
Abstract
Janus kinase 2/signal transducer and activators of transcription 1 (JAK2/STAT1) signaling is a common pathway that contributes to numerous inflammatory disorders, including different forms of acute lung injury (ALI). However, the role of JAK2/STAT1 in ventilator-induced lung injury (VILI) and its underlying mechanism remain unclear. In this study, using lipopolysaccharide (LPS) inhalation plus mechanical ventilation as VILI mouse model, we found that the administration of JAK2 inhibitor AZD1480 markedly attenuated lung destruction, diminished protein leakage, and inhibited cytokine release. In addition, when mouse macrophage-like RAW 264.7 cells were exposed to LPS and cyclic stretch (CS), AZD1480 prevented cell autophagy, reduced apoptosis, and suppressed lactate dehydrogenase release by downregulating JAK2/STAT1 phosphorylation levels and inducing HMGB1 translocation from the nucleus to the cytoplasm. Furthermore, HMGB1 and STAT1 knockdown attenuated LPS+CS-induced autophagy and apoptosis in RAW 264.7 cells. In conclusion, these findings reveal the connection between the JAK2/STAT1 pathway and HMGB1 translocation in mediating lung inflammation and cell death in VILI, suggesting that these molecules may serve as novel therapeutic targets for VILI.
Similar content being viewed by others

Introduction
Acute respiratory distress syndrome (ARDS) has an overall hospital mortality of more than 40% worldwide [1, 2] and there is no clinically proven targeted pharmacological therapy for this disease [3]. Mechanical ventilation (MV), a life-saving support for patients suffering from lung injury, may aggravate injured lungs [4, 5]. Extensive clinical and experimental research has focused on ventilator-associated lung injury and ventilator-induced lung injury (VILI) in patients [6,7,8] and animals [9]. Early on, researchers advocated a protective MV strategy to prevent barotraumas, volutrauma, or atelectrauma; however, the mortality associated with ARDS remains unacceptably high. Thereafter, a number of studies have provided evidence that the inflammatory cytokines secreted by lung cells, such as lung resident cells and recruited cells, may exacerbate lung injury, including biotrauma [10,11,12]. Numerous inflammatory mediators such as tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6, IL-10, interferon-γ, and IL-8 are released by ventilation, which have a wide spectrum of biological actions and may cause cell death [13, 14].
High-mobility group box 1 (HMGB1) has been defined primarily as non-histone nuclear protein and damage-associated molecular pattern molecule [15]. It is believed that HMGB1 can trigger an overwhelming inflammatory response that promotes the progression of lipopolysaccharide (LPS)-induced sepsis [16] and other inflammatory processes [17, 18]. Recent in vivo studies have shown that the concentration of HMGB1 is elevated in pulmonary compartment during lung inflammation [19, 20]. An important role of HMGB1 in VILI is supported by the findings that HMGB1 accumulates in the broncho-alveolar lavage fluid (BALF) of ventilated rabbits and the beneficial effects of anti-HMGB1 antibody [21]. HMGB1 also plays a central role in maintaining the cascade amplification of inflammation and participates in the progression of VILI in a two-hit model with a low dose of LPS 2 h before MV exposure [22]. The biological function of HMGB1 is dependent on its cellular location. For example, nuclear HMGB1 directly binds to DNA and participates in DNA repair, recombination, and replication, whereas extracellular HMGB1 has cytokine-inducing activity that can trigger inflammation and lead to cell death [23].
The Janus kinase (JAK)/signal transducers and activators of transcription (STAT) pathway plays a crucial role in cytokine-mediated biological responses [24, 25]. Moreover, STAT may contribute to the development of acute lung injury (ALI) [26]. A previous study has reported that mechanical stretch activates the JAK/STAT pathway in rat cardiomyocytes [27]. However, whether MV can activate the STAT signaling pathway in the lung remains largely unclear. Given the important role of HMGB1 in ALI-, VILI-, and JAK/STAT-mediated inflammation, this study aimed to investigate the mechanism by which the JAK2/STAT1 pathway is activated and HMGB1 translocation in exaggerating lung inflammation and cell death by using a two-hit VILI model both in vivo and in vitro.
Materials and methods
Reagents
LPS 055: B5 was obtained from Sigma-Aldrich (St. Louis, MO, USA). AZD180 was purchased from SelleckChem (Houston, TX, USA). Mouse HMGB1 antibody was purchased from Abcam (Cambridge, MA, USA). β-Actin and Lamin B1 antibodies were obtained from Proteintech (Wuhan, Hubei, China). Antibodies against phosphorylated JAK2 (p-JAK2; Tyr1007/1008), JAK2, phosphorylated STAT1 (p-STAT1; Tyr701), cleaved caspase 3 (Asp175; 5A1E), caspase 3, and LC3B were obtained from Cell Signaling Technology (Boston, MA, USA). Horseradish peroxidase-conjugated goat anti-rabbit antibody and enhancer chemiluminescent reagent were purchased from Piece Biotechnology (Rockford, IL, USA). Polyvinylidene difluoride membranes were obtained from Millipore (Bedford, MA, USA).
Animal experiment protocols
The study protocol for animal experiments was approved by the Animal Use and Care Committee of Tongji Medical College (Wuhan, China). Male C57BL/6 mice (8- to 10-week-old) were obtained from Hunan SJA Laboratory Animal Co., Ltd (Permit Number: SCKX2011-0003) and housed in a modified specific pathogen-free facility prior to all experiments.
The mice were intraperitoneally anesthetized with 2% pentobarbital sodium (90 mg/kg body weight) and suspended by its front teeth at a 60° angle. After extending the jaw and tongue, 0.3 mg/kg body weight of LPS (1.5 μl/gm body weight of diluted LPS) or an equal volume of pyrogen-free phosphate-buffered saline (PBS) was deposited into the oropharynx through a 24-gauge angio catheter (BD Biosciences). Aspiration of the liquid was visually confirmed. After 2 h, the mice were anesthetized with an intraperitoneal injection of 2% pentobarbital sodium (90 mg/kg body weight). Mice assigned to the control and LPS groups were returned to their cages. Mice assigned to the MV group were placed in supine position on a heating blanket and then received a high tidal volume of MV (Mini Vent, Harvard Biosciences, USA), while breathing room air with zero end-expiratory pressure. Approximately 20 ml/kg of tidal volume was applied to mimic the heterogeneous over-distension of lung injury, resulting from the partially collapsed or fluid-filled lungs. The mice were sustained with 2% pentobarbital sodium (40 mg/kg/h) during MV exposure.
Prior to the experiment, AZD1480 was suspended in saline supplemented with 5% dimethyl sulfoxide and 1% Tween 80. AZD1480 (40 mg/kg body weight) was intraperitoneally injected 30 min before aspiration and only applied to LPS+MV (6 h) group and AZD1480-alone group.
Mice were killed by an overdose of anesthetic agent at 2, 4, and 6 h of ventilation or spontaneous respiration. The lung was exposed and BALF of the left lung was pooled and washed three times with 0.5 ml aliquots of PBS through a tracheal cannula. BALF was spun at 1500 r.p.m./min for 15 min and the resultant supernatant was stored in small aliquots at −80 °C until further analysis. The right lung was removed and one lobe of the right lung was fixed in 4% paraformaldehyde, whereas other lobes of the right lung were immediately stored at −80 °C until further analysis.
Histological analysis
The lung samples were fixed in 4% paraformaldehyde at 4 °C for 48 h, embedded with paraffin wax, and cut into 5 μm-thick sections. The sections were stained with hematoxylin and eosin (Vector Labs, Burlingame, CA, USA) by following a standard protocol. Histopathologic analysis of lung sections was performed as described previously [28]. All the representative images were obtained under ×200 magnification.
ELISA detection of total protein, cytokines, MPO, and HMGB1 levels in BALF
Total protein level in BALF was measured using a bicinchoninic acid (BCA) protein assay kit (Piece Biotechnology, Rockford, IL, USA) according to the manufacturer’s instruction. Enzyme-linked immunosorbent assay (ELISA) was adopted to measure the levels of TNF-α, IL-1β, and IL-6 according to the manufacturer’s protocols (Nanjing Jiancheng Bioengineering Institute, Nanjing, China). The levels of MPO and HMGB1 were determined by the ELISA kits obtained from Bioswamp (Shanghai Jianglin Biological Technology Co., Ltd, China) and Elabscience (Elabscience Biotechnology Co., Ltd, China), respectively.
Cell culture and treatment
Mouse macrophage cells (Raw 264.7 cells) were obtained from American Type Culture Collection (Manassas, VA, USA). The cells were cultured in Dulbecco’s modified Eagle’s medium (GIBCO, Life Technologies, CA, USA) supplemented with 10% fetal bovine serum (GIBCO, Life Technologies, CA, USA) and maintained at 37 °C in a humidified incubator containing 5% CO2.
Prior to treatments, the cells were seeded at 5.0 × 105 cells/well on type I collagen-coated six-well plates and grown to 50% confluence. Then, the cells were divided into six groups as follows: (1) control group without treatment; (2) LPS group, cells were exposed to 1 μg/ml LPS for 24 h; (3) CS (cyclic stretch) group, cells were stretched with the Flexercell Tension Plus TM FX-5000T system (Flexcell International, USA) set at 24% elongation for 12 h at a frequency of 30 cycles/min; (4) LPS+CS group, cells were treated with LPS for 12 h and then subjected to CS exposure; (5) AZD1480+LPS+CS group, cells were pretreated with 5 μM AZD1480 for 30 min before LPS stimulation and CS exposure; and (6) AZD1480 group, cells were only treated with 5 μM AZD1480.
LDH assay
The cells were lysed and centrifuged at 1500 r.p.m./min for 15 min at 4 °C. After centrifugation, the supernatant was collected and stored in small aliquots at −80 °C. The concentrations of lactate dehydrogenase (LDH) in the supernatant were determined using LDH assay kit (KeyGEN Biotech Co., Ltd, China) according to the manufacturer’s protocol.
RNA interference
RAW 264.7 cells were seeded on type I collagen-coated six-well plates at a density of 2.0 × 105 cells/well. Upon reaching confluence, the cells were transfected with scramble small interfering RNA (siRNA), HMGB1-specific, or STAT1-specific siRNA (100 nM, Guangzhou RiboBio Co., Ltd) using HiPerFect transfection reagent (Qiagen, Hilden, German). After 48 h of transfection, the cells were exposed to LPS or/and CS as described above.
Immunofluorescence analysis
The cells were washed twice with PBS and fixed in 4% paraformaldehyde for 10 min at room temperature. Subsequently, the fixed cells were permeabilized with 0.1% Triton X-100 in PBS for 5 min. The cells were then mounted onto coverslips and incubated with primary rabbit polyclonal anti-mouse HMGB1 (Abcam ab18256), followed by incubation with secondary antibodies for 1 h and counterstaining with 4′,6-diamidino-2-phenylindole. The stained slides were observed and photographed using a fluorescence microscope (Olympus, Tokyo, Japan). All the representative images were obtained under ×400 magnification.
Western blot analysis
Lung tissue samples were homogenized and the collected cells were lysed by using the total protein extraction kit (Beyotime Institute of Biotechnology, Shanghai, China) as well as cytoplasm and nuclear extraction kit (Thermo Fisher Scientific, Waltham, MA, USA). Total protein concentration was determined using the BCA protein assay kit (Piece Biotechnology, Rockford, IL, USA) according to the manufacturer’s protocol. Equal amounts (30 μg) of protein were subjected to 10% or 12% SDS-polyacrylamide gel electrophoresis and immunoblotted with the indicated antibodies. The density of protein bands was analyzed using ImageJ software (version 2.1.4.7; National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
Statistical analyses were performed using Graph-Pad Prism V7.0 software (La Jolla, CA, USA). One-way analysis of variance followed by post hoc Tukey’s test was used for multiple group comparisons. All data were presented as means ± SE. P-value < 0.05 was considered statistically significant.
Results
MV enhances LPS-induced lung inflammation and injury
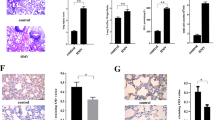
To establish a successful two-hit model of VILI, we selected three different endpoints at 2, 4, and 6 h of ventilation (i.e., 4, 6, and 8 h after vehicle or LPS instillation) for the assessment of lung inflammation and injury. Compared with control group, there were increased alveolar septal thickening and elevated leukocyte infiltration in the alveolar and interstitial spaces of LPS, MV, and LPS+MV groups (Fig. 1a). The lung injury scores of LPS+MV group after 2 and 4 h of ventilation were relatively similar compared with those of the LPS group and MV group (Fig. 1b). For LPS+MV group at 6 h ventilation, there was increased cellularity, septal thickening, as well as perivascular and peribronchial edema. Notably, lung injury was the most severe in this group, along with the highest injury score (Fig. 1a, b).

MV enhances LPS-induced lung inflammation and injury in mice. Mice were subjected to non-ventilated control group and MV with a tidal volume of 20 ml/kg for 2, 4, and 6 h (n = 6 per group). Equal volume of PBS or 0.3 mg/kg LPS was intracheally instilled 2 h before ventilation or spontaneous respiration. a Representative histological images of the lungs in different groups. Scale bar = 100 μm. b Lung injury score in different groups. c Lung permeability was assessed by measuring total BALF protein level. d ELISA assay of TNF-α level in BALF. e ELISA assay of IL-1β level in BALF. f ELISA assay of IL-6 level in BALF. g ELISA assay of MPO level in BALF. h ELISA assay of HMGB1 level in BALF. Values represent the mean ± SEM. *p < 0.05 and **p < 0.01, compared with the control at that time point. ##p < 0.01, compared with the LPS at that time point
Total protein concentrations in BALF were measured as an indicator of lung permeability. In one-hit models, LPS or ventilation alone could increase the total protein level in BALF. Interestingly, LPS plus 6 h of MV elevated the total protein level in BALF and such level was significantly higher than that in the BALF of one-hit group or LPS+MV group at 2 and 4 h (Fig. 1c). However, there was no significant increase in total protein level in the BALF of LPS+MV group at 2 and 4 h ventilation compared with LPS group.
Next, ELISA was used to detect the levels of proinflammatory cytokines and inflammatory enzyme MPO in BALF. LPS or MV increased the levels of TNF-α, IL-1β, IL-6, and MPO in BALF at 2, 4, and 6 h of ventilation (Fig. 1d–g). However, LPS+MV did not synergistically increase the levels of TNF-α, IL-1β, IL-6, and MPO in BALF after 2 and 4 h ventilation. Instead, the synergistic effects of LPS and MV on the increased levels of TNF-α, IL-1β, IL-6, and MPO in BALF were observed after 6 h of ventilation (Fig. 1d–g). Besides, LPS and MV exerted a synergistic effect in increasing the levels of HMGB1 in BALF at all the indicated time points (Fig. 1h).
JAK2 inhibitor prevents LPS+MV-induced lung injury
To assess the role of JAK2/STAT1 in LPS-induced lung inflammation and injury, the mice were treated with JAK inhibitor AZD1480 before the exposure to LPS or/and MV. Histological examination revealed marked increase in neutrophil infiltration and interstitial thickening in the lungs of the mice exposed to LPS+MV, but pretreatment with AZD1480 substantially reduced the severity of lung tissues and lung injury score (Fig. 2a, b). In addition, LPS+MV remarkably increased the levels of total protein, proinflammatory mediators (e.g., TNF-α, IL-1β, IL-6, and HMGB1) and MPO in BALF, but such changes could be attenuated by AZD1480 pretreatment (Fig. 2c–h).

JAK2 inhibitor prevents LPS+MV-induced lung injury in mice. Mice were selected from those in non-ventilated control group or those subjected to 0.3 mg/kg LPS and MV with a tidal volume of 20 ml/kg for 6 h (n = 6 per group). AZD1480 40 mg/kg was administered intraperitoneally 30 min before PBS or LPS aspiration. a Representative histological images of the lungs from mice treated as indicated. Scale bar = 100 μm. b Lung injury score in different groups. c Lung permeability was assessed by measuring total BALF protein level. d ELISA assay of TNF-α level in BALF. e ELISA assay of IL-1β level in BALF. f ELISA assay of IL-6 level in BALF. g ELISA assay of MPO level in BALF. h ELISA assay of HMGB1 level in BALF. i Representative blots showing the levels of phosphorylation of JAK2 and STAT1 in lung lysate from mice treated as indicated. j Desitometry analysis of the phosphorylation of JAK2 level in lung lysate from mice treated as indicated. k Densitometry analysis of phosphorylation of STAT1 level in different groups. l Representative blots showing the levels of LC3- I, LC3-II, cleaved caspase 3, and total caspase 3 in lung lysate from mice treated as indicated. m Densitometry analysis of LC3-II/LC3-I level in different groups. n Densitometry analysis of cleaved caspase 3/caspase 3 in different grops. Data represented the mean ± SEM. *p < 0.05, **p < 0.001, and ***p < 0.001, compared with the control. ##p < 0.01 and ###p < 0.001, compared with the LPS. &p < 0.01, &&p < 0.01, and &&&p < 0.001, compared with the LPS+MV
Furthermore, western blot analysis showed that LPS and MV synergistically increased the phosphorylation levels of JAK2 and STAT1 in lung tissues, but did not alter the total protein levels of JAK2 and STAT1. However, AZD1480 inhibited the phosphorylation levels of JAK2 and STAT1 in lung tissues exposed to LPS and MV (Fig. 2i–k).
Next, we determined whether autophagy and apoptosis are involved in the lung injury induced by LPS and MV. The lungs of mice exposed to LPS+MV demonstrated significant increase in LC3-II and cleaved caspase 3 levels compared with those exposed to LPS or MV alone. However, AZD1480 pretreatment significantly attenuated the increase in LC3-II and cleaved caspase 3 levels in lung tissues exposed to LPS and MV (Fig. 2l–n). Taken together, these data indicate that LPS and MV induce autophagy, increase apoptosis, and cause lung damage via activation of the JAK2/STAT1 signaling pathway.
JAK2 inhibitor suppresses LPS+CS-induced cell death in RAW 264.7 cells
Macrophages are the primary inflammatory cells that exhibit phagocytic function. To further investigate the mechanism by which the JAK2/STAT1 pathway mediates LPS+MV-induced lung injury, we established an experimental model by exposing mouse macrophage-like RAW 264.7 cells to LPS and CS. Not only LPS or CS alone activated the phosphorylation levels of JAK2 and STAT1, LPS+CS also significantly activated the phosphorylation of JAK2 and STAT1. As per our expectation, AZD1480 inhibited the phosphorylation levels of JAK2 and STAT1 in RAW 264.7 cells exposed to LPS and CS (Fig. 3a–c).

JAK2 inhibitor suppresses LPS+CS-induced RAW 264.7 cell death. RAW 264.7 cells were subjected to 1 μg/ml LPS for 24 h and/or 24% elongation stretch for 12 h at a frequency of 30 cycles/min. The cells were pretreated with and without 5 μM AZD1480 30 min before LPS stimulation and CS exposure. a Representative blots showing the phosphorylation of JAK2 and STAT1 in RAW 264.7 cells treated as indicated. b Densitometry analysis of the phosphorylation of JAK2 level in different groups. c Densitometry analysis of phosphorylation of STAT1 level in different groups. d Representative blot showing the levels of LC3-I, LC3-II, cleaved caspase 3, and total caspase 3 in RAW 264.7 cells treated as indicated. e Densitometry analysis of LC3-II/LC3-I level in different groups. f Densitometry analysis of cleaved caspase 3/caspase 3 in different groups. g Measurement of the released LDH in RAW 264.7 cells treated as indicated. Data represented the mean ± SEM from at least two independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 compared with the control. #p < 0.05, ##p < 0.05 compared with the LPS. &p < 0.05 and &&p < 0.01, compared with the LPS+CS
RAW 264.7 cells exposed to LPS and CS showed significant increase in LC3-II and cleaved caspase 3 levels compared with those exposed to LPS or CS alone, but AZD1480 attenuated such changes in the cells exposed to LPS and CS (Fig. 3d–f). LDH is a stable enzyme that can be released upon cell membrane disruption. The level of LDH was measured as another indicator for late apoptosis. LPS or CS could induce the release of LDH, whereas LPS plus CS significantly increased the release of LDH compared with the cells exposed to LPS or CS alone. However, AZD1480 significantly decreased the release of LDH induced by LPS+CS (Fig. 3g). Collectively, these data suggest that LPS and CS induce autophagy, increase apoptosis, and cause cell death in macrophages via activation of the JAK2/STAT1 signaling pathway.
JAK2 inhibitor suppresses LPS+CS-induced HMGB1 translocation to the cytoplasm of RAW 264.7 cells
To investigate the role of HMGB1 in LPS+CS-induced macrophage cell death, HMGB1 translocation was determined by fluorescent immunostaining. In control RAW 264.7 cells, HMGB1 was predominantly localized in the nucleus. LPS and CS induced the cytoplasmic accumulation of HMGB1, but AZD1480 inhibited LPS+CS-induced HMGB1 cytoplasmic accumulation in RAW 264.7 cells (Fig. 4a). Western blot analysis confirmed that LPS+CS increased robust HMGB1 cytoplasmic accumulation and decreased nuclear HMGB1 level, whereas AZD1480 significantly reduced cytoplasmic HMGB1 level and increased nuclear HMGB1 level in RAW 264.7 cells (Fig. 4b–e).

JAK2 inhibitor suppresses LPS+CS-induced HMGB1 translocation to the cytoplasm of RAW 264.7 cells. a Immunofluorescence staining for HMGB1 in RAW 264.7 cells treated as indicated. Scale bar = 100 μm. b Representative blots showing the levels of total HMGB1, cytoplasmic HMGB1, and nuclear HMGB1 in RAW 264.7 cells treated as indicated. β-Actin was loading control for cytoplasmic proteins and Lamin B1 was loading control for nuclear proteins. c Densitometry analysis of total HMGB1 level in different groups. d Densitometry analysis of cytoplasmic HMGB1 level in different groups. e Densitometry analysis of nuclear HMGB1 level in different groups. Data represented the mean ± SEM from at least three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 compared with the control. #p < 0.05 compared with the LPS. &p < 0.05 and &&p < 0.01, compared with the LPS+CS
Next, we transfected RAW 264.7 cells with HMGB1 siRNA, STAT1 siRNA, or scramble control siRNA, and then assessed the knockdown of HMGB1 and STAT1 by western blot analysis (Fig. 5a and 6a). HMGB1 siRNA significantly decreased LC3-II level, but had no significant effect on cleaved caspase 3 level in RAW 264.7 cells (Fig. 5b, c). On the contrary, STAT1 siRNA significantly decreased the levels of LC3-II and cleaved caspase 3 in RAW 264.7 cells exposed to LPS+CS (Fig. 6b, c). Furthermore, siRNA-mediated knockdown of HMGB1 and STAT1 significantly attenuated LPS+CS-induced LDH release in RAW 264.7 cells (Figs. 5d and 6d). These results suggest that HMGB1 and STAT1 mediate LPS+CS-induced cell death in macrophages. In addition, STAT1 siRNA could inhibit the translocation of HMGB1 (Fig. 6e, f). Nevertheless, AZD1480 exerted the same inhibitory effects on HMGB1 translocation with or without STAT1 knockdown (Fig. 6g, h).

HMGB1 mediates LPS+CS-induced RAW 264.7 cell death. RAW 264.7 cells were transfected with 100 nM of control siRNA or HMGB1-specific siRNA for 48 h and then subjected to 1 μg/ml LPS for 24 h plus 24% elongation stretch for 12 h at a frequency of 30 cycles/min. a Western blot analysis of HMGB1 knockdown in RAW 264.7 cells. b Western blot analysis of the levels of LC3-I, LC3-II in RAW 264.7 cells treated with LPS+CS, or/and HMGB1 siRNA or scramble (Ctr) siRNA. c Western blot analysis of the levels of cleaved caspase 3 and total caspase 3 in RAW 264.7 cells treated with LPS+CS or/and HMGB1 siRNA or scramble (Ctr) siRNA. d Measurement of the released LDH in RAW 264.7 cells treated with LPS+CS or/and HMGB1 siRNA or scramble (Ctr) siRNA. Data represented the mean ± SEM from at least three independent experiments. *p < 0.05 and **p < 0.01, compared with control or Ctr siRNA+LPS+CS

STAT1 mediates LPS+CS-induced RAW 264.7 cell death by inhibiting the translocation of HMGB1. RAW 264.7 cells were transfected with 100 nM of control siRNA or STAT1-specific siRNA for 48 h and then subjected to 1 μg/ml LPS for 24 h plus 24% elongation stretch for 12 h at a frequency of 30 cycles/min. AZD1480 (5 μM) was given 30 min before LPS stimulation when necessary. a Western blot analysis of STAT1 knockdown in RAW 264.7 cells. b Western blot analysis of the levels of LC3-I, LC3-II in RAW 264.7 cells treated with LPS+CS or/and HMGB1 siRNA or scramble (Ctr) siRNA. c Western blot analysis of the levels of cleaved caspase 3 and total caspase 3 in RAW 264.7 cells treated with LPS+CS or/and STAT1 siRNA or scramble (Ctr) siRNA. d Measurement of the released LDH in RAW 264.7 cells treated with LPS+CS or/and STAT1 siRNA or scramble (Ctr) siRNA. e Western blot analysis of the levels of cytoplasmic HMGB1 in RAW 264.7 cells treated as indicated. β-Actin was loading control for cytoplasmic proteins. f Western blot analysis of total nuclear HMGB1 in RAW 264.7 cells treated as indicated. Lamin B1 was loading control for nuclear proteins. g Western blot analysis of cytoplasmic HMGB1 in RAW 264.7 cells treated as indicated. β-Actin was loading control for cytoplasmic proteins. h Western blot analysis of nuclear HMGB1 in RAW 264.7 cells treated as indicated. Lamin B1 was loading control for nuclear proteins. Data represented the mean ± SEM from at least two independent experiments. *p < 0.05 and **p < 0.01, ***p < 0.001 compared with control or Ctr siRNA + LPS+CS. &p < 0.05 and &&p < 0.01, compared with the LPS+CS
Discussion
ARDS is a multifactorial disease that is associated with infection, trauma, burns, or aspiration. Patients suffering from severe ALI usually need to receive MV therapy. However, the use of lung-protective ventilation sometimes initiates and accelerates lung injury rather than exerting its curative effect on ARDS. Lung stretching may release proinflammatory cytokines, induce systemic circulation, and trigger and/or exacerbate VILI. Macrophages are the main source of these inflammatory mediators [29]. Moreover, JAK/STAT pathway plays a major role in inflammatory signaling cascades, which has been identified as one of the promising targets [30]. Combining LPS instillation with MV mouse model and CS cell model, we found that JAK2 inhibitor AZD1480 exhibited beneficial effects against LPS and ventilation-induced lung injury and cell death. The activation of the JAK2/STAT1 pathway was verified in the lungs of mice exposed to LPS+MV. The protective effects of AZD1480 on lung inflammatory injury may be associated with its ability to inhibit JAK2/STAT1-induced release of inflammatory mediators.
In most previous animal studies, the degree of MV-induced ALI is largely dependent on VT [31]. Some studies have suggested a “two-hit model” in which the lungs are more susceptible to MV injury after LPS exposure [32, 33]. In order to verify this speculation, we administrated a low amount of LPS (0.3 mg/kg or same volume with control) to induce lung injury. Two hours later, spontaneously breathing or ventilation for 2, 4, and 6 h was performed on the mice. Intriguingly, we observed that a high tidal of volume MV within shorter periods of time did not cause more severe lung injury, such as pulmonary permeability or cytokine release into BALF, compared with LPS up until 6 h ventilation. Hence, the prevention of ALI caused by VILI should pay great attention to the duration of MV and an effective pharmacologic intervention is needed to treat progressive inflammation and injury. In our two-hit mouse model of ALI, JAK2 and STAT1 were found to be tyrosine-phosphorylated and activated in the lungs after intratracheal LPS and MV exposure. In addition, tetrahydroisoquinoline alkaloids reduce inducible nitric oxide synthase (iNOS) via p-STAT-1 inhibition and prevent HMGB1 secretion in LPS-treated lung tissue [34]. These findings reveal the important role of STAT in lung injury and its potential interaction with HMGB1.
At the same time, we noted that the concentration of HMGB1 in BALF was higher in LPS+MV group than in LPS group. These results are consistent with previous findings that HMGB1 in the nucleus can translocate to the cytoplasm and actively promote its own secretion after MV stimulation [20, 21, 35]. Researchers have shown that HMGB1 can regulate autophagy under different lung-related diseases, including asthma, lung fibrosis, lung infections, chronic obstructive pulmonary disease, lung cancer, and ALI [36,37,38]. Tang et al. [39] reported that pharmacological inhibition of HMGB1 translocation could limit starvation-induced autophagy. Moreover, the increased HMGB1 level triggers the stress response of the endoplasmic reticulum and thus promoting the occurrence of autophagy and apoptosis-resistant phenotype in a cigarette smoke model [40]. Autophagy is a process that degrades macromolecules and lysosome-related organelles to balance cellular energy supply, which can be linked to cell survival. However, the role of autophagy in lung inflammation remains controversial, which is largely dependent on the cell type and context. Nakahira et al. [41] showed that autophagy-deficient mouse macrophages increased mitochondrial ROS-induced inflammation and mortality in LPS-treated mice. Autophagy induction also decreased the release of LDH, thus enhancing cell viability and sustaining the integrity of endothelial barrier [42]. Silencing of autophagy-related protein 4 in lung macrophages or pharmacological inhibition of autophagy abolished the high tidal volume ventilation-induced inflammasome activation and lung inflammatory responses [14]. 3-Methyladenine (an autophagy inhibitor) could ameliorate the lung injury and improve cell survival [43]. In the present study, we demonstrated that the conversion rate of cytosolic microtubule-associated protein-1 light chain-3 LC3-I to lapidated LC3-II was increased in our two-hit VILI mouse model. Treatment with JAK2 inhibitor AZD1480 could reduce the levels of LC3-II and diminish lung destruction, lung edema, and cytokine production, indicating that the role of autophagy in promoting pulmonary injuries is dependent on the JAK2/STAT1 pathway. Inappropriate or massive stimulation of autophagy can actually facilitate cell death. Our previous work showed that apoptotic pulmonary cells also plays a critical role during the process of ALI [44]. In addition, MV or CS-induced autophagy and apoptosis in fetal rat lung epithelial cells [45], suggesting that such autophagic cell death exhibits an essential role in MV or CS model. AZD1480 also decreased the levels of cleaved caspase 3, which may be another reason why JAK2 inhibition has protective role in our model.
Macrophages are involved in the initial response to lung inflammation. In order to uncover the underlying mechanism of VILI, we used RAW246.7 cells to investigate the interaction between the JAK2/STAT1 pathway and HMGB1 translocation in mediating lung injury. Consistent with our in vivo findings, the in vitro experiments on mouse macrophage-like RAW 264.7 cells showed that JAK2 inhibitor attenuated cell autophagy and apoptosis by inhibiting JAK2/STAT1 activation. In addition, the loss of macrophage membrane integrity was determined by the level of LDH in the supernatant. When RAW246.7 cells were exposed to LPS and mechanical stretch, the release of LDH from the cells increased significantly, as well as autophagy and apoptosis. Furthermore, the relationship between HMGB1 translocation and apoptosis induction during macrophage activation was revealed. Our experimental findings confirmed the inflammatory role of autophagic cell death in LPS+MV/CS model via HMGB1 translocation.
To further investigate the relationship between the JAK2/STAT1 pathway and HMGB1 translocation, we detected the expression levels of HMGB1 in our LPS+CS cell model. Western blotting results showed that the total HMGB1 expression in RAW 264.7 cells did not change significantly after exposed to LPS and CS. Nonetheless, HMGB1 translocation to the cytoplasm was enhanced by LPS and CS, and AZD1480 could inhibit the translocation of HMGB1 to the cytoplasm. Such protective effects might be attributed to JAK2/STAT pathway instead of HMGB1 alone, as confirmed by the results of RNA interference experiments. Knockdown of HMGB1 by siRNA could decrease the release of LDH as well as autophagy in RAW246.7 cells, suggesting that HMGB1 depletion may protect the cells from LPS+CS-induced cell damage but not completely reverse the cell death. However, autophagic cell death and LDH elevation were reduced by STAT1 knockdown in RAW246.7 cells. STAT1 siRNA also inhibited the HMGB1 translocation from nucleus to cytoplasm, whereas JAK2 inhibition with or without STAT1 siRNA exhibited the same effect. Hence, the contribution of JAK2/STAT1 to VILI depends, at least in part, on its ability to promote cytoplasmic accumulation of HMGB1 as well as induce cell autophagy and apoptosis in the inflamed lungs.
In summary, this study reveals that the JAK/STAT1 signaling pathway plays a pivotal role in VILI by regulating HMGB1 translocation and JAK2 inhibitor AZD1480 may serve as a therapeutic strategy to control VILI.
References
Bellani G, Laffey JG, Pham T, Fan E, Brochard L, Esteban A, et al. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. J Am Med Assoc. 2016;315:788–800.
Villar J, Blanco J, Anon JM, Santos-Bouza A, Blanch L, Ambros A, et al. The ALIEN study: incidence and outcome of acute respiratory distress syndrome in the era of lung protective ventilation. Intensive Care Med. 2011;37:1932–41.
Confalonieri M, Salton F, Fabiano F. Acute respiratory distress syndrome. Eur Res Rev. 2017;26:160116.
Tremblay LN, Slutsky AS. Ventilator-induced lung injury: from the bench to the bedside. Intensive Care Med. 2006;32:24–33.
Gajic O, Dara SI, Mendez JL, Adesanya AO, Festic E, Caples SM, et al. Ventilator-associated lung injury in patients without acute lung injury at the onset of mechanical ventilation. Criti Care Med. 2004;32:1817–24.
Kuchnicka K, Maciejewski D. Ventilator-associated lung injury. Anaest Intensive Ther. 2013;45:164–70.
Brower RG, Matthay MA, Morris A, Schoenfeld D, Thompson BT, Wheeler A. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. New Engl J Med. 2000;342:1301–8.
Ochiai R. Mechanical ventilation of acute respiratory distress syndrome. J Intensive Care. 2015;3:25.
Dreyfuss D, Saumon G. Ventilator-induced lung injury: lessons from experimental studies. Am J Respir Crit Care Med. 1998;157:294–323.
Lex D, Uhlig S. One-hit models of ventilator-induced lung injury: benign inflammation versus inflammation as a by-product. Anesthesiology. 2017;126:909–22.
Jaecklin T, Engelberts D, Otulakowski G, O’Brodovich H, Post M, Kavanagh BP. Lung-derived soluble mediators are pathogenic in ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2011;300:L648–58.
Uhlig S, Ranieri M, Slutsky AS. Biotrauma hypothesis of ventilator-induced lung injury. Am J Respir Crit Care Med. 2004;169:314–5. author reply 5.
Siempos II, Ma KC, Imamura M, Baron RM, Fredenburgh LE, Huh JW, et al. RIPK3 mediates pathogenesis of experimental ventilator-induced lung injury. JCI Insight. 2018;3:97102.
Zhang Y, Liu G, Dull RO, Schwartz DE, Hu G. Autophagy in pulmonary macrophages mediates lung inflammatory injury via NLRP3 inflammasome activation during mechanical ventilation. Am J Physiol Cell Mol Physiol. 2014;307:L173–85.
Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Ann Rev Immunol. 2011;29:139–62.
Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science (New York, NY). 1999;285:248–51.
Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med. 2005;201:1135–43.
Dhupar R, Klune JR, Evankovich J, Cardinal J, Zhang M, Ross M, et al. Interferon regulatory factor 1 mediates acetylation and release of high mobility group box 1 from hepatocytes during murine liver ischemia-reperfusion injury. Shock. 2011;35:293–301.
Ueno H, Matsuda T, Hashimoto S, Amaya F, Kitamura Y, Tanaka M, et al. Contributions of high mobility group box protein in experimental and clinical acute lung injury. Am J Respir Crit Care Med. 2004;170:1310–6.
van Zoelen MA, Ishizaka A, Wolthuls EK, Choi G, van der Poll T, Schultz MJ. Pulmonary levels of high-mobility group box 1 during mechanical ventilation and ventilator-associated pneumonia. Shock. 2008;29:441–5.
Ogawa EN, Ishizaka A, Tasaka S, Koh H, Ueno H, Amaya F, et al. Contribution of high-mobility group box-1 to the development of ventilator-induced lung injury. Am J Respir Crit Care Med. 2006;174:400–7.
Altemeier WA, Matute-Bello G, Gharib SA, Glenny RW, Martin TR, Liles WC. Modulation of lipopolysaccharide-induced gene transcription and promotion of lung injury by mechanical ventilation. JImmunol (Baltimore, MD: 1950). 2005;175:3369–76.
Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–5.
Banerjee S, Biehl A, Gadina M, Hasni S, Schwartz DM. JAK-STAT signaling as a target for inflammatory and autoimmune diseases: current and future prospects. Drugs. 2017;77:521–46.
Wang X, Liu Q, Ihsan A, Huang L, Dai M, Hao H, et al. JAK/STAT pathway plays a critical role in the proinflammatory gene expression and apoptosis of RAW264.7 cells induced by trichothecenes as DON and T-2 toxin. Toxicol Sci. 2012;127:412–24.
Severgnini M, Takahashi S, Rozo LM, Homer RJ, Kuhn C, Jhung JW, et al. Activation of the STAT pathway in acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2004;286:L1282–92.
Pan J, Fukuda K, Saito M, Matsuzaki J, Kodama H, Sano M, et al. Mechanical stretch activates the JAK/STAT pathway in rat cardiomyocytes. Circ Res. 1999;84:1127–36.
Wang L, Wang T, Li H, Liu Q, Zhang Z, Xie W, et al. Receptor interacting protein 3-mediated necroptosis promotes lipopolysaccharide-induced inflammation and acute respiratory distress syndrome in mice. PLoS ONE. 2016;11:e0155723.
Dunn I, Pugin J. Mechanical ventilation of various human lung cells in vitro. Chest. 1999;116:95S–7S.
Aittomaki S, Pesu M. Therapeutic targeting of the Jak/STAT pathway. Basic Clin Pharmacol Toxicol. 2014;114:18–23.
Moriyama K, Ishizaka A, Nakamura M, Kubo H, Kotani T, Yamamoto S, et al. Enhancement of the endotoxin recognition pathway by ventilation with a large tidal volume in rabbits. Am J Physiol Lung Cell Mol Physiol. 2004;286:L1114–21.
Amigoni M, Bellani G, Zambelli V, Scanziani M, Farina F, Fagnani L, et al. Unilateral acid aspiration augments the effects of ventilator lung injury in the contralateral lung. Anesthesiology. 2013;119:642–51.
Lin SM, Lin HC, Lee KY, Huang CD, Liu CY, Wang CH, et al. Ventilator-induced injury augments interleukin-1beta production and neutrophil sequestration in lipopolysaccharide-treated lungs. Shock. 2007;28:453–60.
Ko YS, Park EJ, Kim YM, Kim HJ, Yun-Choi H, Lee DH, et al. A limited series of synthetic tetrahydroisoquinoline alkaloids reduce inflammatory gene iNOS via inhibition of p-STAT-1 and suppress HMGB1 secretion in LPS-treated mice lung tissue. Int Immunopharmacol. 2017;52:297–304.
Ding N, Wang F, Xiao H, Xu L, She S. Mechanical ventilation enhances HMGB1 expression in an LPS-induced lung injury model. PLoS ONE. 2013;8:e74633.
Jessop F, Hamilton RF, Rhoderick JF, Shaw PK, Holian A. Autophagy deficiency in macrophages enhances NLRP3 inflammasome activity and chronic lung disease following silica exposure. Toxicol Appl Pharmacol. 2016;309:101–10.
Xia F, Deng C, Jiang Y, Qu Y, Deng J, Cai Z, et al. IL4 (interleukin 4) induces autophagy in B cells leading to exacerbated asthma. Autophagy. 2018;14:450–64.
Wen Z, Fan L, Li Y, Zou Z, Scott MJ, Xiao G, et al. Neutrophils counteract autophagy-mediated anti-inflammatory mechanisms in alveolar macrophage: role in posthemorrhagic shock acute lung inflammation. J Immunol (Baltimore, MD: 1950). 2014;193:4623–33.
Tang D, Kang R, Livesey KM, Cheh CW, Farkas A, Loughran P, et al. Endogenous HMGB1 regulates autophagy. J Cell Biol. 2010;190:881–92.
Petrusca DN, Van Demark M, Gu Y, Justice MJ, Rogozea A, Hubbard WC, et al. Smoking exposure induces human lung endothelial cell adaptation to apoptotic stress. Am J Respir Cell Mol Biol. 2014;50:513–25.
Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12:222–30.
Zhang D, Zhou J, Ye LC, Li J, Wu Z, Li Y, et al. Autophagy maintains the integrity of endothelial barrier in LPS-induced lung injury. J Cell Physiol. 2018;233:688–98.
Li C, Liu H, Sun Y, Wang H, Guo F, Rao S, et al. PAMAM nanoparticles promote acute lung injury by inducing autophagic cell death through the Akt-TSC2-mTOR signaling pathway. J Mol Cell Biol. 2009;1:37–45.
Xie W, Wang H, Liu Q, Li Y, Wang J, Yao S, et al. ResolvinD1 reduces apoptosis and inflammation in primary human alveolar epithelial type 2 cells. Lab Invest. 2016;96:526–36.
Yeganeh B, Lee J, Bilodeau C, Lok I, Ermini L, Ackerley C, et al. Acid sphingomyelinase inhibition attenuates cell death in mechanically ventilated newborn rat lung. Am J Respir Crit Care Med. 2019;199:760–72.
Acknowledgements
This study was supported by the grant from the National Natural Science Foundation of China (Number 81370112 and Number 81873952).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Liu, Q., Xie, W., Wang, Y. et al. JAK2/STAT1-mediated HMGB1 translocation increases inflammation and cell death in a ventilator-induced lung injury model. Lab Invest 99, 1810–1821 (2019). https://doi.org/10.1038/s41374-019-0308-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-019-0308-8
This article is cited by
-
Inhibition of importin-7 attenuates ventilator-induced lung injury by targeting nuclear translocation of p38
Inflammation Research (2023)
-
IL-9 blockade attenuates inflammation in a murine model of mechanical ventilation-induced lung injury by inhibiting the NLRP3 inflammasome pathway
Inflammopharmacology (2022)
-
Ventilator-induced lung-injury in mouse models: Is there a trap?
Laboratory Animal Research (2021)
-
Inhibition of MicroRNA-214 Alleviates Lung Injury and Inflammation via Increasing FGFR1 Expression in Ventilator-Induced Lung Injury
Lung (2021)
