamine D. Ann Med Intern (Paris). 1980;131:422–6.
Sweet RA, Males JL, Hamstra AJ, DeLuca HF. Vitamin D metabolite levels in oncogenic osteomalacia. Ann Intern Med. 1980;93:279–80.
Hioco J, Chanzy MO, Hioco F, Voisin MC, Villiaumey J. Osteomalacia with mesenchymatous tumors—two new cases. XVe Congres International de Rhumatologie, Paris, 1981. Rev Rhum Mal Osteoartic. 1981;48:890.
Nortman DF, Brautbar N, Coburn JW, Haussler MR, Singer FR, Brickman AS, et al. Response of tumor-associated osteomalacia to l,25(OH)2-vitamin D3. Clin Res. 1981;27:92A.
Nomura G, Koshino Y, Morimoto H, Kida H, Nomura S, Tamai K. Vitamin D resistant hypophosphatemic osteomalacia-associated with osteosarcoma of the mandible: report of a case. Jpn J Med. 1982;21:35–9.
Linsey M, Smith W, Yamauchi H, Bernstein L. Nasopharyngeal angiofibroma presenting as adult osteomalacia: case report and review of the literature. Laryngoscope. 1983;93:1328–31.
Ryan EA, Reiss E. Oncogenous osteomalacia. Review of the world literature of 42 cases and report of two new cases. Am J Med. 1984;77:501–12.
Shenker Y, Grekin RJ. Oncogenic osteomalacia. Isr J Med Sci. 1984;20:739–41.
Seshadri MS, Cornish CJ, Mason RS, Posen S. Parathyroid hormone-like bioactivity in tumours from patients with oncogenic osteomalacia. Clin Endocrinol (Oxf). 1985;23:689–97.
Jefferis AF, Taylor PC, Walsh-Waring GP. Tumour-associated hypophosphataemic osteomalacia occurring in a patient with an odontogenic tumour of the maxilla. J Laryngol Otol. 1985;99:1011–7.
Papotti M, Foschini MP, Isaia G, Rizzi G, Betts CM, Eusebi V. Hypophosphatemic oncogenic osteomalacia: report of three new cases. Tumori. 1988;74:599–607.
Nitzan DW, Horowitz AT, Darmon D, Friedlaender MM, Rubinger D, Stein P, et al. Oncogenous osteomalacia: a case study. Bone Miner. 1989;6:191–7.
Harvey JN, Gray C, Belchetz PE. Oncogenous osteomalacia and malignancy. Clin Endocrinol (Oxf). 1992;37:379–82.
Wilkins GE, Granleese S, Hegele RG, Holden J, Anderson DW, Bondy GP. Oncogenic osteomalacia: evidence for a humoral phosphaturic factor. J Clin Endocrinol Metab. 1995;80:1628–34.
Lee HK, Sung WW, Solodnik P, Shimshi M. Bone scan in tumor-induced osteomalacia. J Nucl Med. 1995;36:247–9.
Avila NA, Skarulis M, Rubino DM, Doppman JL. Oncogenic osteomalacia: lesion detection by MR skeletal survey. Am J Roentgenol. 1996;167:343–5.
Kim YG, Choi YS, Lee SC, Ryu DM. Tumor-induced osteomalacia-associated with lesions in the oral and maxillofacial region: report of two cases. J Oral Maxillofac Surg. 1996;54:1352–7.
Catalano PJ, Brandwein M, Shah DK, Urken ML, Lawson W, Biller HF. Sinonasal hemangiopericytomas: a clinicopathologic and immunohistochemical study of seven cases. Head Neck. 1996;18:42–53.
David K, Revesz T, Kratimenos G, Krausz T, Crockard HA. Oncogenic osteomalacia-associated with a meningeal phosphaturic mesenchymal tumor. Case Report J Neurosurg. 1996;84:288–92.
Yang IM, Park YK, Hyun YJ, Kim DY, Woo JT, Kim SW. Oncogenic osteomalacia caused by a phosphaturic mesenchymal tumor of the oral cavity: a case report. Korean J Intern Med. 1997;12:89–95.
Gonzalez-Compta X, Manos-Pujol M, Foglia-Fernandez M, Peral E, Condom E, Claveguera T, et al. Oncogenic osteomalacia: case report and review of head and neck associated tumours. J Laryngol Otol. 1998;112:389–92.
Ohashi K, Ohnishi T, Ishikawa T, Tani H, Uesugi K, Takagi M. Oncogenic osteomalacia presenting as bilateral stress fractures of the tibia. Skelet Radiol. 1999;28:46–8.
Clunie GP, Fox PE, Stamp TC. Four cases of acquired hypophosphataemic (“oncogenic”) osteomalacia. Problems of diagnosis, treatment and long-term management. Rheumatol (Oxf). 2000;39:1415–21.
Sandhu FA, Martuza RL. Craniofacial hemangiopericytoma associated with oncogenic osteomalacia: case report. J Neurooncol. 2000;46:241–7.
Reyes-Mugica M, Arnsmeier SL, Backeljauw PF, Persing J, Ellis B, Carpenter TO. Phosphaturic mesenchymal tumor-induced rickets. Pediatr Dev Pathol. 2000;3:61–9.
John MR, Wickert H, Zaar K, Jonsson KB, Grauer A, Ruppersberger P, et al. A case of neuroendocrine oncogenic osteomalacia-associated with a PHEX and fibroblast growth factor-23 expressing sinusidal malignant schwannoma. Bone. 2001;29:393–402.
Kawai Y, Morimoto S, Sakaguchi K, Yoshino H, Yotsui T, Hirota S, et al. Oncogenic osteomalacia secondary to nasal tumor with decreased urinary excretion of cAMP. J Bone Miner Metab. 2001;19:61–4.
Reis-Filho JS, Paiva ME, Lopes JM. Pathologic quiz case. A 36-year-old woman with muscle pain and weakness. Phosphaturic mesenchymal tumor (mixed connective tissue variant)/oncogenic osteomalacia. Arch Pathol Lab Med. 2002;126:1245–6.
Fuentealba C, Pinto D, Ballesteros F, Pacheco D, Boettiger O, Soto N, et al. Oncogenic hypophosphatemic osteomalacia-associated with a nasal hemangiopericytoma. J Clin Rheumatol. 2003;9:373–9.
Kaylie DM, Jackson CG, Gardner EK. Oncogenic osteomalacia caused by phosphaturic mesenchymal tumor of the temporal bone. Otolaryngol Head Neck Surg. 2006;135:653–4.
Koriyama N, Nishimoto K, Kodama T, Nakazaki M, Kurono Y, Yoshida H, et al. Oncogenic osteomalacia in a case with a maxillary sinus mesenchymal tumor. Am J Med Sci. 2006;332:142–7.
Inokuchi G, Tanimoto H, Ishida H, Sugimoto T, Yamauchi M, Miyauchi A, et al. A paranasal tumor associated with tumor-induced osteomalacia. Laryngoscope. 2006;116:1930–3.
Elston MS, Stewart IJ, Clifton-Bligh R, Conaglen JV. A case of oncogenic osteomalacia with preoperative secondary hyperparathyroidism: description of the biochemical response of FGF23 to octreotide therapy and surgery. Bone. 2007;40:236–41.
Beech TJ, Rokade A, Gittoes N, Johnson AP. A haemangiopericytoma of the ethmoid sinus causing oncogenic osteomalacia: a case report and review of the literature. Int J Oral Maxillofac Surg. 2007;36:956–8.
Kenealy H, Holdaway I, Grey A. Occult nasal sinus tumours causing oncogenic osteomalacia. Eur J Intern Med. 2008;19:516–9.
Yun KI, Kim DH, Pyo SW. A phosphaturic mesenchymal tumor of the floor of the mouth with oncogenic osteomalacia: report of a case. J Oral Maxillofac Surg. 2009;67:402–5.
Uramoto N, Furukawa M, Yoshizaki T. Malignant phosphaturic mesenchymal tumor, mixed connective tissue variant of the tongue. Auris Nasus Larynx. 2009;36:104–5.
Savage CR, Zimmer LA. Oncogenic osteomalacia from pterygopalatine fossa mass. J Laryngol Otol. 2009;123:1052–4.
Gupta R, Sharma A, Ksh A, Khadgawat R, Dinda AK. Phosphaturic mesenchymal tumor of the sinonasal tract. Acta Endocrinol Buchar. 2009;5:537–42.
Peterson NR, Summerlin DJ, Cordes SR. Multiple phosphaturic mesenchymal tumors associated with oncogenic osteomalacia: case report and review of the literature. Ear Nose Throat J. 2010;89:E11–5.
Shelekhova KV, Kazakov DV, Michal M. Sinonasal phosphaturic mesenchymal tumor (mixed connective tissue variant): report of 2 cases. Am J Surg Pathol. 2010;34:596–7.
Parshwanath HA, Kulkarni PR, Rao R, Joshi SK, Patil P. Phosphaturic mesenchymal tumor of ethmoid sinus. Indian J Pathol Microbiol. 2010;53:384–5.
Kurien R, Manipadam MT, Rupa V. Oncogenic osteomalacia in a patient with an ethmoid sinus tumour. J Laryngol Otol. 2010;124:799–803.
Pedrazzoli M, Colletti G, Ferrari M, Rossetti G, Moneghini L, Autelitano L. Mesenchymal phosphaturic neoplasm in the maxillary sinus: a case report. Int J Oral Maxillofac Surg. 2010;39:1027–32.
Guglielmi G, Bisceglia M, Scillitani A, Folpe AL. Oncogenic osteomalacia due to phosphaturic mesenchymal tumor of the craniofacial sinuses. Clin Cases Miner Bone Metab. 2011;8:45–9.
Uno T, Kawai K, Kunii N, Fukumoto S, Shibahara J, Motoi T, et al. Osteomalacia caused by skull base tumors: report of 2 cases. Neurosurgery. 2011;69:E239–44. discussion E44
Xian-Ling W, Jian-Ming B, Wen-Wen Z, Zhao-Hui L, Jing-Tao D, Ju-Ming L, et al. Osteomalacia caused by tumors in facies cranii mimicking rheumatoid arthritis. Rheumatol Int. 2012;32:2573–6.
Munoz J, Michel Ortega R, Celzo F, Donthireddy V. Tumour-induced osteomalacia. BMJ Case Rep. 2012;2012:bcr0320125975. https://doi.org/10.1136/bcr.03.2012.5975.
Battoo AJ, Salih S, Unnikrishnan AG, Jojo A, Bahadur S, Iyer S, et al. Oncogenic osteomalacia from nasal cavity giant cell tumor. Head Neck. 2012;34:454–7.
Bower RS, Daugherty WP, Giannini C, Parney IF. Intracranial phosphaturic mesenchymal tumor, mixed connective tissue variant presenting without oncogenic osteomalacia. Surg Neurol Int. 2012;3:151.
Brandwein-Gensler M, Siegal GP. Striking pathology gold: a singular experience with daily reverberations: sinonasal hemangiopericytoma (glomangiopericytoma) and oncogenic osteomalacia. Head Neck Pathol. 2012;6:64–74.
Mathis DA, Stehel EJ Jr, Beshay JE, Jojo A, Bahadur S, Iyer S, et al. Intracranial phosphaturic mesenchymal tumors: report of 2 cases. J Neurosurg. 2013;118:903–7.
Tarasova VD, Trepp-Carrasco AG, Thompson R, Recker RR, Chong WH, Collins MT, et al. Successful treatment of tumor-induced osteomalacia due to an intracranial tumor by fractionated stereotactic radiotherapy. J Clin Endocrinol Metab. 2013;98:4267–72.
Okamiya T, Takahashi K, Kamada H, Hirato J, Motoi T, Fukumoto S, et al. Oncogenic osteomalacia caused by an occult paranasal sinus tumor. Auris Nasus Larynx. 2015;42:167–9.
Fernandez-Cooke E, Cruz-Rojo J, Gallego C, Romance AI, Mosqueda-Pena R, Almaden Y, et al. Tumor-induced rickets in a child with a central giant cell granuloma: a case report. Pediatrics. 2015;135:e1518–23.
Fathalla H, Cusimano M, Di Ieva A, Karamchandani J, Fung R, Kovacs K. Osteomalacia-inducing tumors of thebrain: a case report, review and a hypothesis. World Neurosurg. 2015;84:189 e1–5.
Qari H, Hamao-Sakamoto A, Fuselier C, Cheng YS, Kessler H, Wright J. Phosphaturic mesenchymal tumor: 2 new oral cases and review of 53 cases in the head and neck. Head Neck Pathol. 2016;10:192–200.
Wasserman JK, Purgina B, Lai CK, Gravel D, Mahaffey A, Bell D, et al. Phosphaturic mesenchymal tumor involving the head and neck: a report of five cases with FGFR1 fluorescence in situ hybridization analysis. Head Neck Pathol. 2016;10:279–85.
Nitzan DW, Marmary Y, Azaz B. Mandibular tumor-induced muscular weakness and osteomalacia. Oral Surg Oral Med Oral Pathol. 1981;52:253–6.
Dupond JL, Mahammedi H, Prie D, Collin F, Gil H, Blagosklonov O, et al. Oncogenic osteomalacia: diagnostic importance of fibroblast growth factor-23 and F-18 fluorodeoxyglucose PET/CT scan for the diagnosis and follow-up in one case. Bone. 2005;36:375–8.
Woo VL, Landesberg R, Imel EA, Singer SS, Folpe AL, Econs MJ, et al. Phosphaturic mesenchymal tumor, mixed connective tissue variant, of the mandible: report of a case and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2009;108:925–32.
Bergwitz C, Collins MT, Kamath RS, Rosenberg AE. Case records of the Massachusetts General Hospital. Case 33-2011. A 56-year-old man with hypophosphatemia. N Engl J Med. 2011;365:1625–35.
Monappa V, Naik AM, Mathew M, Rao L, Rao SK, Ramachandra L, et al. Phosphaturic mesenchymal tumour of the mandible—the useful criteria for a diagnosis on fine needle aspiration cytology. Cytopathology. 2014;25:54–6.
Deep NL, Cain RB, McCullough AE, Hoxworth JM, Lal D. Sinonasal phosphaturic mesenchymal tumor: case report and systematic review. Allergy Rhinol (Provid). 2014;5:162–7.
Lamont EB, Cavaghan MK, Brockstein BE. Oncogenic osteomalacia as a harbinger of recurrent osteosarcoma. Sarcoma. 1999;3:95–9.
White KE, Waguespack SG, Econs MJ. Case 29-2001: oncogenic hypophosphatemic osteomalacia. N Engl J Med. 2002;346:381–2.
Agaimy A, Michal M, Chiosea S, Petersson F, Hadravsky L, Kristiansen G, et al. Phosphaturic mesenchymal tumors: clinicopathologic, immunohistochemical and molecular analysis of 22 cases expanding their morphologic and immunophenotypic spectrum. Am J Surg Pathol. 2017;41:1371–80.
Houang M, Clarkson A, Sioson L, Elston MS, Clifton-Bligh RJ, Dray M, et al. Phosphaturic mesenchymal tumors show positive staining for somatostatin receptor 2A (SSTR2A). Hum Pathol. 2013;44:2711–8.
Clugston E, Gill AC, Graf N, Bonar F, Gill AJ. Use of immunohistochemistry for SSTR2A to support a diagnosis of phosphaturic mesenchymal tumour. Pathology. 2015;47:173–5.
Shiba E, Matsuyama A, Shibuya R, Yabuki K, Harada H, Nakamoto M, et al. Immunohistochemical and molecular detection of the expression of FGF23 in phosphaturic mesenchymal tumors including the non-phosphaturic variant. Diagn Pathol. 2016;11:26.
Tajima S, Fukayama M. CD56 may be a more useful immunohistochemical marker than somatostatin receptor 2A for the diagnosis of phosphaturic mesenchymal tumors. Int J Clin Exp Pathol. 2015;8:8159–64.
Andreopoulou P, Dumitrescu CE, Kelly MH, Brillante BA, Cutler Peck CM, Wodajo FM, et al. Selective venous catheterization for the localization of phosphaturic mesenchymal tumors. J Bone Miner Res. 2011;26:1295–302.
Bahrami A, Weiss SW, Montgomery E, Horvai AE, Jin L, Inwards CY, et al. RT-PCR analysis for FGF23 using paraffin sections in the diagnosis of phosphaturic mesenchymal tumors with and without known tumor induced osteomalacia. Am J Surg Pathol. 2009;33:1348–54.
Carter JM, Caron BL, Dogan A, Folpe AL. A novel chromogenic in situ hybridization assay for FGF23 mRNA in phosphaturic mesenchymal tumors. Am J Surg Pathol. 2015;39:75–83.
Chong WH, Molinolo AA, Chen CC, Collins MT. Tumor-induced osteomalacia. Endocr Relat Cancer. 2011;18:R53–77.
Miederer M, Seidl S, Buck A, Scheidhauer K, Wester HJ, Schwaiger M, et al. Correlation of immunohistopathological expression of somatostatin receptor 2 with standardised uptake values in 68Ga-DOTATOC PET/CT. Eur J Nucl Med Mol Imaging. 2009;36:48–52.
Kaemmerer D, Peter L, Lupp A, Schulz S, Sänger J, Prasad V, et al. Molecular imaging with (6)(8)Ga-SSTR PET/CT and correlation to immunohistochemistry of somatostatin receptors in neuroendocrine tumours. Eur J Nucl Med Mol Imaging. 2011;38:1659–68.
Acknowledgements
This research is supported by Chinese Academy of Medical Sciences (CAMS) Initiative for Innovative Medicine (CAMS-I2M) (Project No. 2017-I2M-1-001). We thank Rasa Hamilton (Moffitt Cancer Center) for editorial assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Wu, H., Bui, M.M., Zhou, L. et al. Phosphaturic mesenchymal tumor with an admixture of epithelial and mesenchymal elements in the jaws: clinicopathological and immunohistochemical analysis of 22 cases with literature review. Mod Pathol 32, 189–204 (2019). https://doi.org/10.1038/s41379-018-0100-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-018-0100-0
This article is cited by
-
Persistence and recurrence in tumor-induced osteomalacia: A systematic review of the literature and results from a national survey/case series
Endocrine (2022)
-
Phosphaturic Mesenchymal Tumors in the Head and Neck Demonstrate a Broad Clinical and Morphologic Spectrum
Head and Neck Pathology (2022)
-
Oncogenic Osteomalacia Due to Recurrent Phosphaturic Mesenchymal Tumour in the Obturator Canal
Indian Journal of Surgery (2022)
-
Extended Whole-body Ga-68 DOTATATE PET-CT in evaluating Tumour-Induced Osteomalacia: Case report and review of literature
Nuclear Medicine and Molecular Imaging (2021)
-
Ameloblastic Fibroodontoma of Mandible Causing Tumor Induced Osteomalacia: A Case Report with Review of 88 Phosphaturic Oral Neoplasms
Head and Neck Pathology (2021)
