
Abstract
Dermatofibrosarcoma protuberans is underlined by recurrent collagen type I alpha 1 chain-platelet-derived growth factor B chain (COL1A1-PDGFB) fusions but ~ 4% of typical dermatofibrosarcoma protuberans remain negative for this translocation in routine molecular screening. We investigated a series of 21 cases not associated with the pathognomonic COL1A1-PDGFB fusion on routine fluorescence in situ hybridization (FISH) testing. All cases displayed morphological and clinical features consistent with the diagnosis of dermatofibrosarcoma protuberans. RNA-sequencing analysis was successful in 20 cases. The classical COL1A1-PDGFB fusion was present in 40% of cases (n = 8/20), and subsequently confirmed with a COL1A1 break-apart FISH probe in all but one case (n = 7/8). 55% of cases (n = 11/20) displayed novel PDGFD rearrangements; PDGFD being fused either to the 5′ part of COL6A3 (2q37.3) (n = 9/11) or EMILIN2 (18p11) (n = 2/11). All rearrangements led to in-frame fusion transcripts and were confirmed at genomic level by FISH and/or array-comparative genomic hybridization. PDGFD-rearranged dermatofibrosarcoma protuberans presented clinical outcomes similar to typical dermatofibrosarcoma protuberans. Notably, the two EMILIN2-PDGFD cases displayed fibrosarcomatous transformation and homozygous deletions of CDKN2A at genomic level. We report the first recurrent molecular variant of dermatofibrosarcoma protuberans involving PDGFD, which functionally mimic bona fide COL1A1-PDGFB fusions, leading presumably to a similar autocrine loop-stimulating PDGFRB. This study also emphasizes that COL1A1-PDGFB fusions can be cytogenetically cryptic on FISH testing in a subset of cases, thereby representing a diagnostic pitfall that pathologists should be aware of.
Similar content being viewed by others
Introduction
Dermatofibrosarcoma protuberans is a superficial and locally invasive mesenchymal tumor, which undergoes fibrosarcomatous transformation, portending a metastatic potential in 10% of cases [1]. Giant cell fibroblastoma represents a morphological variant of dermatofibrosarcoma protuberans occurring in children. Both dermatofibrosarcoma protuberans and giant cell fibroblastoma are molecularly defined by the presence of translocation (17;22)(q21.3;q13.1), which fuses collagen type I alpha 1 chain (COL1A1) with platelet-derived growth factor B chain (PDGFB) [2]. Most dermatofibrosarcomas protuberans display typical morphological features including a dense storiform proliferation of cells expressing diffusely and strongly CD34 [1]. Fibrous morphological variants of dermatofibrosarcoma protuberans may be confused with benign cellular fibrous histiocytomas or dermatofibromas [1]. A diagnosis of dermatofibrosarcoma protuberans warrants complete surgical excision that may require large resection owing to the propensity of dermatofibrosarcoma protuberans to infiltrate adjacent tissues [3]. Imatinib and pazopanib therapies, which target PDGF downstream signaling, may be used in unresectable or metastatic cases [3, 4]. Molecular confirmation of dermatofibrosarcoma protuberans is therefore strongly recommended. COL1A1-PDGFB fusion gene is mostly screened with dual fusion or break-apart fluorescence in situ hybridization (FISH) in routine practice [5]. Reverse transcription polymerase chain reaction (RT-PCR) assays are slightly less sensitive than FISH owing to the variability of COL1A1 breakpoints and require non-degraded RNA, although some studies report comparable sensitivity [6, 7]. Cytogenetic studies have shown that t(17;22) is mostly unbalanced, associated with supernumerary ring chromosomes, and may therefore be evidenced by array-comparative genomic hybridization [8, 9]. Altogether, routine molecular screening remains negative in ~ 4% of otherwise typical dermatofibrosarcomas protuberans, raising clinical uncertainties and leading to render a diagnosis of dermatofibrosarcoma protuberans lacking the canonical translocation and therefore “molecularly unconfirmed” [5]. Single reports of molecular variants of dermatofibrosarcoma protuberans have been reported but no systematic study has been performed so far [10,11,12,13].
In order to identify novel fusion genes in dermatofibrosarcoma protuberans, we investigated by RNA-sequencing and array-comparative genomic hybridization a series of 21 morphologically typical dermatofibrosarcomas protuberans, negative for COL1A1-PDGFB fusion on routine molecular testing.
Materials and methods
Sample selection
We identified 25 cases in our records diagnosed as dermatofibrosarcoma protuberans between 2006 and 2017 but negative on molecular testing. Cases were retrieved from the archives of the department of Genetics of University Hospital of Nice (Nice, France), and departments of Pathology of Centre Léon Bérard (Lyon, France) and Institut Bergonié (Bordeaux, France). Notably, all three participating institutions used FISH for molecular testing, but they applied two different screening strategies. First, COL1A1-PDGFB fusion probe followed by PDGFB break-apart probe in case of negative or equivocal result. Second, a screening with PDGFB break-apart probe in the first place, followed if negative or ambiguous by a screening with a COL1A1 break-apart probe. Owing to the evolution of molecular testing strategies over time, seven cases of our series had not been previously tested with a PDGFB break-apart probe. Hence, FISH analysis was completed in these cases, rendering positive results in four cases, which were therefore removed from the final cohort (n = 21).
All cases from the series were registered in the RREPS sarcoma database, a national clinical database approved by the Commission Nationale Informatique et Libertés (CNIL) and the Comité national d'éthique.
The material used in this publication was provided by the SarcomaBCB, the Conticanet database (https://auth.sarcomabcb.org).
Clinical review
Clinical follow-up was obtained from the medical records of patients either available at our institutions or through corresponding clinicians. Follow-up duration was calculated from the date of the clinical detection of the lesion. Clinical data are provided in Supplementary Table 1.
Histopathological analyses
All cases of the final series were reviewed blindly by soft tissue expert pathologists (FLL, JMC, DRV). Fibrosarcomatous transformation was defined by the presence of a fascicular component with increased mitotic activity contrasting with the storiform pattern of the DFSP component [1]. All samples were studied with a panel of antibodies available in routine including AE1/E3, CD34, S100 protein, EMA, and Ki67. Pathological data are summarized in Supplementary Table 1.
FISH analyses
FISH analyses were performed on formalin-fixed paraffin-embedded tissue sections using commercial and custom bacterial artificial chromosome (BAC) probes. Commercial FISH probes used in the study included dual fusion probe COL1A1/PDGFB (#Z-2116-200, Zytovision, Bremerhaven, Germany), PDGFB Break-Apart probe (#Z-2119-200, Zytovision), and COL1A1 Break Apart Probe (#Z-2121-200, ZytoVision). PDGFD, EMILIN2, and COL6A3 break-apart probes were prepared with BAC listed in Supplementary Table 2. FISH analysis was performed by assessing at least 100 non-overlapping intact nuclei by two independent operators. The positive threshold to call the FISH assay positive was 15%. BACs were cultured and labeled as previously described [14].
Array-comparative genomic hybridization analyses
Genomic DNA was extracted from formalin-fixed paraffin-embedded tissue using QIAamp, DNA micro kit (Qiagen, Hilden, Germany). Genomic DNA and human reference DNA (Promega) were labeled with cyanin 5 (Cy5) and cyanine 3 (Cy3), respectively, using the Genomic DNA High-Throughput ULS Labeling Kit (Agilent Technologies, Santa Clara, California) and co-hybridized onto a Sureprint G3 Human CGH microarray 4 × 180K (Agilent) following manufacturer’s recommendations. Data were analyzed by Agilent Genomic Workbench software v7.0 or by Cytogenomics software (v2.9.2.4, Agilent) and expressed according to the human reference hg19 (GRCh37, Genome Reference Consortium Human Reference 36). The identification of aberrant copy number segments was based on ADM-2 segmentation algorithm with a threshold of 6.0.
RNA-sequencing analyses
RNA sequencing was performed with formalin-fixed paraffin-embedded material in all cases. Total RNA was extracted from formalin-fixed paraffin-embedded tissue section using Trizol reagent (Thermo Fisher Scientific, Courtaboeuf, France) following manufacturers’ recommendations. DNA was removed using RNase-free DNase set (Qiagen) followed by second Trizol extraction. Quantity and quality of total RNA were evaluated using NanoDrop (Thermo Fisher Scientific) and Tape Station with Hs RNA Screen Tape (Agilent) using a cutoff of DV200 (defined as the percentage of RNA fragments above 200 nucleotides) above 13%. All samples passed quality criteria. Libraries were prepared with 100 ng of total RNA using TruSeq RNA Access Library Prep Kit (Illumina, San Diego, USA). Libraries were pooled by group of 14 samples at 4 nM with 1% PhiX. Sequencing was performed (75 cycles paired end) using a NextSeq 500/550 High Output V2 kit on Illumina NextSeq 500 platform (Illumina, San Diego, CA). RNA-sequencing was successful in 20 out of 21 cases. The case with failure was ruled out from the final series as no material was available to complete its characterization by array-comparative genomic hybridization and FISH (final series n = 20, Table 1).
Sequencing data (up to 15 million reads per sample) were analyzed with BaseSpace sequence Hub (Illumina). Reads were aligned with STAR and TopHat2 on GRCh38 reference genome. The fusion transcripts were called with Manta, STAR-Fusion, FusionMap and TopHat2 fusion and validated if the Manta score was up to 0.7 and present in fusion list with TopHat2 fusion and/or STAR-Fusion and/or FusionMap [15,16,17].
To perform the clustering analysis, gene expression values were extracted using Kallisto v0.42.5 tool [18] with GENECODE release 23 genome annotation based on GRCh38 genome reference. Kallisto TPM expression values were transformed in log2(TPM + 2) and all samples were normalized together using the quantile method from the R limma package within R (version 3.1.1) environment. Clustering was performed with the R package Cluster v2.0.3 using Pearson correlation distance and Ward’s clustering method. Significance of clusters was assessed using SigClust as previously described [19].
RT-PCR and Sanger sequencing
In 11 cases, we performed reverse transcriptase-polymerase chain reaction (RT-PCR) to confirm either the COL6A3-PDGFD fusion transcript or the EMILIN2-PDGFD fusion transcript, using the following custom primers (designed using Primer 3 program): COL6A3 forward primer 5′-GCAAGGTCAGCTTCTAGTTCA-3', PDGFD reverse primer 5′-TGGCCAACTTCAGCTCTTCT-3′ (in case number 1, 2, 4, 5, 6, 7, 8, 19), COL6A3 forward primer 5′-CAGCAAGGTCAGCTTCTAGTT-3′ and PDGFD reverse primer 5'-CAGTTCCACAGCCACAATTTC-3′ (in case number 3) and EMILIN2 forward primer 5′-GCCACGTCTTCCAGATTTCTA-3′ and PDGFD reverse primer 5′-CAGTTCCACAGCCACAATTTC-3′ (in case number 9, 10). Fusion transcripts were sequenced by Sanger after extraction and enzymatic purification of RT-PCR product using High pure PCR product purification kit (Roche Diagnostics, Meylan, France).
Results
Clinicopathological features
All cases were located in classical anatomical sites including trunk (n = 11/20), limbs (n = 6/20), and head and neck (n = 3/20) (Table 1). Patients’ age ranged from 4 to 73 years old. Clinically, lesions presented with a classical scar-like appearance (n = 2), slow-growing cutaneous plaques (n = 1) or cutaneous plaques with red discoloration (n = 2). In addition, one case had a multinodular presentation. All tumors displayed outcomes in line with the local malignant potential of dermatofibrosarcoma protuberans with late local recurrences occurring up to 12 years after initial resection (Supplementary Table 1). All cases presented morphological features either typical of dermatofibrosarcoma protuberans (n = 12/20) (Figs. 1 and 2) or consistent with a morphological variant of dermatofibrosarcoma protuberans including fibrous pattern (n = 5/20), pigmented pattern (also referred to as Bednar tumor) (n = 1/20) or hybrid Bednar/dermatofibrosarcoma protuberans variant with focal presence of pigments or giant cells (n = 2/20) (Supplementary Table 1). Four cases displayed fibrosarcomatous transformation (Fig. 3). All but two cases showed classical honeycomb infiltrative borders. Five cases were deeply located in hypodermis without dermal infiltration.

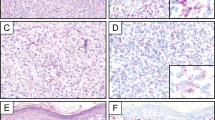
Cryptic COL1A1-PDGFB fusion in dermatofibrosarcoma protuberans. a–b Band-like infiltration of hypodermis and subcutaneous adipose tissue (case number 15, HES staining, × 2.5 and × 6.5 magnifications, respectively) c Storiform proliferation infiltrating hypodermis with a honeycomb pattern (HES, × 200 magnification). d–e High-power field view of tumor cell nuclei displaying ovoid shape and monomorphism d and focal interspersed multinucleated cells e (HES, × 200 magnification) f Fluorescence in situ hybridization (FISH) using COL1A1 break-apart probe. FISH analysis shows an unbalanced translocation with a gain of the 5′ part of COL1A1 (probe labeled in red). g Array-comparative genomic hybridization profile harbors a genomic breakpoint at COL1A1 locus with a 5′COL1A1 gain but no breakpoint at PDGFB locus (chromosome 22 view not shown). Additional copy number alterations include gains of 4q28-q35, 6p, and whole chromosome 7

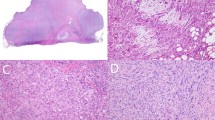
COL6A3-PDGFD dermatofibrosarcoma protuberans. a Biopsy specimen highlighting deep-seated dense nodular proliferation (case number 6, HES staining, × 20 magnification). b Storiform proliferation with honeycomb infiltrative pattern typical of dermatofibrosarcoma protuberans (HES, × 200). c Fibrous stromal changes focally interposed between tumor cells (HES, × 250) d Tumor cells display ovoid monomorphic nuclei (HES, × 400). e Array-comparative genomic hybridization profile showing deletions and breakpoints involving COL6A3 and PDGFD. Additional copy number alteration includes gain of chromosome 12. f–g Fluorescence in situ hybridization (FISH) using COL6A3 break-apart probe f and PDGFD break-apart probe g showing an unbalanced rearrangement with loss of the 3′COL6A3 and the 5′PDGFD, respectively. h Structure of COL6A3-PDGFD fusion transcript. From top to bottom: scheme representing locus and chromosomal position of COL6A3 and PDGFD; schematic of breakpoints positions involving COL6A3 exon 42 and PDGFD exon 6; nucleotide sequence of adjoined sequences

EMILIN2-PDGFD dermatofibrosarcoma protuberans. a Biopsy specimen highlighting densely cellular tumor infiltrating hypodermis (case number 9, HES staining, × 2 magnification). b Focal collagenized stroma arranged in elongated fascicles suspicious for fibrosarcomatous transformation (× 200 magnification). c Tumor cells display ovoid monomorphic nuclei embedded in a collagenous stroma (× 400 magnification). d CD34 staining in this case (× 250 magnification). e Array-comparative genomic hybridization profile showing a homozygous deletion of CDKN2A (P16) but no breakpoint within EMILIN2 or PDGFD loci. f–g Fluorescence in situ hybridization (FISH) using EMILIN2 break-apart probe f and PDGFD break-apart probe g showing a rearrangement of the EMILIN2 and PDGFD loci, respectively. h Structure of EMILIN2-PDGFD fusion. From top to bottom scheme representing locus and chromosomal position of EMILIN2 and PDGFD; schematic of breakpoints positions which constantly occurred in exon 4 of EMILIN2 and in exon 6 of PDGFD; nucleotide sequence of adjoined sequences
RNA sequencing
Classical COL1A1-PDGFB fusions were found in eight cases (n = 8/20). PDGFB breakpoint was constantly found in exon 2, whereas breakpoints in COL1A1 varied from exons 11 to 45 (Supplementary Table 3).
PDGFD rearrangements were found in 11 cases (n = 11/20), PDGFD being fused either to collagen type VI alpha 3 chain (COL6A3) at 2q37 (n = 9/11) or elastin microfibril Interface 2 (EMILIN2) at 18p11 (n = 2/11). Breakpoints in COL6A3 involved either exon 42 (n = 8/9) or exon 43 (n = 1/9), whereas breakpoint in PDGFD constantly involved exon 6 (Fig. 2) and breakpoints in EMILIN2 were located within exon 4 (Fig. 3).
One case (case number 20) failed to show a fusion transcript by RNA-sequencing with the different algorithms.
FISH and RT-PCR
COL6A3-PDGFD transcript and EMILIN2-PDGFD transcript were confirmed by RT-PCR in all cases (case number 1–10 and 19). Regarding cases with COL1A1-PDGFB fusions, FISH analyses were completed with a COL1A1 break-apart probe, which was positive in 7/8 cases (Fig. 1). Regarding cases with PDGFD fusions, FISH analyses were carried out with PDGFD break-apart probe, which was positive in nine cases out of 11 (n = 9/11). Gene partners fused to PDGFD were also assessed with COL6A3 and EMILIN2 break-apart probes (Figs. 2 and 3, respectively), showing rearrangements in 5/7 and 2/2 cases, respectively (Supplementary Table 3).
Array-comparative genomic hybridization
On profiles, all cases with cryptic COL1A1-PDGFB highlighted genomic breakpoints within COL1A1 with a genomic gain in flanking region 17q21.33-qter (n = 5/5). Additional non-recurrent copy number alterations were present in all cases. Genomic gains were seen in 1q21-q25.3, 4q28-q35, 6p, 7pter, 22q11 in one case each. Whole-chromosome gains involved chromosomes 6, 7, 10, and genomic loss were seen in 1q22–23 6q12-q26 and 8p in one case each (Supplementary Table 3).
Array-comparative genomic hybridization analysis of the dermatofibrosarcomas protuberans with COL6A3-PDGFD fusion highlighted no breakpoint within PDGFB nor COL1A1 but within COL6A3 and PDGFD in 4/7 and 6/8 cases, respectively. Copy number alterations were present in the regions flanking the breakpoints in all but one case (n = 6/7). Additional non-recurrent copy number alterations included gain of chromosome 12 (n = 1/7), loss of 3q (n = 1/7), gains of chromosomes 1, 7, and 17 (n = 1/7). The genomic profiles of the two cases of EMILIN2-PDGFD dermatofibrosarcoma protuberans displayed breakpoints within PDGFD (n = 1/2), without copy number alteration involving EMILIN2. Interestingly, both profiles presented homozygous deletion of CDKN2A locus (n = 2/2). None of these cases displayed copy number alterations in the genomic regions flanking the breakpoints.
Clustering analysis
To assess whether PDGFD-rearranged dermatofibrosarcoma protuberans were biologically similar to PDGFB-rearranged dermatofibrosarcoma protuberans, we compared the RNA-seq expression profiles of samples of our cohort (including five PDGFB- and nine PDGFD-rearranged dermatofibrosarcoma protuberans) to other spindled cutaneous neoplasms including infantile fibrosarcomas (n = 7), clear cell sarcomas of soft tissue (n = 2), ALK-rearranged Spitzoid neoplasms (n = 5), NTRK1/3-rearranged Spitzoid neoplasms (n = 7) and conventional PDGFB-rearranged dermatofibrosarcomas protuberans positive for gene rearrangement upon routine molecular screening (n = 3) (Supplementary Table 4). All cases clustered together with transcriptional profiles distinct from ALK- and NTRK1/3-rearranged Spitzoid proliferations and infantile fibrosarcomas. The dermatofibrosarcoma protuberans cluster lumped together all dermatofibrosarcoma protuberans including those with PDGFB fusions (both cryptic and overt rearrangements) and PDGFD fusions (Fig. 4).

Transcriptomic classification of dermatofibrosarcoma protuberans. Unsupervised hierarchical clustering, using the top 10% most variant genes based on interquantile range, comparing PDGFB-rearranged dermatofibrosarcoma protuberans (n = 8, including 3 “conventional” dermatofibrosarcomas protuberans positive for PDGFB rearrangement using FISH analysis), PDGFD-rearranged dermatofibrosarcoma protuberans (n = 9), infantile fibrosarcomas (n = 7), clear cell sarcomas of soft tissue (superficially seated) (n = 2), ALK-rearranged Spitzoid neoplasms (n = 5), and NTRK1/3-rearranged Spitzoid neoplasms (n = 7). Branches depicted with colors within the dendrogram were found significant (p value < 10e-5) by SigClust clustering significance assessment. Samples are listed in Table 1 and Supplementary Table 4. Infantile fibrosarcomas are highlighted in purple, low grade fibromyxoid sarcomas in orange, spitzoid neoplasms in yellow, clear cell sarcomas in light red, and dermatofibrosarcoma protuberans in red
Discussion
Dermatofibrosarcoma protuberans has historically been defined by the presence of recurrent COL1A1-PDGFB fusions, which lead to the upregulation of PDGFRB signaling through an autocrine activating loop [2,20,21,22]. However, ~ 4% of dermatofibrosarcoma protuberans prove negative for the translocation upon routine FISH testing [5].
We evidenced by RNA-sequencing that molecularly unconfirmed dermatofibrosarcoma protuberans displayed “cryptic” COL1A1-PDGFB fusions in 40% of cases (n = 8/20). All dermatofibrosarcomas protuberans associated with “cryptic” fusions involved previously reported COL1A1 breakpoints, which position have been shown to vary considerably from exon 7 to exon 49. Interestingly, all cases displayed visible genomic breakpoints within COL1A1 on quantitative genomic profiles along with and/or gains of the 17q locus flanking the breakpoint (n = 5/5) (Fig. 1). Notably, subsequent FISH analyses with COL1A1 break-apart probe were positive in all but one case (n = 7/8). Altogether, our findings support that array-comparative genomic hybridization or screening with COL1A1 break-apart probe may support the diagnosis of cryptic dermatofibrosarcoma protuberans. This complementary screening strategy remains cost-effective as compared with the cost of RNA-sequencing performed in all cases suspicious of dermatofibrosarcoma protuberans. Cryptic rearrangements have also been reported in other translocation-related sarcomas and are thought to be related to structural variations of the translocation or induction of neo-exons secondary to splicing variations [23,24,25,26,27].
Second, we report herein that 55% (n = 11/20) of cytogenetically negative- dermatofibrosarcoma protuberans are associated with alternative rearrangements involving PDGFD. Two different types of fusion transcripts were evidenced with the 3′ part of PDGFD being fused to the 5′ part either of COL6A3 (n = 9/11) or EMILIN2 (n = 2/11). PDGFD, located at 11q22.3, encodes a protein belonging to the same family of platelet-derived growth factor than PDGFB [28]. PDGFD is able to bind to PDGF receptor B (PDGFRB) [29, 30]. PDGFD has oncogenic properties through promotion of cell proliferation and angiogenesis [28] and its overexpression has been linked to a variety of malignancies [31,32,33,34]. The breakpoint was constantly located within exon 6, preserving the binding domain to PDGF receptors in a similar manner than translocations involving PDGFB. Interestingly, the loss of exon 6 have been shown to induce truncation owing to a premature stop codon in mice, therefore a breakpoint located downstream exon 6 might jeopardize PDGFD signaling [35]. COL6A3, located at 2q37.3, encodes a protein of extracellular matrix involved in cellular adhesion belonging to the same superfamily as COL1A1 [36]. COL6A3 contains 3152 amino acids-long alpha 3 chains, which have the same sequence as alpha 1 chains and harbor the same triple-helical and von Willebrand factor A domains [36]. Notably, somatic rearrangements of COL6A3 have been previously reported in tenosynovial giant cell tumors in which COL6A3 is fused to colony stimulating factor 1 (CSF1) [37, 38]. In addition, overexpression of COL6A3 has been described in a wide array of malignancies including gastric [39], ovarian [40], colorectal [41, 42], and pancreatic cancers [43]. It has also been correlated to resistance to chemotherapy [40, 44]. The breakpoint occurred in exon 42 of COL6A3 in all but one case (n = 8/9), in contrast to the frequent variations seen in COL1A1. All COL6A3-PDGFD cases displayed genomic imbalances in regions flanking the breakpoints of COL6A3 and/or PDGFD and three cases displayed additional copy number alterations (n = 3/7). The morphological and clinical features of COL6A3-rearranged dermatofibrosarcoma protuberans did not differ from those seen in bona fide dermatofibrosarcoma protuberans. The second fusion partner EMILIN2 was involved in 2/11 cases a. EMILIN2, located at 18p11.32–p11.31, encodes a glycoprotein related to the superfamily of collagen, which contains collagen-like domains [36]. EMILIN2 assembles into multimers, involved in the composition of extracellular matrix [45, 46]. The genomic breakpoints were located constantly in exons 4 and 6 of EMILIN2 and PDGFD, respectively. The fusion preserved preserves key structural domains of EMILIN2, including EMI domain, coiled-coil structures and leucine zippers and gC1q domain [45, 46]. EMILIN2-PDGFD dermatofibrosarcoma protuberans displayed homozygous deletion of CDKN2A in both cases (n = 2/2). Interestingly, both cases displayed fibrosarcomatous transformation and were deep-seated within hypodermis without dermal connection. It is notable that homozygous deletions of CDKN2A are linked to malignancy in varied tumor types including mesenchymal, melanocytic, and epithelial neoplasms [47,48,49]. Therefore, this alteration may portend an increased malignant potential in this subset of dermatofibrosarcoma protuberans, which warrants further studies on larger cohorts with longer follow-up to assess whether the behavior of cases of EMILIN2-PDGFD dermatofibrosarcoma protuberans is similar or not to classical and COL6A3-PDGFD-associated cases.
Altogether, PDGFD rearrangements may functionally mimic the biology of COL1A1-PDGFB fusions as it involves functionally related genes and display a similar structure. Rearrangements might therefore be targeted by imatinib as classical dermatofibrosarcoma protuberans.
In conclusion, we report herein new and recurrent molecular variants of dermatofibrosarcoma protuberans underlined by PDGFD rearrangements, which are clinically, morphologically and molecularly indistinguishable from COL1A1-PDGFB dermatofibrosarcoma protuberans. Furthermore, we showed that roughly half of cases of dermatofibrosarcoma protuberans negative upon routine FISH screening are associated with cryptic COL1A1-PDGFB rearrangements that may be identified by array-comparative genomic hybridization or FISH with a COL1A1 break-apart probe. The functional similarities between the classical COL1A1-PDGFB and the alternative COL6A3-PDGFD or EMILIN2-PDGFD transcripts suggest they activate an analogous oncogenic autocrine loop involving PDGFRB signaling.
References
Mentzel T, Pedeutour F, Lazar A and Coindre JM. Dermatofibrosarcoma protuberans. In: Fletcher CDM, Bridge JA, Hogendoorn P and Mertens F, editors. WHO classification of tumours of soft tissue and bone. 4th ed. Lyon: IARC; 2013. p. 77-79
Simon MP, Pedeutour F, Sirvent N, et al. Deregulation of the platelet-derived growth factor B-chain gene via fusion with collagen gene COL1A1 in dermatofibrosarcoma protuberans and giant-cell fibroblastoma. Nat Genet. 1997;15:95–98.
Saiag P, Grob J-J, Lebbe C, et al. Diagnosis and treatment of dermatofibrosarcoma protuberans. European consensus-based interdisciplinary guideline. Eur J Cancer. 2015;51:2604–8.
Stacchiotti S, Pantaleo MA, Negri T, et al. Efficacyand biological activity of imatinib in metastatic dermatofibrosarcoma protuberans (DFSP). Clin Cancer Res. 2016;22:837–46.
Karanian M, Pérot G, Coindre J-M, et al. Fluorescence in situ hybridization analysis is a helpful test for the diagnosis of dermatofibrosarcoma protuberans. Mod Pathol. 2015;28:230–7.
Salgado R, Llombart B, M Pujol R, et al. Molecular diagnosis of dermatofibrosarcoma protuberans: a comparison between reverse transcriptase-polymerase chain reaction and fluorescence in situ hybridization methodologies. Genes Chromosomes Cancer. 2011;50:510–7.
Patel KU, Szabo SS, Hernandez VS, et al. Dermatofibrosarcoma protuberans COL1A1-PDGFB fusion is identified in virtually all dermatofibrosarcoma protuberans cases when investigated by newly developed multiplex reverse transcription polymerase chain reaction and fluorescence in situ hybridization assays. Hum Pathol. 2008;39:184–93.
Linn SC, West RB, Pollack JR, et al. Gene expression patterns and gene copy number changes in dermatofibrosarcoma protuberans. Am J Pathol. 2003;163:2383–95.
Sirvent N, Maire G, Pedeutour F. Genetics of dermatofibrosarcoma protuberans family of tumors: from ring chromosomes to tyrosine kinase inhibitor treatment. Genes Chromosomes Cancer. 2003;37:1–19.
Nakamura I, Kariya Y, Okada E, et al. A novel chromosomal translocation associated With COL1A2-PDGFB gene fusion in dermatofibrosarcoma protuberans: PDGF expression as a new diagnostic tool. JAMA Dermatol. 2015;151:1330–7.
Bianchini L, Maire G, Guillot B, et al. Complex t(5;8) involving the CSPG2 and PTK2B genes in a case of dermatofibrosarcoma protuberans without the COL1A1-PDGFB fusion. Virchows Arch Int J Pathol. 2008;452:689–96.
Saab J, Rosenthal IM, Wang L, et al. Dermatofibrosarcoma protuberans-like tumor with COL1A1 copy number gain in the absence of t(17;22). Am J Dermatopathol. 2017;39:304–9.
Sinovic J, Bridge JA. Translocation (2;17) in recurrent dermatofibrosarcoma protuberans. Cancer Genet Cytogenet. 1994;75:156–7.
Sirvent N, Coindre J-M, Maire G, et al. Detection of MDM2-CDK4 amplification by fluorescence in situ hybridization in 200 paraffin-embedded tumor samples: utility in diagnosing adipocytic lesions and comparison with immunohistochemistry and real-time PCR. Am J Surg Pathol. 2007;31:1476–89.
Trapnell C, Roberts A, Goff L, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–78.
Ge H, Liu K, Juan T, et al. FusionMap: detecting fusion genes from next-generation sequencing data at base-pair resolution. Bioinformatics. 2011;27:1922–8.
Lågstad S, Zhao S, Hoff AM, et al. chimeraviz: a tool for visualizing chimeric RNA. Bioinformatics. 2017;33:2954–6.
Bray NL, Pimentel H, Melsted P, et al. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol. 2016;34:525–7.
Le Loarer F, Watson S, Pierron G, et al. SMARCA4 inactivation defines a group of undifferentiated thoracic malignancies transcriptionally related to BAF-deficient sarcomas. Nat Genet. 2015;47:1200–5.
Greco A, Fusetti L, Villa R, et al. Transforming activity of the chimeric sequence formed by the fusion of collagen gene COL1A1 and the platelet derived growth factor b-chain gene in dermatofibrosarcoma protuberans. Oncogene. 1998;17:1313–9.
Shimizu A, O’Brien KP, Sjöblom T, et al. The dermatofibrosarcoma protuberans-associated collagen type Ialpha1/platelet-derived growth factor (PDGF) B-chain fusion gene generates a transforming protein that is processed to functional PDGF-BB. Cancer Res. 1999;59:3719–23.
Sjöblom T, Shimizu A, O’Brien KP, et al. Growth inhibition of dermatofibrosarcoma protuberans tumors by the platelet-derived growth factor receptor antagonist STI571 through induction of apoptosis. Cancer Res. 2001;61:5778–83.
Brassesco MS, Cortez MA, Valera ET, et al. Cryptic SYT/SXX1 fusion gene in high-grade biphasic synovial sarcoma with unique complex rearrangement and extensive BCL2 overexpression. Cancer Genet Cytogenet. 2010;196:189–93.
Torres L, Lisboa S, Cerveira N, et al. Cryptic chromosome rearrangement resulting in SYT-SSX2 fusion gene in a monophasic synovial sarcoma. Cancer Genet Cytogenet. 2008;187:45–49.
Lestou VS, O’Connell JX, Robichaud M, et al. Cryptic t(X;18), ins(6;18), and SYT-SSX2 gene fusion in a case of intraneural monophasic synovial sarcoma. Cancer Genet Cytogenet. 2002;138:153–6.
Kovar H, Jugovic D, Melot T, et al. Cryptic exons as a source of increased diversity of Ewing tumor-associated EWS-FLI1 chimeric products. Genomics. 1999;60:371–4.
Chen S, Deniz K, Sung Y-S, et al. Ewing sarcoma with ERG gene rearrangements: a molecular study focusing on the prevalence of FUS-ERG and common pitfalls in detecting EWSR1-ERG fusions by FISH. Genes Chromosomes Cancer. 2016;55:340–9.
Wang Z, Kong D, Li Y, et al. PDGF-D signaling: a novel target in cancer therapy. Curr Drug Targets. 2009;10:38–41.
Bergsten E, Uutela M, Li X, et al. PDGF-D is a specific, protease-activated ligand for the PDGF beta-receptor. Nat Cell Biol. 2001;3:512–6.
LaRochelle WJ, Jeffers M, McDonald WF, et al. PDGF-D, a new protease-activated growth factor. Nat Cell Biol. 2001;3:517–21.
Torres-Martin M, Lassaletta L, Isla A, et al. Global expression profile in low grade meningiomas and schwannomas shows upregulation of PDGFD, CDH1 and SLIT2 compared to their healthy tissue. Oncol Rep. 2014;32:2327–34.
Ustach CV, Kim H-RC. Platelet-derived growth factor D is activated by urokinase plasminogen activator in prostate carcinoma cells. Mol Cell Biol. 2005;25:6279–88.
Wang Z, Kong D, Banerjee S, et al. Down-regulation of platelet-derived growth factor-D inhibits cell growth and angiogenesis through inactivation of Notch-1 and nuclear factor-kappaB signaling. Cancer Res. 2007;67:11377–85.
Xu L, Tong R, Cochran DM, et al. Blocking platelet-derived growth factor-D/platelet-derived growth factor receptor beta signaling inhibits human renal cell carcinoma progression in an orthotopic mouse model. Cancer Res. 2005;65:5711–9.
Reigstad LJ, Varhaug JE, Lillehaug JR. Structural and functional specificities of PDGF-C and PDGF-D, the novel members of the platelet-derived growth factors family. FEBS J. 2005;272:5723–41.
Ricard-Blum S. The collagen family. Cold Spring Harb Perspect Biol. 2011;3:a004978.
West RB, Rubin BP, Miller MA, et al. A landscape effect in tenosynovial giant-cell tumor from activation of CSF1 expression by a translocation in a minority of tumor cells. Proc Natl Acad Sci USA. 2006;103:690–5.
Möller E, Mandahl N, Mertens F, et al. Molecular identification of COL6A3-CSF1 fusion transcripts in tenosynovial giant cell tumors. Genes Chromosomes Cancer. 2008;47:21–25.
Xie X, Liu X, Zhang Q, et al. Overexpression of collagen VI α3 in gastric cancer. Oncol Lett. 2014;7:1537–43.
Sherman-Baust CA, Weeraratna AT, Rangel LBA, et al. Remodeling of the extracellular matrix through overexpression of collagen VI contributes to cisplatin resistance in ovarian cancer cells. Cancer Cell. 2003;3:377–86.
Qiao J, Fang C-Y, Chen S-X, et al. Stroma derived COL6A3 is a potential prognosis marker of colorectal carcinoma revealed by quantitative proteomics. Oncotarget. 2015;6:29929–46.
Yu J, Wu WKK, Li X, et al. Novel recurrently mutated genes and a prognostic mutation signature in colorectal cancer. Gut. 2015;64:636–45.
Arafat H, Lazar M, Salem K, et al. Tumor-specific expression and alternative splicing of the COL6A3 gene in pancreatic cancer. Surgery. 2011;150:306–15.
Park J, Morley TS, Scherer PE. Inhibition of endotrophin, a cleavage product of collagen VI, confers cisplatin sensitivity to tumours. EMBO Mol Med. 2013;5:935–48.
Mongiat M, Mungiguerra G, Bot S, et al. Self-assembly and supramolecular organization of EMILIN. J Biol Chem. 2000;275:25471–80.
Colombatti A, Spessotto P, Doliana R, et al. The EMILIN/Multimerin family. Front Immunol. 2011;2:93.
Hwang HC, Sheffield BS, Rodriguez S, et al. Utility of BAP1 immunohistochemistry andp16 (CDKN2A) FISH in the diagnosis of malignant mesothelioma in effusion cytology specimens. Am J Surg Pathol. 2016;40:120–6.
Li Z, Gonzalez CL, Wang B, et al. Cdkn2a suppresses metastasis in squamous cell carcinomas induced by the gain-of-function mutantp53(R172H). J Pathol. 2016;240:224–34.
Rebouissou S, Hérault A, Letouzé E, et al. CDKN2A homozygous deletion is associated with muscle invasion in FGFR3-mutated urothelial bladder carcinoma. J Pathol. 2012;227:315–24.
Acknowledgements
We wish to thank pathologists and clinicians who provided samples and clinical follow-up: M Chargeboeuf (Lons le Saunier), C Charon-Barra (Dijon), C Delfour (Montpellier), S Depardieu (Metz), A Leroux (Vandoeuvre les Nancy), A Lipandy (Nimes), E Maubec (Paris), C de Mauroy (Rouen), YM Robin (Lille), JF Titton (Besancon), M Battistella (Paris), and MP Wissler (Villeurbanne). We are grateful to A Bazin, AC Peyron, and A Ribeiro for their insights and participation in this study. This work was supported by grants from La Ligue contre le Cancer de l'Ain et de Savoie, Institut National du Cancer (NETSARC, RREPS, RESOS).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Dadone-Montaudié, B., Alberti, L., Duc, A. et al. Alternative PDGFD rearrangements in dermatofibrosarcomas protuberans without PDGFB fusions. Mod Pathol 31, 1683–1693 (2018). https://doi.org/10.1038/s41379-018-0089-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-018-0089-4
This article is cited by
-
Molekularpathologie bei ausgewählten Weichgewebssarkomen: diagnostisch und therapeutisch relevante Aberrationen
Die Onkologie (2023)
-
Detection of sarcoma fusions by a next-generation sequencing based–ligation-dependent multiplex RT-PCR assay
Modern Pathology (2022)
-
Coamplification of 12q15 and 12p13 and homozygous CDKN2A/2B deletion: synergistic role of fibrosarcomatous transformation in dermatofibrosarcoma protuberans with a cryptic COL1A1-PDGFB fusion
Virchows Archiv (2022)
-
Novel TNC-PDGFD fusion in fibrosarcomatous dermatofibrosarcoma protuberans: a case report
Diagnostic Pathology (2021)
-
PDGFB RNA in situ hybridization for the diagnosis of dermatofibrosarcoma protuberans
Modern Pathology (2021)
