
Abstract
We used single cell Q-PCR on a micro-fluidic platform (Fluidigm) to analyse clonal, genetic architecture and phylogeny in acute myeloid leukaemia (AML) using selected mutations. Ten cases of NPM1c mutant AML were screened for 111 mutations that are recurrent in AML and cancer. Clonal architectures were relatively simple with one to six sub-clones and were branching in some, but not all, patients. NPM1 mutations were secondary or sub-clonal to other driver mutations (DNM3TA, TET2, WT1 and IDH2) in all cases. In three of the ten cases, single cell analysis of enriched CD34+/CD33− cells revealed a putative pre-leukaemic sub-clone, undetectable in the bulk CD33+ population that had one or more driver mutations but lacked NPM1c. Cells from all cases were transplanted into NSG mice and in most (8/10), more than one sub-clone (#2-5 sub-clones) transplanted. However, the dominant regenerating sub-clone in 9/10 cases was NPM1+ and this sub-clone was either dominant or minor in the diagnostic sample from which it was derived. This study provides further evidence, at the single cell level, for genetic variegation in sub-clones and stem cells in acute leukaemia and demonstrates both a preferential order of mutation accrual and parallel evolution of sub-clones.
Similar content being viewed by others


Introduction
Although almost all cancers originate in a single cell, the sequential acquisition of necessary additional mutations fuels sub-clonal diversity which is then a substrate for positive or negative selection within the tissue ecosystems and with therapy [1, 2]. This process frequently results in complex cell population structures and highly variegated genetics [3,4,5].
The genomics of AML have been described in considerable detail, revealing multiple sub-types [6, 7] and sequential transition between clinically silent pre-leukaemia and overt disease [8, 9]. Sub-clonal architectures in diagnostic samples have been inferred from allele burdens [10] and appear to be relatively simple compared to that observed in many common cancers [11, 12].
The extent of sub-clonal diversity and phylogenetic architecture is, however, best derived from single cell analysis. This is challenging however in terms of accuracy and depth, but has proven illuminating in some solid tumours [13] and ALL [3].
Only a few studies to date have reported single cell genetics and inferred sub-clonal phylogenies in AML. Paguirigan et al. [14] used single cell, multiplexed Q-PCR to investigate patterns of segregation of two concurrent mutations in AML–FLT3−ITD and NPM1c mutations. The data revealed significantly more sub-clonal diversity than could be inferred from analysis of the bulk population. Klco et al. [15] fractionated immuno-phenotypically distinct cell populations from a patient with AML and sequenced the amplified DNA from single cells for ten known mutations. From these data, they could infer a branching sub-clonal architecture. Jan et al. used a Q-PCR assay on colonies derived from sorted single cells derived from two cases of AML and were able to infer an ordered sequence of mutations [8]. Quek et al. screened single cells for targeted mutations in immunophenotypically-defined subsets and identified putative clonal sequences and mutation order in six cases [16].
In our previous studies in ALL, we used multi-colour FISH or multi-plexed Q-PCR in a micro-fluidic platform (Fluidigm) to detect sub-clonal variegation and clonal architecture [3, 17]. In this study, we sought to replicate our observations on ALL for AML, selecting the subset of cases with NPM1c mutations. NPM1c+ cases constitute around 27% of adult AML with a variable but overall intermediate risk [7]. The questions posed included the extent of sub-clonal complexity that was discernible, sequential order of mutations and whether stem cells or leukaemia propagating cells, assayed by xeno-transplantation, were genetically variable.
Materials and methods
Sample cohort
A total of ten well-characterised NPM1 mutant AML samples [18] were selected for further study according to engraftment potential. Blood and marrow samples were collected from patients with AML after written informed consent at St Bartholomew’s Hospital. The protocol was approved by the East London and City Research Ethics Committee. All studies comply with the rules of the revised Helsinki protocol. These had all been found to successfully transplant in NOD/SCID mice [18]. This selection criterion may have biased our analysis towards poorer prognosis cases [7, 18]. Available peripheral blood was collected prior to treatment at presentation (n = 10) and from matched relapse (n = 3). Mononuclear cells were obtained by density gradient centrifugation. Details of the patient samples are listed (Table 1).
FACS cell sorting according to immunophenotype details can be found in Supplementary Information.
Mutation analysis
A targeted screening approach investigating 111 genes (Table 2) was used to identify mutations and DNA coding region alterations in each NPM1c AML as previously described [7] that could potentially be tracked in single cells. The analysis is based on variants that can be classified as recurrent driver mutations, using widely accepted genetic criteria. These included non-synonymous base substitutions and small (<200-bp) insertions or deletions (indels). Table 3 lists the probes used for mutant versus wild type sequences and PCR primers.
Sequencing data
For the targeted mutation screening of each leukaemia, two populations of interest were stained and sorted as described in Supplementary Information and DNA extracted (Qiagen® DNA blood kit according to manufacturers’ instructions): peripheral blood T-cells (CD3+/CD33−) (as a control) and mononuclear blast cells (CD3−/CD33+). The latter had <1% CD34+ cells and we refer to this population as CD34−.
For details of library preparation, sequencing, alignment and analysis, please refer to Supplementary Information.
Xeno-transplantation
NOD/SCID (Il2rg−/−) mice (Jackson Laboratory, Bar Harbor, ME) were injected intravenously (3 mice per AML sample) with 9–10 million AML cells after T-cell depletion by Easysep T-cell enrichment cocktail (Stem Cell Technologies). Mice were bled by tail veins at 12–14 weeks and blood leucocytes investigated by FACS (as described in Supplementary Information and Supplementary Figs. 2 and 3) using anti-human and anti-mouse CD45 antibodies to determine the percentage of leukaemic cell engraftment. For details of how successful/undetectable/minimal grafts were managed and serial transplantations were carried out, please refer to Supplementary Information.
Single cell sorting and multiplex Q-PCR analysis
Single cell sorting was carried out (see Supplementary Information and Supplementary Fig. 1) according to our established published Q-PCR single cell (Fluidigm) protocol [17]. Briefly, from each case single AML cells (either CD33+/CD34−/CD3− (blast population), CD3+/CD33− (internal control), CD34+/CD33− (putative stem cell), CD45+ (human cells post-transplant) or cord blood cells (normal diploid control) were sorted into individual wells of a 96 well plate, lysed and DNA target amplification completed for regions of interest encompassing patient specific mutations or DNA alterations. Allelic discrimination Q-PCR assays were designed specifically for each mutation in every patient. Standard Q-PCR assays targeting unique FLT3−ITDs were designed for each positive patient. Genes targeted in each case are listed in Table 1. The ß2M locus, located in a diploid region of the genome, was used as a control. Q-PCR completed using the 48 × 48 dynamic array and the BioMark™ HD from Fluidigm.
Several approaches were adopted during this experiment to optimise and confirm the presence of a single cell and ensure all assays performed efficiently under experimental conditions [17]; a brief description can be found in Supplementary Information and Supplementary Fig. 4.
Maximum parsimony
Maximum parsimony searches for sub-clonal phylogenies were conducted using heuristic searches as previously described [17]; a brief description can be found in Supplementary Information.
Results
Our targeted exomic screening approach identified a number of common or recurrent driver SNV mutations in each patient’s diagnostic sample (Table 1); similar to those previously described for NPM1c AML [7, 19,20,21]. Five of the ten cases had both DNM3TA and TET2 mutations, reflecting the selection of driver mutations that cooperate to confer fitness advantage of haemopoietic stem cells [22]. Allele frequencies varied greatly suggested that many mutations were probably sub-clonally distributed.
Individual cells sorted as CD34+/CD33− or CD33+/CD3− were assayed by multiplex Q-PCR for each driver mutation identified in that patient’s sample. We similarly assessed individual cells (unsorted) from NSG mice in which T-cell depleted AML cells from each patient had been transplanted. From those single cell data, we are able to infer a probable clonal phylogeny for each case with genetically distinct sub-clones, the immunophenotype and the clonal derivation of leukaemia that regenerated in NSG mice. We take the latter as a read-out of sub-clones with self-renewal or stem cell activity.
Clonal architectures
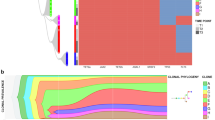
Figure 1 summarises the data from all ten cases (see Supplementary Information for more detailed data). This includes an identifier (t = transplant) of sub-clones that successfully transplanted into mice (t1, t2 and t3 refer to individually transplanted mice using diagnostic material from each patient). The phylogenetic or sub-clonal architectures inferred are relatively simple and either linear or branching (three patients). The analyses are relatively insensitive however with minor clones below 5% being difficult to detect. It is very likely that we are significantly under-estimating clonal complexity and will have missed minor sub-clones that could be clinically relevant, emerging at relapse [23].

Clonal phylogenies, inferred by maximum parsimony, and sub-clone genotypes in 10 patients. Genetically distinct sub-clone percentages (as a fraction of the total population) are indicated next to each clone; e.g., patient 1, most primitive sub-clone, CD34+/CD33− first and CD33+/CD34-/CD3− second percentages indicated as 18%/5%, respectively. This indicates that this sub-clone was found in 18% of the total CD34+/CD33- cells investigated and 5% of the total CD33+/CD34−/CD3−cells investigated (for the relapse samples of patients 9 and 10 only bulk cells without phenotype consideration could be sorted, as the samples available were from fixed cytogenetic preparations; the sub-clone is shown as a single percentage). Those sub-clones that grew in mice are indicated with horizontal black arrows. t1-3 (%). T, transplant. 1-3 individual mice. % fraction of human cells in mouse bone marrow. Sub-clone denoted by dotted circle is below detection limit in diagnostic sample but present in mouse transplant read-out. Dotted arrows lines between sub-clones (case #1 and #3) indicates alternative clonal phylogenies. In case #3, there are 4 possible equally parsimonious phylogenetic trees (details in Supplementary Information Figs. 5 and 6). Further details on each of the individual 10 patients’ clonal analyses are given in Supplementary Information
In two patients (#1 and #3; Fig. 1), there were more than one equally parsimonious phylogenetic trees (illustrated by alternative dotted lines connecting sub-clones). We depict all equally parsimonious trees for patients #1 and #3 in Supplementary Figs. 5 and 6.
The number of identifiable sub-clones varied from one to six. In four patients (#4, #5, #7 and #8) the small, putative stem cell CD34+/CD33− fractions contained a genetic sub-clone that was not discernible in the large CD33+ blast population. These cells had fewer mutations, lacked NPM1c mutation and could represent pre-leukaemic cells [8, 9].
We did not detect DNM3TA or other putative founder mutations in the T cells by single cell analysis. However, in most cases reported by Shlush et al. [24], the mutant DNM3TA allele frequency in T cells was low and so could have been missed in our samples in which only a maximum of 48 single T cells were assayed. In the total or bulk population of NPM1c AML-derived T cells that were subject to targeted sequencing in our series of patients, the calculated allele frequency for DNM3TA mutations and other putative driver mutations in AML cells ranged from 0.64 to 4.35% in the T cell population.
NPM1c mutations were always preceded by mutations previously considered as possible founders; DNMT3A, IDH2, WT1, TET2, as well as some additional mutations that are less well validated as early events in NPM1c AML including NRAS, ZRSR2 and CBL. FLT3 mutations and FLT3−ITDs were found to occur both before and after the acquisition of NPM1c but were always sub-clonal to putative founder mutations.
Match relapsed cases
In the three NPM1c AMLs with matched relapse samples (#8, 9, 10), we found high levels of NPM1c sub-clones in the CD34+/CD33− population at diagnosis ranging from 43–100%. In the single case in which the CD34+/CD33− population could be assessed at relapse (patient #8), the size of the NPM1c sub-clone had increased from 43 to 95%. In these AMLs it was also possible to identify sub-clones at relapse or in the mice after transplant of the diagnostic or relapsed material that had acquired more mutations in addition to those found in the major clone at diagnosis (in #9, #10; see Table 1). Some of these mutations could not be tracked by Q-PCR but were identified by direct sequencing (Table 1). Patient #10 had two FLT3 sub-clonal mutations (detected by sequencing), one at diagnosis (10.03%) rising to 41.64% whilst the other was only detected at relapse (41.16%). Neither of these FLT3 mutations could be tracked, so they do not appear in patient #10 clonal structure (Fig. 1).
Reiterative mutations
Reiterated mutations in individual driver genes were identified in some cases. In patient #3, the two distinctive FLT3 mutations were segregated in distinctive sub-clones. Similarly, in patient #1, the two distinctive TET2 mutations were present in separate (minor/major) sub-clones. In contrast, in patient #7 the two TET2 mutations were in the same sub-clone and probably bi-allelic. Phylogenetic architectures suggested that NPM1c mutations may also have been reiterative in some cases, for example with patient #3 (and possibly patient #1) but the invariant nature of this mutation makes this more ambiguous.
Stem cell read-outs in transplants
The single cell genetics of regenerated leukaemias in mice (see t1,t2,t3 % in Fig. 1) allowed us to infer the sub-clonal origins of leukaemias and hence the genetic composition and its variation in the stem or leukaemia propagating cell compartment of these AML. The clonal read-outs in the transplants were diverse but some patterns emerged.
In eight cases (patients #1, #2, #3, #4, #5, #6, #8 at diagnosis, #10 at relapse) two to five sub-clones present in the diagnostic sample regenerated in the mice. However in each case, one sub-clone was dominant, proportionally and this sub-clone always contained NPM1c.
In one patient (#7), only one sub-clone was present at low levels (0.39% CD34+ cells) in a single mouse and, surprisingly, this corresponded to the most ancestral sub-clone in the diagnostic sample which had DNMT3A as its sole identifier mutation. These are most likely pre-leukaemic cells. In patient #8, two sub-clones read-out in mice from the diagnostic sample. The dominant or largest sub-clone in all three mice harboured not only a NPM1c but also a TET2 mutation; this clone was below the detection limit in the diagnostic sample itself (indicated by dotted circle in Fig. 1). The relapse sample from patient #8 contained only one NPM1c sub-clone corresponding to the major sub-clone seen at diagnosis. However, in the transplant of this sample, a NPM1c-negative sub-clone, ancestral to the relapse sub-clone, represented 100% of the regenerated leukaemia.
Finally, in patient #9, there was only one clone discernible both at diagnosis and relapse and this clone read-out consistently in transplants of diagnostic and relapse samples.
Discussion
These single cell data provide definitive identification of clonal architectures and preferential order of mutations, furthering endorse the concept of sub-clonal complexity in myeloid leukaemia [7, 14,15,16]. However, the current limits of single cell screening means that we will have under-estimated the extent of sub-clonal genetic diversity that can be revealed by ultra-deep sequencing [25] and by new technologies that allow interrogation of thousands of cells [26]. This has implications for clonal architecture and phylogeny. For example, in diagnostic samples from several patients (#2, #3, #4, #8, #9), the sub-clone with the most simple genetic composition at the base of the phylogenetic tree harboured more than one mutation. The phylogenetic structure is therefore likely to have missed earlier, sequential (pre-leukaemic) clones [8].
Different driver mutations have epistatic or synergistic functional impacts in AML [7, 22, 27] and the order of mutation accrual may impact on stem/progenitor cell function and clinical features [28]. Our data provides direct evidence that NPM1c mutation is a sub-clonal and therefore secondary mutation rather than a truncal or initiating lesion, as previously suggested [29]. This concurs with the observations of Shlush et al. [9] who found (in ten patients with AML) that DNMT3A mutations in AML were present in differentiation competent haemopoietic stem cells and putative pre-leukaemic clones. NPM1 mutations, in contrast, were absent from such cells but present in blasts cells with a myeloid progenitor cell phenotype presumed to be descended from the DNMT3A mutant clones. Similarly, Corces-Zimmerman et al. [30] found that NPM1c mutations were absent in purified haemopoietic stem cells, in contrast to putative founder mutations including DNMT3A, IDH1, IDH2 and ASXL1. In cases of AML analysed at the single cell level, Jan et al. [8] (one case) and Quek et al. [16] (three cases) documented that NPM1c was sub-clonal or secondary to TET2 mutations. However, Quek et al. [16] also identified, in two cases, very rare CD34+ cells that had NPM1 mutations but not other mutations found in the bulk leukaemic cells raising the possibility that NPM1 might occasionally be a founder mutation in pre-leukaemic cells. The preservation of diagnostic DNMT3A but not NPM1c mutations in remission [9, 31] and in a small minority of relapses is also commensurate with the predominantly secondary, sub-clonal nature of NPM1c [32, 33]. As is the presence of DNMT3A and TET2 but not NPM1c mutations in covert pre-malignant clones of normal, ageing adults [34].
A preferential order of mutation may reflect genetic network or cell context dependencies. NPM1c (and FLT3 mutations) might be potent drivers only when arising in myeloid progenitor cells with enhanced self-renewal provided by mutations in epigenetic mutations such as DNM3TA or TET2.
In the bulk blast cell population, DNMT3A and NPM1c mutations were present at similar high allele burden suggesting these were concurrent in the same cells [9]. In another study however, NPM1c allele burden was consistently less than that of other drivers including DNMT3A commensurate with a sub-clonal origin [19]. In our series, the allele burden for NPM1c was consistently less than that of other putative founder mutations including DNMT3A, TET2 and IDH2 (Fig. 1). The existence of clones ancestral to those with NPM1c mutations was clearly evident (in 8/10 cases) in the minor population sub-fractionated as CD34+/CD33-. This again accords with the data of Shlush et al. [9].
Mouse models with transgene or knock-in NPM1c have been developed to assess the role of NPM1 in leukaemogenesis [35]. By itself NPM1 expressed in haemopoietic stem cells produces a myeloproliferative disorder and a low penetrance of late occurring AML. A high frequency of AML does develop in NPM1c mice subjected to insertional mutagenesis [36] or in compound mutant mice with both NPM1c and FLT3-ITD [37, 38]. These modelling data testify to the functional impact of NPM1c on myeloid cells and leukaemogenesis but underscore that it is, at best, a weak initiating or founder lesion for AML.
The order of mutations and their position in the phylogenic tree is relevant to the selection of mutated gene for targeted therapy [4]. In the cases of NPM1c+ AML, the phylogenetic studies highlight DNMT3A and TET2 as truncal mutations as reported previously [8, 9]. Effective therapeutic targeting of either NPM1c or FLT3 mutations might be expected to debulk the leukaemia but with only transient benefit. However, persistence or increase of MRD in AML via detection of NPM1c transcripts is strongly predictive of relapse [39] and in the great majority (>95%) of cases of NPM1c+ AML that relapse, the relapsing clone is NPM1c+ [40]. In contrast, persistence of founder mutations (DNMT3A, TET2, ASXL1) or pre-leukaemic clones, is not predictive of relapse [41] This reflects the strong driver status of NPM1c mutations and the malignant potential of NPM1c sub-clones which is likely contingent upon the genetic background of founder (truncal) mutations (i.e., by epistasis) and additional co-existing sub-clonal mutations (e.g., in FLT3). Effective targeting of NPM1c could, therefore, be very beneficial in restraining progression of disease.
There was evidence for reiterated driver mutations in sub-clones of several cases in this study. This has been described before in ALL [3] and other cancers [42]. Mutations that are highly recurrent between patients with a sub-type of leukaemia (or any cancer) might be expected to occur more than once within a leukaemia from single patients. Functionally, this could reflect either the fitness advantage of bi-allelic mutations of the same gene in the same cells or convergent evolution of sub-clones contingent upon prevalent selective pressures or preferential, epistatic partnership with earlier, common mutations [43].
A comparison of clonal structures in three cases of matched diagnosis and relapse samples (#8, #9, #10) allowed us to infer the possible sub-clonal origins of the relapses. In one patient (#9), there was only one clone detectable at diagnosis and that same clone was the only clone observed at relapse. In case #8, the single relapse detected corresponded to one of two clones present at diagnosis. However, sequencing also revealed a TET2 mutation at low allele burden (1.03%) at relapse. The allele burden for this mutation at diagnosis was undetectable. However, when the diagnostic sample was transplanted into mice, a sub-clone with that ‘relapse’ TET2 mutation was the dominant clone (refer to Fig. 1 for case #8).
In patient #10, there were three sub-clones at diagnosis and all three were present in the relapse sample. These data raise the possibility that relapse in AML is not necessarily monoclonal and this should be further explored as it has important implications for the basis of drug resistance.
Xeno-transplant read-outs depend upon the genetic background of the immuno-deficient mice [15] and may not faithfully reflect the true diversity of propagating cells in AML. Furthermore, we made no attempt to titrate leukaemia propagating activity by varying the number of cells transplanted or by serial transplantation (except in patient #1). We note however that replicate mice provide very similar read-outs which suggest intrinsic, functional properties of AML sub-clones are being registered. The only conclusion we wish to draw from these limited transplant experiments is that multiple sub-clones from individual patients transplant indicating, as we showed previously for ALL [3] and glioblastoma [44], that individual leukaemia’s contain several, genetically distinct cells with self-renewing or leukaemia propagating activity. These cells will provide a diverse pool of cells distributed throughout the phylogenetic tree and from which relapse or drug resistance can emerge as recently demonstrated by Shlush et al. [24]. As such they function as cellular units of evolutionary selection [45, 46]. However, sub-clones have variable repopulating capacity [47] and as previously reported in AML [15], one NPM1c sub-clone dominated leukaemia regeneration in mice. This may reflect the increased malignant potential of this sub-clone and the contribution of NPM1c+ cells to relapse in >95% of cases [40]. In all our six cases where the diagnostic clone had both NPM1c and FLT3 ITD or FLT3 mutations, the dominant sub-clone in transplant readouts had both mutations. Competitiveness of sub-clones with this genotype in a xenotransplant context might be relevant to the very poor prognosis of AML cases that harbour a combination of mutants in DNMT3A, NPM1c and FLT3 [7].
References
Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481:306–13.
Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–28.
Anderson K, Lutz C, van Delft FW, Bateman CM, Guo Y, Colman SM, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature. 2011;469:356–61.
McGranahan N, Swanton C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell. 2015;27:15–26.
Yates LR, Campbell PJ. Evolution of the cancer genome. Nat Rev Genet. 2012;13:795–806.
Cancer Genome Atlas Research N, Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–74.
Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374:2209–21.
Jan M, Snyder TM, Corces-Zimmerman MR, Vyas P, Weissman IL, Quake SR, et al. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci Transl Med. 2012;4:149ra118.
Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 2014;506:328–33.
Miller CA, Wilson RK, Ley TJ. Genomic landscapes and clonality of de novo AML. N Engl J Med. 2013;369:1472–3.
de Bruin EC, McGranahan N, Mitter R, Salm M, Wedge DC, Yates L, et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science. 2014;346:251–6.
Suzuki H, Aoki K, Chiba K, Sato Y, Shiozawa Y, Shiraishi Y, et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nat Genet. 2015;47:458–68.
Wang Y, Waters J, Leung ML, Unruh A, Roh W, Shi X, et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature. 2014;512:155–60.
Paguirigan AL, Smith J, Meshinchi S, Carroll M, Maley C, Radich JP. Single-cell genotyping demonstrates complex clonal diversity in acute myeloid leukemia. Sci Transl Med. 2015;7:281re282.
Klco JM, Spencer DH, Miller CA, Griffith M, Lamprecht TL, O’Laughlin M, et al. Functional heterogeneity of genetically defined subclones in acute myeloid leukemia. Cancer Cell. 2014;25:379–92.
Quek L, Otto GW, Garnett C, Lhermitte L, Karamitros D, Stoilova B, et al. Genetically distinct leukemic stem cells in human CD34- acute myeloid leukemia are arrested at a hemopoietic precursor-like stage. J Exp Med. 2016;213:1513–35.
Potter NE, Ermini L, Papaemmanuil E, Cazzaniga G, Vijayaraghavan G, Titley I, et al. Single-cell mutational profiling and clonal phylogeny in cancer. Genome Res. 2013;23:2115–25.
Taussig DC, Vargaftig J, Miraki-Moud F, Griessinger E, Sharrock K, Luke T, et al. Leukemia-initiating cells from some acute myeloid leukemia patients with mutated nucleophosmin reside in the CD34(-) fraction. Blood. 2010;115:1976–84.
Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366:1079–89.
Peterlin P, Renneville A, Ben Abdelali R, Nibourel O, Thomas X, Pautas C, et al. Impact of additional genetic alterations on the outcome of patients with NPM1-mutated cytogenetically normal acute myeloid leukemia. Haematologica. 2015;100:e196–199.
Rose D, Haferlach T, Schnittger S, Perglerova K, Kern W, Haferlach C. Subtype-specific patterns of molecular mutations in acute myeloid leukemia. Leukemia. 2017;31:11–17.
Zhang X, Su J, Jeong M, Ko M, Huang Y, Park HJ, et al. DNMT3A and TET2 compete and cooperate to repress lineage-specific transcription factors in hematopoietic stem cells. Nat Genet. 2016;48:1014–23.
Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481:506–10.
Shlush LI, Mitchell A, Heisler L, Abelson S, Ng SWK, Trotman-Grant A, et al. Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature. 2017;547:104–8.
Griffith M, Miller CA, Griffith OL, Krysiak K, Skidmore ZL, Ramu A, et al. Optimizing cancer genome sequencing and analysis. Cell Syst. 2015;1:210–23.
Pellegrino M, Sciambi A, Treusch S, Durruthy-Durruthy R, Gokhale K, Jacob J, et al. High-throughput single-cell DNA sequencing of acute myeloid leukemia tumors with droplet microfluidics. Genome Res. 2018;28:1345–52.
Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150:264–78.
Ortmann CA, Kent DG, Nangalia J, Silber Y, Wedge DC, Grinfeld J, et al. Effect of mutation order on myeloproliferative neoplasms. N Engl J Med. 2015;372:601–12.
Martelli MP, Pettirossi V, Thiede C, Bonifacio E, Mezzasoma F, Cecchini D, et al. CD34 + cells from AML with mutated NPM1 harbor cytoplasmic mutated nucleophosmin and generate leukemia in immunocompromised mice. Blood. 2010;116:3907–22.
Corces-Zimmerman MR, Hong WJ, Weissman IL, Medeiros BC, Majeti R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc Natl Acad Sci, USA. 2014;111:2548–53.
Parkin B, Londono-Joshi A, Kang Q, Tewari M, Rhim AD, Malek SN. Ultrasensitive mutation detection identifies rare residual cells causing acute myelogenous leukemia relapse. J Clin Invest. 2017;127:3484–95.
Kim T, Yoshida K, Kim YK, Tyndel MS, Park HJ, Choi SH, et al. Clonal dynamics in a single AML case tracked for 9 years reveals the complexity of leukemia progression. Leukemia. 2016;30:295–302.
Kronke J, Bullinger L, Teleanu V, Tschurtz F, Gaidzik VI, Kuhn MW, et al. Clonal evolution in relapsed NPM1-mutated acute myeloid leukemia. Blood. 2013;122:100–8.
Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488–98.
Sportoletti P, Varasano E, Rossi R, Mupo A, Tiacci E, Vassiliou G, et al. Mouse models of NPM1-mutated acute myeloid leukemia: biological and clinical implications. Leukemia. 2015;29:269–78.
Vassiliou GS, Cooper JL, Rad R, Li J, Rice S, Uren A, et al. Mutant nucleophosmin and cooperating pathways drive leukemia initiation and progression in mice. Nat Genet. 2011;43:470–5.
Mallardo M, Caronno A, Pruneri G, Raviele PR, Viale A, Pelicci PG, et al. NPMc + and FLT3_ITD mutations cooperate in inducing acute leukaemia in a novel mouse model. Leukemia. 2013;27:2248–51.
Mupo A, Celani L, Dovey O, Cooper JL, Grove C, Rad R, et al. A powerful molecular synergy between mutant Nucleophosmin and Flt3-ITD drives acute myeloid leukemia in mice. Leukemia. 2013;27:1917–20.
Shayegi N, Kramer M, Bornhauser M, Schaich M, Schetelig J, Platzbecker U, et al. The level of residual disease based on mutant NPM1 is an independent prognostic factor for relapse and survival in AML. Blood. 2013;122:83–92.
Ivey A, Hills RK, Simpson MA, Jovanovic JV, Gilkes A, Grech A, et al. Assessment of minimal residual disease in standard-risk AML. N Engl J Med. 2016;374:422–33.
Jongen-Lavrencic M, Grob T, Hanekamp D, Kavelaars FG, Al Hinai A, Zeilemaker A, et al. Molecular minimal residual disease in acute myeloid leukemia. N Engl J Med. 2018;378:1189–99.
Gerlinger M, McGranahan N, Dewhurst SM, Burrell RA, Tomlinson I, Swanton C. Cancer: evolution within a lifetime. Annu Rev Genet. 2014;48:215–36.
Greaves M. Evolutionary determinants of cancer. Cancer Discov. 2015;5:806–20.
Piccirillo SGM, Colman S, Potter NE, van Delft FW, Lillis S, Carnicer M-J, et al. Genetic and functional diversity of propagating cells in glioblastoma. Stem Cell Rep. 2015;4:7–15.
Greaves M. Cancer stem cells as ‘units of selection’. Evol Appl. 2013;6:102–8.
Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14:275–91.
Wang K, Sanchez-Martin M, Wang X, Knapp KM, Koche R, Vu L, et al. Patient-derived xenotransplants can recapitulate the genetic driver landscape of acute leukemias. Leukemia. 2017;31:151–8.
Acknowledgements
This work was supported by Bloodwise, the Kay Kendall Leukaemia Fund and Gabrielle’s Angel Foundation UK.
Author contributions
NP completed all laboratory work and analysis except initial patient sample sequencing and animal experiments/care/transplants, assisted with manuscript writing and editing. FM-M completed all animal experiments/care/transplants. IT assisted with FACS experiments. LE completed all phylogenetic analysis. GV assisted with FACS experiments. EP completed the targeted sequencing and analysis of each patient sample with support of PC. JG provided access to patient samples. DT co-designed experiments, managed animal experiments and contributed to writing of the manuscript. MG co-designed experiments, supervised primary work and co-wrote the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Potter, N., Miraki-Moud, F., Ermini, L. et al. Single cell analysis of clonal architecture in acute myeloid leukaemia. Leukemia 33, 1113–1123 (2019). https://doi.org/10.1038/s41375-018-0319-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-018-0319-2
This article is cited by
-
The new diagnostic criteria for myelodysplasia-related acute myeloid leukemia is useful for predicting clinical outcome: comparison of the 4th and 5th World Health Organization classifications
Annals of Hematology (2022)
-
The contribution of single-cell analysis of acute leukemia in the therapeutic strategy
Biomarker Research (2021)
-
Clinical relevance of extracellular vesicles in hematological neoplasms: from liquid biopsy to cell biopsy
Leukemia (2021)
-
Clonal dominance is an adverse prognostic factor in acute myeloid leukemia treated with intensive chemotherapy
Leukemia (2021)
-
Single-cell analysis based dissection of clonality in myelofibrosis
Nature Communications (2020)
