
Abstract
Chordoma is a rare malignant tumor of bone with high morbidity and mortality. Recently, aggressive pediatric poorly differentiated chordoma with SMARCB1 loss has been described. This study summarizes the clinicopathologic features of poorly differentiated chordoma with SMARCB1 loss in the largest series to date. A search of records between 1990–2017 at MGH identified 19 patients with poorly differentiated chordoma. Immunohistochemical stains were evaluated. Kaplan–Meier survival statistics and log-rank (Mantel Cox) tests compared survival with other subtypes. The patients (n = 19) were diagnosed at a median age of 11 years (range: 1–29). Tumors arose in the skull base and clivus (n = 10/19; 53%); cervical spine (n = 6/19; 32%); and sacrum or coccyx (n = 3/19; 16%). The clinical stage of these patients (AJCC 7e) was stage 2A (n = 7/16; 44%); stage 2B (n = 6/16; 38%); stage 4A (n = 1/16; 6%); and stage 4B (n = 2/16; 13%). The tumors were composed of sheets of epithelioid cells with nuclear pleomorphism, abundant eosinophilic cytoplasm, and increased mitoses. Tumors were positive for cytokeratin (n = 18/18; 100%) and brachyury (n = 18/18; 100%). Patients were treated with a combination of excision, radiation therapy, and chemotherapy. No difference in overall survival, progression free survival, local control time, and metastasis free survival was identified between poorly differentiated chordoma of the skull base and of the spine. Compared to other chordoma subtypes, poorly differentiated chordoma has a significantly decreased mean overall survival after stratification by site (p = 0.037). Pediatric poorly differentiated chordoma has a distinct clinical and immunohistochemical profile, with characteristic SMARCB1 loss and decreased survival compared to conventional/chondroid chordoma. Recognition of this subtype is important because these malignancies should be treated aggressively with multimodality therapy.
Similar content being viewed by others


Introduction
Chordoma is a rare primary malignant tumor of bone demonstrating notochordal differentiation. It almost always arises in the axial skeleton, most commonly the sacrum, followed by the skull base and the mobile spine [1,2,3,4,5]. There is a slight male predominance with a peak incidence between 50–60 years of age. Across all subtypes, chordoma has a low incidence below the age of 40 years old, and it is very rare in children and adolescents (<5% of all chordoma cases) [6, 7]. Although population-based studies using the Surveillance, Epidemiology, and End Results (SEER) database suggest that the tumor overall is quite rare with an annual incidence of 0.08 per 100,000, it has a high morbidity and mortality with a median survival of 6.29 years. Comprehensive survival analysis of 400 cases in the SEER database showed that survival at 5, 10, and 20 years drops steeply over time at 98%, 40%, and 13% respectively [6].
The current World Health Organization (WHO) “Classification of Soft Tissues and Bone” continues to classify chordoma into three subtypes: conventional, chondroid, and dedifferentiated [8]. The morphology varies between these subtypes, from the “physaliphorous cells” and myxoid matrix of the conventional type, to matrix that mimics hyaline cartilage in the chondroid type, to biphasic tumors with juxtaposed high grade undifferentiated sarcomas in the dedifferentiated type. All subtypes (except the dedifferentiated component in dedifferentiated chordoma) typically show positive immunohistochemical staining for keratins, T brachyury, S100, and epithelial membrane antigen (EMA) [5]. Recently, however, a subset of chordoma with an absence of SMARCB1 (SNF5-homolog/SMARCB1, also known as INI1) expression has been described in the pediatric population with a particularly aggressive clinical course [9,10,11,12].
SMARCB1 is a chromatin remodeling member of the SWI/SNF complex. It is known to be lost in a number of sarcomas including malignant rhabdoid tumor, epithelioid sarcoma, epithelioid malignant peripheral nerve sheath tumor (MPNST), and myoepithelial carcinoma [13]. Although loss of SMARCB1 expression has been previously described in a minority of chordomas [11, 12], a closer look at the clinical features associated with these tumors has led to the suggestion that they may comprise a distinct subset of chordoma separate from the three histological types in the current WHO classification system [9, 10, 14, 15]. The clinical features of this subset of chordoma, referred to as “poorly differentiated chordoma,” have not yet been well characterized. In this study, we aim to summarize the clinicopathologic features of the largest series of poorly differentiated chordoma to date.
Materials and methods
A comprehensive search of an IRB approved Sarcoma Data Repository at the Massachusetts General Hospital, as well as the clinical and pathology records at the Stephan L. Harris Center for Chordoma Care at the Massachusetts General Hospital (MGH) between 1990 and 2017 identified a total of 19 patients with a diagnosis of “poorly differentiated chordoma” as rendered by expert bone and soft tissue pathologists on the bases of morphology and immunohistochemistry for S100, keratin, T brachyury, and SMARCB1. Retention of nuclear SMARCB1 by immunohistochemistry was used as an exclusion criteria. Of these 19 patients, a total of 3 patients were pathology cases seen in consultation and have limited clinical data; for the remaining 16 patients, clinical data was gathered from the electronic medical record as available, and Kaplan–Meier survival statistics were performed compared to outcomes for all chordoma patients seen at our institution since the year 2000. A log-rank (Mantel Cox) test was used to compare survival with other chordoma subtypes from patients seen at the Stephan L. Harris Center for Chordoma Care at MGH.
Results
Demographics
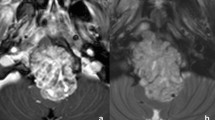
The patients (n = 19) were diagnosed at an average age of 11.9 years (range: 1 to 29 years; median: 11 years; standard deviation: 8.4 years) with a M:F ratio of 0.46. Most of the tumors arose in the skull base and clivus (n = 10/19; 53%) [Fig. 1a]; cervical spine (n = 6/19; 32%); and sacrum or coccyx (n = 3/19; 16%). The most common symptoms at presentation were pain and discomfort (n = 12/16; 75%), as well as neurologic symptoms (n = 9/16; 56%); other less common complaints included weight loss, neck stiffness, respiratory symptoms, and compression fracture.

Radiology and histopathology of poorly differentiated chordoma. The radiology and histopathology for patient 5 are presented in this figure. An axial MRI of the brain demonstrates a large enhancing skull base lesion measuring approximately 4.5 cm with extensive mass effect on the third ventricle and pons (a). On histology, the tumor is composed of sheets of mitotically active epithelioid cells with oval and irregular nuclei, variably prominent nucleoli, and eosinophilic cytoplasm (b). No myxoid stroma was present, but areas of necrosis were readily identified (c). On immunohistochemistry, the lesional cells were strongly positive for keratin (in this case, MNF116) (d) and nuclear T brachyury (e), with nuclear INI-1 loss (f)
The patients were given an AJCC TNM 7th edition pathologic stage of T1 (n = 9/16; 56%) and T2 (n = 7/16; 44%). Of the T1 patients, no patients were identified to have nodal metastases, but two patients were found to have distant metastasis. Of the T2 patients, a single patient was found to have both lymph node and distant metastases. Overall, the clinical stage of these patients per the guidelines given by the AJCC 7th edition was Stage 2A (n = 7/16; 44%); Stage 2B (n = 6/16; 38%); Stage 4A (n = 1/16; 6%); and Stage 4B (n = 2/16; 13%). [Table 1]
Histology and immunohistochemistry
Each of the patients in this cohort underwent either a core biopsy or excisional biopsy for definitive diagnosis, and the slides were reviewed at our institution. Morphologically, the tumors were composed of epithelioid cells with a cohesive and often sheet-like pattern of growth. [Fig. 1b] The cells were remarkable for prominent nuclear pleomorphism and relatively abundant eosinophilic cytoplasmic, with rhabdoid morphology being described at least focally in 3 tumors. The canonical physaliferous cells of conventional chordoma were not identified, and extracellular myxoid stroma was generally not present; in the single case where myxoid stroma was present, it was very minimal. Necrosis was present at least focally in 5 tumors, [Fig. 1c] and extraosseous extension was identified in all cases. Mitoses were typically increased (up to 12 mitoses/10 HPFs). Two cases also had a Ki67 immunostain performed, and both showed an elevated Ki67 proliferation index in the range of 10–15%.
Immunohistochemical analysis showed that the tumors were positive for cytokeratin (n = 18/18; 100%) and T brachyury (n = 18/18; 100%) and generally negative for S100 (n = 14/17; 82%). SMARCB1 was lost in all tumors that were tested (n = 17/17; 100%). [Fig. 1d–f]
Treatment and clinical follow-up
Patients were treated with a combination of surgical excision, radiation therapy, and chemotherapy as shown in Table 1. Two patients were treated with definitive radiation therapy and chemotherapy alone. Thirteen patients were treated with surgical excision; of these, 5 patients were given adjuvant radiation therapy, 4 patients were given both radiation therapy and chemotherapy, and 4 patients were treated with surgery alone. Notably, two patients were treated with multimodality therapy including surgery, chemotherapy, and both preoperative and postoperative radiation. Chemotherapy agents included doxorubicin, ifosfamide, etoposide, vincristine, and cyclophosphamide. Radiation therapy consisted of proton-based radiation therapy to a median dose of 72 Gy (range: 66.6 to 79.4 Gy). Seven patients had radiation therapy postoperatively only, and another two patients (patients #6 and 7) had radiation therapy preoperatively and postoperatively.
Clinical follow-up reveals that six patients were found to have local recurrence at a median of 11 months (range: 3 to 26 months) from the initial diagnosis. Among the 13 non-metastatic cases at diagnosis, three patients developed distant metastases at 3, 39, and 40 months from the initial diagnosis. In this cohort, three patients are deceased with disease (DWD); five patients currently have no evidence of disease (NED); and four patients are alive with disease (AWD). An additional three patients are alive with no clinical follow-up, and another patient is deceased with an unknown disease status.
The cumulative mean overall survival (OS) for poorly differentiated chordoma regardless of site is 53 months (standard error, SE = 9 months; median = 51 months). No statistically significant difference was identified in mean overall survival, mean progression free survival, mean local control time, and mean metastasis free survival between poorly differentiated chordoma of the skull base and of the spine. [Fig. 2a–d] At our institution, 681 patients with conventional or chondroid chordoma (irrespective of site) had a cumulative mean overall survival of 109 months (SE = 5 months) and 96 months (SE = 5 months), respectively. A log-rank (Mantel–Cox) test shows that poorly differentiated chordoma has a statistically significant decreased mean overall survival (p = 0.025) when compared overall to a cumulative institutional database of 526 patients with conventional chordoma (n = 526; 77%) and 155 patients with chondroid chordoma (n = 155; 23%). [Fig. 3a] Compared with patients at our institution with conventional or chondroid chordoma, a significantly decreased overall survival of poorly differentiated chordoma (p = 0.037) was found after stratification by site (skull base vs. spine). [Fig. 3b, c] Data on progression free survival, local control time, and metastasis free survival was not available for the entire cohort of base of skull chordomas for more rigorous comparison to other histological subtypes.

Survival curves for poorly differentiated chordoma by site. Kaplan-Meier survival statistics were performed on poorly differentiated chordoma comparing survival of patients with tumors arising in the skull base and those with tumors arising in the spine. For the skull base and the spine, respectively, mean overall survival (a) is 62 months (SE = 11 months) and 46 months (SE = 7 months); mean progression free survival (b) is 34 months (SE = 13 months) and 23 months (SE = 7 months); mean local control time (c) is 41 months (SE = 13 months) and 31 months (SE = 8 months); and mean metastasis free survival (d) is 49 months (SE = 13 months) and 30 months (SE = 7 months). A log-rank (Mantel–Cox) test shows that there is no difference in survival between poorly differentiated chordoma of the skull base and the spine for mean overall survival (p = 0.63), mean progression free survival (p = 0.88), mean local control time (p = 0.88), and mean metastasis free survival (p = 0.50)

Comparison of survival curves for poorly differentiated chordoma to a cumulative institutional database. Kaplan–Meier survival statistics were performed on poorly differentiated chordoma compared to conventional and chondroid chordoma patients seen at the Massachusetts General Hospital. Mean overall survival of poorly differentiated chordoma (a) is 53 months (SE = 9 months). When stratified by site (b, c), a significantly decreased overall survival was identified (p = 0.037) when compared to both conventional chordoma and chondroid chordoma of the comparable sites
In the spine, poorly differentiated chordoma had a mean overall survival of 46 months (SE = 7 months; median = 51 months); mean progression free survival of 23 months (SE = 10 months; median = 68 months); mean local control time of 31 months (SE = 8 months; median = 26 months); and mean metastasis free survival of 30 months (SE = 7 months; median = 39 months). At our institution, 126 patients with spinal conventional and chondroid chordoma respectively had a mean overall survival of 129 months (SE = 13 months) and 101 months (SE = 9 months); mean progression free survival of 100 months (SE = 12 months) and 60 months (SE = 6 months); mean local control time of 121 months (SE = 14 months) and 63 months (SE = 6 months); and mean metastasis free survival of 165 months (SE = 15 months) and 83 months (SE = 6 months). A log-rank (Mantel-Cox) test shows that spinal poorly differentiated chordoma has a significantly decreased mean overall survival (p = 0.035; Fig. 4a), mean progression free survival (p < 0.0005; Fig. 4b), mean local control time (p = 0.006; Fig. 4c), and mean metastasis free survival (p < 0.005; Fig. 4d) when compared overall to 126 patients with spinal conventional chordoma (n = 98; 78%) and chondroid chordoma (n = 28; 22%) at our institution.

Comparison of survival curves for spinal poorly differentiated chordoma to a spinal institutional database. Kaplan–Meier survival statistics were performed on spinal poorly differentiated chordoma compared to spinal conventional and chondroid chordoma patients seen at the Massachusetts General Hospital. For spinal poorly differentiated chordoma, mean overall survival is 46 months (SE = 7 months); mean progression free survival is 23 months (SE = 10 months); mean local control time is 31 months (SE = 8 months); and mean metastasis free survival is 30 months (SE = 7 months). A log-rank (Mantel–Cox) test shows that spinal poorly differentiated chordoma has a significantly decreased mean overall survival (p = 0.035; a), mean progression free survival (p < 0.0005; b), mean local control time (p = 0.006; c), and mean metastasis free survival (p < 0.0005; d) when compared to both conventional chordoma and chondroid chordoma of the spine
Discussion
Poorly differentiated chordoma is a rare subset of chordoma with distinct clinical features compared to those defined by the most recent WHO classification. In our series, compared to conventional chordoma, poorly differentiated chordoma has a marked tendency to occur in a younger population (children and adolescents, though not universally) with a very slight female predominance, and have an increased predilection for the skull base and cervical spine. Like conventional tumors, the cases in this series were all positive for T brachyury and wide-spectrum keratin; however, in 14 of 17 cases, they had negative S100 staining. Clinically, compared to historical controls at our institution, these tumors appear to be more aggressive than conventional and chondroid chordoma in both the skull base and the spine, with a decreased mean overall survival.
In general, poor survival may be partially attributable to the fact that chordoma is often indolent and slow-growing, and therefore commonly diagnosed at a late stage, making distant metastasis at presentation more likely [16, 17]. However, patient survival appears to be more affected by the tumor’s locally aggressive nature, leading to a high rate of local recurrence [18, 19]. This appears to be particularly true in poorly differentiated chordoma, which in our cohort appears to have predilection for aggressive behavior, with rapid onset of both local recurrence and distant metastatic disease. Similar results were seen in smaller series from Yadav et al and a small subset of skull base chordoma from Hoch et al [11, 20]. Consequently, clinical management in these cases can be quite difficult, and while en-bloc resection with wide negative margins is generally considered the primary treatment modality for conventional chordoma [19, 21], this is not clinically feasible in patients with aggressive skull base tumors. Currently, the use of adjuvant radiation therapy for treatment of chordoma is institution-dependent but has been described to have therapeutic and palliative utility in certain cases [22,23,24].
For conventional chordoma, the data for systemic therapy in the adjuvant or metastatic setting remains underwhelming. Most published pilot or phase 2 studies of small kinase inhibitors show only modest benefit [25,26,27,28,29], and the consensus is that conventional chemotherapy is not effective in this subtype. In contrast, there are case reports that conventional sarcoma chemotherapy agents may have activity in poorly differentiated chordoma, dedifferentiated chordoma, or pediatric chordoma that is not further classified [1, 30, 31]. It is hypothesized that the presence of the extracellular matrix acts as a barrier for many chemotherapeutic agents, and that concomitant use of drugs targeting the extracellular matrix might be used in conjunction with more standard agents. However, because poorly differentiated chordoma lacks extracellular matrix, more conventional therapies for sarcoma management may be effective. In our series, patients did receive chemotherapy; however, it is impossible to make meaningful conclusions on activity given the heterogeneity of patient management and agents utilized. We are encouraged by the outcomes of two patients treated only with chemoradiation that are currently without evidence of disease at 12 and 11 months from initial diagnosis.
Perhaps more exciting is whether this unique subset of chordoma could be targeted through novel molecular therapies. Enhancer of Zeste homologue 2 (EZH2) is the catalytic methyl transferase of the polycomb repressive complex 2 (PRC2), an epigenetic regulator of transcription [32]. SMARCB1 inactivation is believed to lead to loss of inhibition of EZH2 and oncogenesis [33]. Moreover, targeting EZH2 in models of malignant rhabdoid tumor, a SMARCB1 negative sarcoma, leads to cell death [34,35,36], and early activity has been reported in SMARCB1 negative epithelioid sarcoma [37]. Tazemetostat, a novel EZH2 inhibitor, has entered early phase clinical trials for patients with SMARCB1 loss, including poorly differentiated chordoma (NCI# NCT02601950 and NCT02601937). We eagerly await data from these clinical trials on the SMARCB1 negative chordoma patients.
A limitation to the survival findings reported in this study is that this cohort is composed of a heterogeneous population of patients, who have been managed variably either as patients treated at our institution as a primary tumor and those who are managed at our institution as a locally recurrent disease after prior intervention elsewhere. In our experience, prior surgical procedures at an outside facility generally have an unfavorable impact on local control. Nearly all the patients diagnosed with poorly differentiated chordoma in this study had prior interventions (predominantly surgical) at outside institutions, and consequently, it is difficult to compare the outcomes of this population to patients treated solely at our institution. While the survival data in this cohort certainly points to overall worsened outcomes, a larger cohort may be necessary to confirm these findings.
As poorly differentiated chordoma lacks the classic morphologic characteristics of conventional chordoma (such as myxoid extracellular matrix and physaliphorous cells), the differential diagnosis is broad and immunohistochemical results may be misleading. Since these tumors often have a cohesive epithelioid morphology and are positive for keratin, the most concerning and probably most common misdiagnosis is metastatic carcinoma, particularly in adults, which can extremely rarely show SMARCB1 loss [38]. In children, the most problematic entity is atypical teratoid/rhabdoid tumor, which also has SMARCB1 loss, as described above, but will be negative for T brachyury. Other entities in the differential diagnosis include sarcomas; in particular, epithelioid sarcoma may present a difficulty, as this entity is also SMARCB1 negative and positive for cytokeratin [13]. Similarly, a subset of epithelioid malignant peripheral nerve sheath tumors (MPNST) and myoepithelial carcinomas also show an immunophenotype with positive cytokeratin and negative nuclear SMARCB1 [13, 39, 40]. In these cases, patient demographics, clinical history, and radiographic findings may be helpful, and immunohistochemistry for T brachyury is essential in making the appropriate diagnosis. Although other tumors have been described to be positive for T brachyury, nuclear T brachyury immunoreactivity is a fairly sensitive and specific marker for notochordal origin [41].
Poorly differentiated chordoma has a distinct clinical and immunohistochemical profile compared to conventional chordoma, and its morphologic characteristics lead to a broad differential that includes metastatic carcinoma and sarcoma. Regardless of morphology, chordoma should remain on the differential for all midline tumors of the axial skeleton, and the use of immunohistochemistry for T brachyury and SMARCB1 may be helpful in distinguishing chordoma from its mimics. In our series, the largest poorly differentiated chordoma cohort to date, we confirm this is a disease of young adults and children, with a predominance of skull base and cervical spine origin. The outcome of this cohort was inferior to historical controls of conventional and chondroid chordoma. We believe that without further evidence these patients should be treated with aggressive multimodality therapy, when feasible. For those patients with advanced disease, we strongly support participation in clinical trials.
References
Dahlin DC, Maccarty CS. Chordoma. Cancer . 1952;5:1170–8.
Healey JH, Lane JM. Chordoma: a critical review of diagnosis and treatment. Orthop Clin North Am. 1989;20:417–26.
Horten BC, Montague SR. In vitro characteristics of a sacrococcygeal chordoma maintained in tissue and organ culture systems. Acta Neuropathol. 1976;35:13–25.
Shen J, Li CD, Yang HL, Lu J, et al. Classic chordoma coexisting with benign notochordal cell rest demonstrating different immunohistological expression patterns of brachyury and galectin-3. J Clin Neurosci. 2011;18:96–9.
Vujovic S, Henderson S, Presneau N, et al. Brachyury, a crucial regulator of notochordal development, is a novel biomarker for chordomas. J Pathol. 2006;209:157–65.
McMaster ML, Goldstein AM, Bromley CM, et al. Chordoma: incidence and survival patterns in the United States, 1973-1995. Cancer Causes Control. 2001;12:1–11.
Wold LE, Laws ER Jr.. Cranial chordomas in children and young adults. J Neurosurg. 1983;59:1043–7.
Fletcher CDM, Bridge JA, Hogendoorn PCW, et al., editors. WHO Classification of Tumours of Soft Tissue and Bone Geneva: WHO Press; 2013.
Cha YJ, Hong CK, Kim DS et al. Poorly differentiated chordoma with loss of SMARCB1/INI1 expression in pediatric patients: a report of two cases and review of the literature. Neuropathology.
Hasselblatt M, Thomas C, Hovestadt V, et al. Poorly differentiated chordoma with SMARCB1/INI1 loss: a distinct molecular entity with dismal prognosis. Acta Neuropathol. 2016;132:149–51.
Hoch BL, Nielsen GP, Liebsch NJ, et al. Base of skull chordomas in children and adolescents: a clinicopathologic study of 73 cases. Am J Surg Pathol. 2006;30:811–8.
Mobley BC, McKenney JK, Bangs CD, et al. Loss of SMARCB1/INI1 expression in poorly differentiated chordomas. Acta Neuropathol. 2010;120:745–53.
Hornick JL, Dal Cin P, Fletcher CD. Loss of INI1 expression is characteristic of both conventional and proximal-type epithelioid sarcoma. Am J Surg Pathol. 2009;33:542–50.
Antonelli M, Raso A, Mascelli S, et al. SMARCB1/INI1 involvement in pediatric chordoma: a mutational and immunohistochemical analysis. Am J Surg Pathol. 2017;41:56–61.
Renard C, Pissaloux D, Decouvelaere AV, et al. Non-rhabdoid pediatric SMARCB1-deficient tumors: overlap between chordomas and malignant rhabdoid tumors? Cancer Genet. 2014;207:384–9.
Bjornsson J, Wold LE, Ebersold MJ, et al. Chordoma of the mobile spine. a clinicopathologic analysis of 40 patients. Cancer . 1993;71:735–40.
Chambers PW, Schwinn CP. Chordoma. A clinicopathologic study of metastasis. Am J Clin Pathol. 1979;72:765–76.
Bergh P, Kindblom LG, Gunterberg B, et al. Prognostic factors in chordoma of the sacrum and mobile spine: a study of 39 patients. Cancer . 2000;88:2122–34.
Boriani S, Chevalley F, Weinstein JN, et al. Chordoma of the spine above the sacrum. Treatment and outcome in 21 cases. Spine. 1996;21:1569–77.
Yadav R, Sharma MC, Malgulwar PB, et al. Prognostic value of MIB-1, p53, epidermal growth factor receptor, and INI1 in childhood chordomas. Neuro Oncol. 2014;16:372–81.
Kayani B, Sewell MD, Hanna SA, et al. Prognostic factors in the operative management of dedifferentiated sacral chordomas. Neurosurgery . 2014;75:269–75.
Chen YL, Liebsch N, Kobayashi W, et al. Definitive high-dose photon/proton radiotherapy for unresected mobile spine and sacral chordomas. Spine. 2013;38:E930–6.
DeLaney TF, Liebsch NJ, Pedlow FX, et al. Long-term results of Phase II study of high dose photon/proton radiotherapy in the management of spine chordomas, chondrosarcomas, and other sarcomas. J Surg Oncol. 2014;110:115–22.
Hamamoto S, Matsuoka T, Okuma T, et al. Effective palliative radiofrequency ablation for tumors causing pain, numbness and motor function disorders: case series. BMC Res Notes. 2014;7:765.
Azzarelli A, Quagliuolo V, Cerasoli S, et al. Chordoma: natural history and treatment results in 33 cases. J Surg Oncol. 1988;37:185–91.
Patel SS, Schwab JH. Immunotherapy as a potential treatment for chordoma: a review. Curr Oncol Rep. 2016;18:55.
Stacchiotti S, Casali PG. Systemic therapy options for unresectable and metastatic chordomas. Curr Oncol Rep. 2011;13:323–30.
Stacchiotti S, Longhi A, Ferraresi V, et al. Phase II study of imatinib in advanced chordoma. J Clin Oncol. 2012;30:914–20.
Stacchiotti S, Tamborini E, Lo Vullo S, et al. Phase II study on lapatinib in advanced EGFR-positive chordoma. Ann Oncol. 2013;24:1931–6.
Al-Rahawan MM, Siebert JD, Mitchell CS, et al. Durable complete response to chemotherapy in an infant with a clival chordoma. Pediatr Blood Cancer. 2012;59:323–5.
Scimeca PG, James-Herry AG, Black KS, et al. Chemotherapeutic treatment of malignant chordoma in children. J Pediatr Hematol Oncol. 1996;18:237–40.
Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature . 2011;469:343–9.
Wilson BG, Wang X, Shen X, et al. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell. 2010;18:316–28.
Alimova I, Birks DK, Harris PS, et al. Inhibition of EZH2 suppresses self-renewal and induces radiation sensitivity in atypical rhabdoid teratoid tumor cells. Neuro Oncol. 2013;15:149–60.
Knutson SK, Warholic NM, Wigle TJ, et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci USA. 2013;110:7922–7.
Kurmasheva RT, Sammons M, Favours E et al. Initial testing (stage 1) of tazemetostat (EPZ-6438), a novel EZH2 inhibitor, by the pediatric preclinical testing program. Pediatr Blood Cancer
Gounder MM, Stacchiotti S, Schöffski P, et al. Phase 2 multicenter study of the EZH2 inhibitor tazemetostat in adults with INI1 negative epithelioid sarcoma (NCT02601950). J Clin Oncol. 2017;35:abstr 11058. suppl
Bishop JA, Antonescu CR, Westra WH. SMARCB1 (INI-1)-deficient carcinomas of the sinonasal tract. Am J Surg Pathol. 2014;38:1282–9.
Gleason BC, Fletcher CD. Myoepithelial carcinoma of soft tissue in children: an aggressive neoplasm analyzed in a series of 29 cases. Am J Surg Pathol. 2007;31:1813–24.
Laskin WB, Weiss SW, Bratthauer GL. Epithelioid variant of malignant peripheral nerve sheath tumor (malignant epithelioid schwannoma). Am J Surg Pathol. 1991;15:1136–45.
Miettinen M, Wang Z, Lasota J, et al. Nuclear Brachyury expression is consistent in chordoma, common in germ cell tumors and small cell carcinomas, and rare in other carcinomas and sarcomas: an immunohistochemical study of 5229 cases. Am J Surg Pathol. 2015;39:1305–12.
Acknowledgements
We would like to acknowledge Al Ferreira, RN, of the UCLA Chordoma Center for his contributions to clinical follow up in this study.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Shih, A.R., Cote, G.M., Chebib, I. et al. Clinicopathologic characteristics of poorly differentiated chordoma. Mod Pathol 31, 1237–1245 (2018). https://doi.org/10.1038/s41379-018-0002-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-018-0002-1
This article is cited by
-
SMARCB1-Deficient Skull Base Chondrosarcoma with 12p Duplication Presenting as Somatic-Type Malignancy Arising from Metastatic Seminoma
Head and Neck Pathology (2024)
-
Imaging of pituitary tumors: an update with the 5th WHO Classifications—part 2. Neoplasms other than PitNET and tumor-mimicking lesions
Japanese Journal of Radiology (2023)
-
Update of pediatric bone tumors—notochordal tumors, chondrogenic tumors, and vascular tumors of the bone
Skeletal Radiology (2023)
-
Virus-like particle-based delivery of Cas9/guide RNA ribonucleoprotein efficiently edits the brachyury gene and inhibits chordoma growth in vivo
Discover Oncology (2023)
-
Lynch syndrome-associated chordoma with high tumor mutational burden and significant response to immune checkpoint inhibitors
Brain Tumor Pathology (2023)
