
Abstract
Myocyte enhancer-binding factor 2B (MEF2B) has been implicated as a transcriptional regulator for BCL6. However, details about the interaction between MEF2B and BCL6 during expression, as well as the relationship of MEF2B to the expression of other germinal center (GC) markers, have not yet been fully explained. Using germinal center B-cell-like diffuse large B-cell lymphoma (GC-DLBCL) and activated B-cell diffuse large B-cell lymphoma (ABC-DLBCL) cell lines, we analyzed the expression of MEF2B and its associations with BCL6, CD10, and ERK. Furthermore, small interfering RNA (siRNA) was used to study the possible effects of MEF2B knockdown on these proteins and cell growth. Analysis of the BCL6 transcriptional complex was performed using electrophoretic mobility shift assay. The correlation between MEF2B expression and the genetic type of DLBCL was assessed using immunohistochemistry on 111 patient samples, and via in silico analysis of publicly available microarray (Gene Expression Omnibus (GEO)) datasets. Our results indicate that the expression of MEF2B protein is important for the growth of GC-DLBCL cells, as evidenced by MEF2B knockdown inhibition of cell growth and the subsequent suppression of BCL6, CD10, and ERK phosphorylation. Analysis of BCL6 transcription factors in nuclear extracts of MEF2-expressing DLBCL cells showed involvement of MEF2B with AP-2α and BCL6 proteins in the formation of the BCL6 gene transcriptional complex. Indeed, differential expression of MEF2B in the GC-DLBCL is statistically significant compared to the ABC-DLBCL in the GEO datasets, as well as in tissue microarray, as indicated via immunohistochemistry (Visco–Young algorithm). Our findings indicate that MEF2B is an essential component of the BCL6 gene transcriptional complex for the regulation of DLBCL growth via the promotion of BCL6 expression. Beyond its regulatory role in DLBCL growth, MEF2B expression correlated positively with BCL6 and CD10 expression, and was preferentially expressed in the GBC-DLBCL group.
Similar content being viewed by others

Introduction
Diffuse large B-cell lymphoma (DLBCL) is a morphologically and genetically heterogeneous disease [1]. Using gene expression data, DLBCL is classified into cell-of-origin (COO) categories, primarily including the germinal center B-cell-like group (GBC) and the activated B-cell group (ABC) [2]. BCL6 is a master regulator in GC phenotype, as it is essential for GC formation [3, 4]. The expression of wild-type MEF2B is a supposed independent regulator of BCL6 expression [5]. GC-derived lymphoma cells are dependent on MEF2B for growth and proliferation, which is partially related to MEF2B control over BCL6 expression [6]. However, the exact relationship between MEF2B and BCL6 is still poorly defined; while MEF2B may bind to the BCL6 promoter region at a site upstream of the BCL6 transcription start site [6]. MEF2B is not included in the direct transcriptional targets bound by BCL6 [7]. Mutations in MEF2B are also detected in DLBCL and follicular lymphoma cases. MEF2B mutations, in particular homozygous deletions of MEF2B, are detected in DLBCL, indicating its possible role as a tumor suppressor gene. More recent data suggest that it acts as an oncogene as well [8,9,10]. The limited data concerning the expression of MEF2B with immunohistochemistry in lymphomas indicate that MEF2B is expressed in almost all cases of follicular lymphoma and Burkitt lymphoma, the majority of GC-DLBCL, and a subset of non-GC-DLBCL [11, 12].
This study is designed to address the mechanistic role of MEF2B protein in the regulation of BCL6 expression and subsequent development and progression of DLBCL. We performed functional analysis of MEF2B in both MEF2B-expressing and MEF2B-deficient DLBCL lymphoma cell lines, which revealed that MEF2B binds with AP-2-α and BCL6 proteins while acting as a transcriptional complex for BCL6 gene. We further showed that MEF2B induces the expression of BCL6 when transfected into originally MEF2B-deficient cells. Knocking down the MEF2B gene ultimately suppressed BCL6 and CD10 expression, as well as ERK1/2 phosphorylation. We found that MEF2B expression is correlated with both BCL6 and CD10 expression, and using tissue microarray, we found that it is differentially expressed in the GC-DLBCL group (TMA) and in six Gene Expression Omnibus (GEO) datasets of DLBCL.
Materials and methods
Human cell lines and treatments
GC-DLBCL cell lines (SU-DHL-4, SU-DHL-5, and SU-DHL-6) and ABC-DLBCL (OCI-LY 3, HBI-1, and SU-DHL-2) cell lines were used [13, 14, 15].
Immune blotting analysis
Protein analysis was performed using standard Western blotting. The following antibodies were used at the indicated dilution: anti-MEF2B (#HPA004734, Atlas Antibodies), 1:1000; anti-BCL6 (#ab8666, Abcam), 1:1000; anti-CD10 (#ab73409, Abcam), 1:1000; ERK1/2 (Sc-154-G), 1:2000; and anti-p-ERK (Sc-7383), 1:1000; anti-actin (Sc-1615), 1:2000, each from Santa Cruz Biotechnology.
Assessment of cell growth rate
The cell lines were seeded in 96-microwell plates (1 × 104 cells per well) (Nunc, Waltham, MA, USA) and allowed to grow for the indicated time periods. The growth rate of the MEF2B-deficient cell lines (OCI-LY 3, HBI-1, and SU-DHL-2) was compared to that of the MEF2B-expressing cell lines (SU-DHL-4, SU-DHL-5, and SU-DHL-6) using the colorimetric MTT (3-(4,5-dimethythiazol-2-yl)-2,5-diphenyl tetrazolium bromide) assay (Roche, Bâle, Switzerland) as previously described [16].
Cell cycle distribution assay
The analysis of cell cycle distribution of GC-DLBCL cell lines (SU-DHL-4 andSU-DHL-5) and ABC-DLBCL (SU-DHL-2) cell lines before and after the transfection with MEF2B-specific small interfering RNA (siRNA) was performed using flow cytometry [17]. Briefly, the cells (1 × 105 cells per well in six-well plate) were washed in warm phosphate-buffered saline, and then incubated in propidium iodide (PI, 2.5 µg/mL (Invitrogen, USA)) and RNase 1 (Invitrogen) for 30 min. The cell cycle distribution was analyzed via flow cytometry. G1, S, and G2 phase percentages were recorded.
Analysis of apoptosis using the annexin V/PI
The analysis of apoptosis in GC-DLBCL cell lines (SU-DHL-4 andSU-DHL-5) and ABC-DLBCL cell lines (SU-DHL-2) was performed via flow cytometry using annexin V-FITC/PI (Vybrant; Invitrogen, Karlsruhe, Germany) for 15 min at room temperature with protection from light [18].
RNA silencing
The knockdown of MEF2B and BCL6 genes was performed according to our established protocol [19]. Briefly, the MEF2B-expressing cell lines (SU-DHL-4, SU-DHL-5, and SU-DHL-6) were plated for 24 h before the transfection. Cells were transfected in serum-free RPMI with 50 nmol/L control (siRNA negative control #SR-CL000-005 from Eurogentec, Seraing, Belgium), MEF2B siRNAs (predesigned Silencer select siRNA #S8650 from Ambion, Darmstadt, Germany), or BCL6 siRNA (sc-29791, Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA). The knockdown of MEF2B in MEF2B-expressing cells was performed via transfection of the siRNAs (5′-CCC TGC GCC GGC TGC CCT TG-3′ and 5′-GCC GAC GGC TGG CCC CGG TA-3′) directed to the 3′-terminal region of MEF2B gene (Eurofins, Germany) using Lipofectamine 2000 (Invitrogen) for the indicated time period [20]. After assessment of MEF2B knockdown efficiency via Western blot analysis, the cells then underwent MTT assay.
Electrophoretic mobility shift assay
The DNA-binding activity of the transcription factors was analyzed as described previously [21]. Briefly, we used double-stranded synthetic oligonucleotides that represent the specific binding site of BCL6 promoter to BCL6 protein (5′-TGT CCT GGA AAG AA-3′) and the transcription factor AP-2α (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) . The double-stranded DNA consensus sequences were end-labeled with [γ-32P]ATP (Hartmann Analytika, Munich, Germany) using T4 polynucleotide kinase (Genecraft, Lüdinghausen, Germany). The DNA-binding activity of each transcription factor was measured via incubation of 4 µg of nuclear extracts with a labeled probe of the transcription factor of interest in a total reaction volume of 30 µl containing electrophoretic mobility shift assay (EMSA)-binding buffer (10 mM Tris, pH 7.5; 50 mM NaCl, 1 mM EDTA; 1 mM MgCl2; 0.5 mM DTT, and 4% glycerol). The supershift assay was performed via incubation of the corresponding antibodies of AP-2α, BCL6, or MEF2B with the nuclear extracts for 1 h prior to the incubation with probe. After 30 min of incubation at room temperature, the DNA-binding activity of the transcription factors was analyzed via electrophoresis for 3 h at 100 V in 0.5× Tris-borate-EDTA running buffer at room temperature. The dried gel was visualized via exposure to high-performance autoradiography film.
Luciferase reporter assays
For the activity of BCL6 promoter assays, SU-DHL-2 and SU-DHL-6 cells were transiently transfected with 0.25 μg of either pGL-BCL6 combined with PCMV-MEF2B or with its transcription-defective mutant with deletion of the DNA-binding and dimerization domains pCMV-ΔMADSMEF2, as used previously [6]. After 48 h, the cells were extracted in 150 ml of luciferase lysis buffer.The collected cell lysates were then analyzed in a MicroBeta Luminescence counter (Perkin-Elmer Life Sciences) using a Luciferase Reporter Assay Kit (Promega). All luciferase activities were normalized to the transfection efficiency [22].
Tissue samples
This study was performed in accordance with the Institutional Review Board policies of the University of Louisville. A TMA was constructed from formalin-fixed, paraffin-embedded tissue (FFPET) sections of 27 de novo DLBCL, NOS (not otherwise specified) cases as per the World Health Organization classification obtained from the University of Louisville [1]. In addition, previously prepared commercial TMA slides for 102 tissue cores were acquired from Abcam (#ab178212, Cambridge, MA, USA): 97 cores from DLBCL cases and 5 reactive non-lymphoma cores. Two board-certified pathologists (SME and ZG) reviewed the hematoxylin and eosin- and CD20 immunohistochemical (IHC)-stained slides from the TMA to verify that each met the criteria for DLBCL and verified tissue adequacy in each core. Of these 97 DLBCL cases in the TMA, 84 cases met the criteria for diagnosis and adequacy. In total, 111 tissue samples were included in the study (27 FFPETs and 84 TMA cores). We were able to perform NanoString analysis on 24 of the in-house FFPET cases to validate the COO classification of DLBCL, as detailed below.
Validation of the IHC COO classifier using NanoString gene expression assay
Given the relatively small number of the cases available to perform the NanoString assay, we elected to use the cases as a validation set for an IHC-based classifier. We chose the Visco–Young IHC algorithm, given its highest concordance rate with gene expression profiling (GEP) data among IHC algorithms [23]. In a separate study, we validated the Visco–Young algorithm in our cohort and profiled 24 of the in-house DLBCL cases using the research-only version of the NanoString Lymphoma Subtyping Test algorithm. This was meant to determine the COO molecular subtype. We measured the coefficient of agreement between the NanoString and the Visco–Young IHC classifier [24, 25].
Immunohistochemistry
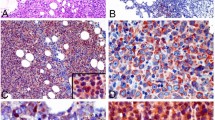
IHC staining was performed on Leica Bond III autostainer according to the previously published protocol [26]. Sections from each of the TMAs were cut and immunostained with CD20, CD10, BCL6, and FOXP-1 (Table 1) to classify according to Visco–Young algorithm [23]. We repeated the IHC staining for CD20, CD10, and BCL6 on the TMA from the 27 in-house cases to remain consistent during technical procedures and while interpreting the commercial TMA cases. As discussed, MEF2B is a nuclear transcription factor with nuclear localization by immunohistochemistry [12, 27]. We found that the polyclonal rabbit antibody from Atlas antibodies (HPA004734; Stockholm, Sweden) is more suitable than the monoclonal mouse antibody from Abnova (M24/clone 4B5; Taipei, Taiwan), the latter showed background staining in stromal cells, plasma cells, and interfollicular lymphocytes. With benign tonsils used as positive controls, MEF2B is expressed strongly in the GCs with similar staining patterns to those of CD10 and BCL6 in the control slides (Fig. 5a).
Staining assessment and scoring for MEF2B
For MEF2B IHC evaluation in this study, we used a slightly modified Histoscore (H-score) system which is a semiquantitative approach to score for each tissue core, considering both the intensity and proportion of the staining [28, 29]. A final H-score was calculated via adding the sum of scores obtained for each intensity (I) and proportion (P) of each respective tumor. The intensity score is based on a scale of three levels (0—negative, 1—weakly positive, and 2—strongly positive). A formula of H = I1 × P1 + I2 × P2 + I3 × P3 is used. The cutoff point for MEF2B stain positivity was calculated using the online tool Cut-off Finder (http://molpath.charite.de/cutoff/) [30]. In summary, GC-DLBCL cases were the supposed defining variable in a receiver operating characteristic (ROC) curve, which plotted MEF2B sensitivity versus 1-specificity by calculating the respective area under the ROC (AUROC). The optimal cutoff point of MEF2B expression for the diagnosis of GC-DLBCL is when both sensitivity and specificity are the highest [31].
GEO gene expression profile datasets and analysis
Six gene expression profile datasets from the GEO database (http://www.ncbi.nlm.nih.gov/geo) were analyzed for comparison of MEF2B expression in GC-DLBCL versus ABC-DLBCL cases. The datasets used included GSE32918, GSE19246 [32], GSE23501 [33], GSE4475 [34], GSE22470 [35], and GSE31312 [23] (Table 2). We used the GEO2R tool to assess the differential expression of MEF2B in the two DLBCL subgroups in each dataset, applying p value calculation without adjustment.
Statistical analysis
Statistical analysis was performed using PRISM (GraphPad Software, La Jolla, CA, USA) and IBM SPSS Statistics Subscription (Armonk, NY, USA). Spearman’s ρ test was used to evaluate the correlation of MEF2B positivity with the expression of BCL6 and CD10 via immunohistochemistry. Cohen's κ coefficient was used to study the agreement between Visco–Young IHC and NanoString COO DLBCL subgroups. Paired t tests were used to assess the statistical significance of MEF2B expression in the TMA IHC and NanoString DLBCL subgroups. Non-paired t test was used to assess the differential expression of MEF2B in the GEO datasets.
Results
Analysis of MEF2B expression in ABC- and GC-DLBCL-derived cell lines
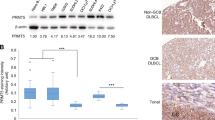
We analyzed the MEF2B expression in the GC-DLBCL cell lines (SU-DHL-4, SU-DHL-5, and SU-DHL-6) and the ABC-DLBCL cell lines (OCI-LY-3, HBL-1, and SU-DHL-2) using Western blot. The cell lines were allowed to grow under normal conditions. Seventy-two hours later, the cells were harvested and the total cell lysates were prepared. Analysis of the cell lysates using Western blot revealed that MEF2B is expressed in GC-DLBCL cells, but not in ABC-DLBCL cells (Fig. 1a). We also noted that the expression of MEF2B, BCL6, and CD10 was associated with the phosphorylation of the ERK1/2 proteins, suggesting the involvement of MEF2B protein in the regulation of BCL6 and CD10 expression and the activation of ERK pathway.

a. Western blot analysis of total cell lysates of MEF2B in MEF2B-deficient cell lines (OCI-LY-3, HBL-1, and SU-DHL-2) and MEF2B-expressing cell lines (SU-DHL-4, SU-DHL-5, and SU-DHL-6) demonstrates the expression of BCL6, CD10, and phosphorylation of ERK1/2 in the MEF2B-expressing cell lines. Actin was used as an internal control for loading and transfer. Data are representative of three independent experiments. b Time-course experiment demonstrates the growth rate of MEF2B-deficient cell lines (OC-LY-3, HBL-1, and SU-DHL-2) and MEF2B-expressing cell lines (SU-DHL-4, SU-DHL-5, and SU-DHL-6). MTT assay demonstrated the growth rate of MEF2B-deficient (OCI-LY-3, HBL-1, and SU-DHL-2) and MEF2B-expressing cell lines (SU-DHL-4, SU-DHL-5, and SU-DHL-6) cell lines. The values are expressed as the means ± SD of three independent experiments performed in duplicate
MEF2B promotes the growth of DLBCL cell lines
To demonstrate whether the growth rate of human lymphoma cells is associated with the expression of MEF2B, the human lymphoma MEF2B-deficient (ABC-DLBCL) (OCI-LY-3, HBI-1, and SU-DHL-2) and MEF2B-expressing (GC-DLBCL) (SU-DHL-4, SU-DHL-5, and SU-DHL-6) cell lines were allowed to grow for regulated time intervals up to 72 h. Cell viability was then assessed via MTT assay. Analysis of the MTT assay data (Fig. 1b) displayed a growth advantage in the cells expressing MEF2B as compared to the MEF2B-deficient cells after 24 h.
We noted that the growth rate of SU-DHL-4, SU-DHL-5, and SU-DHL-6 cells increased thereafter at 48 h to reach a maximum at 72 h when compared with OCI-LY-3, HBI-1, and SU-DHL-2 cells. This suggests that MEF2B protein helps to modulate DLBCL cell growth. Next, we confirmed the growth advantage of MEF2B-expressing cells over the MEF2B-deficient cells using two independent techniques, namely cell cycle assay and apoptosis-specific assay. We tested the cell cycle distribution in SU-DHL-2 (MEF2B-deficient) and SU-DHL-4 (MEF2B-expressing) cells. Images from the PI FACS assay displayed a time-dependent increased percentage of G1 in both cell lines, which was markedly visible in SU-DHL-4 cells when compared to those noted in SU-DHL-2 cells (Fig. 2a). While no marked decrease in the S-phase percentage was noted in SU-DHL-2 cells, there was a marked decrease in the S-phase percentage in SU-DHL-4 cells. Furthermore, the increased percentage of the G1 phase in both cell lines was correlated with the decreased percentage of the G2 phase at all time periods. Quantified results in Fig. 2a further confirmed the time-dependent induction of the G1 phase in SU-DHL-4 cells compared to SU-DHL-2 cells. In contrast, FACS analysis of annexin V/PI (Fig. 2b) did not reveal significant apoptotic effects in the MEF2B-deficient cells when compared to the MES2B-expressing cells. Our data suggest that the growth advantage of SU-DHL-4 cells over SU-DHL-2 cells is attributed to the cell cycle delay or growth inhibition of SU-DHL-2 cells. The noted variation of apoptosis among SU-DHL-2 cells at different time periods (Fig. 2b) is expected to result from the physical damage of the cells during the performance of annexin staining. These results are somewhat similar to what was observed by Ying et al. [6].

a Cell cycle analysis demonstrates G1, S, and G2 phases in MEF2B-deficient (SU-DHL-2) and MEF2B-expressing (SU-DHL-4) cell lines. Cell populations represent each phase of cell cycle for SU-DHL-2 and SU-DHL-4 cell lines. Data represent the mean±standard deviation (SD) (n = 3). b Flow cytometry analysis of apoptosis demonstrates the portion of annexin V-stained cells in SU-DHL-2 and SU-DHL-4 cell lines. Apoptotic cells are represented in the lower right corner. Data are representative of three independent experiments performed separately. c Western blot analysis demonstrates that the knockdown of MEF2B by its specific siRNA in SU-DHL-4, SU-DHL-5, and SU-DHL-6 cell lines is associated with the inhibition of the expression of BCL6 and CD10 as well as the phosphorylation of ERK proteins. Actin was used as an internal control for loading and transfer. Data are representative of three independent experiments. d MTT assay demonstrates in a time-course experiment the inhibition of the growth rate of SU-DHL-4, SU-DHL-5, and SU-DHL-6 cell lines by the knockdown of MEF2B by its specific siRNA. The values are expressed as the means ± SD of three independent experiments performed in duplicate
Expression of MEF2B is vital for GC-DLBCL cell growth
To address the role of MEF2B in the regulation of lymphoma cell growth, the MEF2B-expressing lymphoma cell lines were transfected with MEF2B-specific siRNA, and then allowed to grow for regulated time intervals up to 72 h. First, we analyzed the siRNA efficiency of MEF2B knockdown expression using Western blot. MEF2B knockdown is associated with the suppression of BCL6 and CD10 expression, as well as the suppression of ERK phosphorylation in SU-DHL4, SU-DHL5, and SU-DHL6 cell lines (Fig. 2c). We then examined whether MEF2B knockdown is associated with lymphoma cell growth inhibition. The same cells underwent MTT assay to analyze viability. As expected, MEF2B knockdown was found to inhibit growth of all tested cells (Fig. 2d). Taken together, the expression of MEF2B is vital for the growth and maintenance of lymphoma cells in the CG-DLBCL group. Next, we confirmed the MTT assay data using flow cytometry analysis of cell cycle and apoptosis in SU-DHL-4 and SU-DHL-5 cells both before and after MEF2B knockdown (Fig. 3a). PI FACS assay images confirmed that MEF2B knockdown by its respective siRNA induced time-dependent decreased percentages of the G1 phase, and increases in the percentage of S and G2 phases. Quantified results further confirmed the noted time-dependent reduction in the G1 phase in response to the knockdown of MEF2B in both SU-DHL-4 and SU-DHL-5 cells. A minor time-dependent induction of apoptosis is seen in the same cell lines (Fig. 3b). Thus, we concluded that the knockdown of MEF2B leads to disruption of SU-DHL-4 and SU-DHL-5 cell cycles, with arrest in the G2/S phases and mild apoptosis induction.

a Cell cycle analysis demonstrates G1, S, and G2 phase in control (Ctr) and siRNA-transfected MEF2B-expressing (SU-DHL-4 and SU-DHL-5) cell lines. Cell populations represent each phase of cell cycle for SU-DHL-4 and SU-DHL-5 cell lines before and after the transfection with siRNA specific to MEF2B. b Flow cytometry analysis of apoptosis demonstrates the portion of annexin V-stained cell in SU-DHL-4 and SU-DHL-5 cell lines before and after transfection with siRNA specific to MEF2B for the indicated time periods. Apoptotic cells are represented in the lower right corner. Data are representative of three independent experiments performed separately
BCL6 expression is induced by transfecting MEF2B into MEF2B-deficient cells
We subsequently aimed to determine the inhibitory effect of the accumulated BCL6 on its expression. We thus performed a time-course experiment up to 96 h following the transfection of SU-DHL-2 (MEF2-deficient) with luciferase reporter BCL6 promoter construct PGL-BCL6-Luc in combination with construct of the MEF2B-WT together with siRNA specific to the complementary DNA region encoding for the C-terminal domain of MEF2B. First, we assessed the expression level of both MEF2B and BCL6 by Western blot analysis (Fig. 4b). We successfully induced the expression of both MEF2B and BCL6 at 24 h post transfection, with an increase thereafter at 48 h. Although no changes were noted on the induced expression of MEF2B over time, the increase of BCL6 expression suddenly decreased at both 72-h and 96-h intervals post transfection (Fig. 4b). This suggests a negative effect for the induced BCL6 expression upon transcriptional activation, a finding in accordance with previous reports [36,37,38]. As expected, knocking down MEF2B using siRNA led to the suppression of BCL6 protein expression (Fig. 4a). Similar results were obtained using the defective mutant ΔMADSMEF2 (Fig. 4d).

a Western blot demonstrates the expression level of BCL6 in SU-DHL-6 (MEF2B-positive) before and after the knockdown of MEF2B by its specific siRNA. b Western blot analysis of MEF2B and BCL6 proteins using the total cell lysates derived from SU-DHL-2 (MEF2B-deficient) following the transfection with MEF2-WT together with ΔMADSMEF2 and PGL-BCL6-promoter/Luc before and after the transfection with siRNA specific to cDNA encoding region of the C-terminal domain of MEF2B. Actin was used as an internal control for loading and transfer. c Electrophoretic mobility shift assay (EMSA) demonstrates the DNA-binding activity of the transcription factors BCL6 and AP-2α supershift analysis using specific anti-AP-2α, BCL6, and MEF2B antibodies demonstrates the involvement of MEF2B, BCL6, and AP-2α proteins in the formation of the transcriptional complex of BCL6 gene. The competitor was used as a negative control for the DNA-binding activity of the transcription factors. Data are representative of three independent experiments. d Lucieferase assay demonstrates the activity of the BCL6 promoter in response to the expression of MEF2B-WT or C-terminal truncated MEF2B (ΔMADSMEF2) in SU-DHL-2 cells
MEF2B forms a transcriptional complex with AP-2α and BCL6 proteins to mediate its effect on the BCL6 gene
Next, we specified the role of MEF2B in BCL6 induction. First the lymphoma cell lines SU-DHL-6 were allowed to grow for 72 h.The nuclear extracts were then prepared for analysis of the transcriptional complex of the BCL6 via EMSA. Our analysis was based on the potential consensus sequences of BCL6 promoter reported by Ohashi et al. [39]. We analyzed the DNA-binding sites of AP-2α and BCL6 in the context of MEF2B as a transcriptional activator of BCL6 [6]. We then analyzed the DNA-binding activity of BCL6 and AP-2α using the nuclear extracts of SU-DHL-6. EMSA data (Fig. 4c) demonstrated the enhancement of the AP-2α and BCL6 proteins' DNA activities at their specific DNA-binding sites. Next, we set out to identify the detected transcriptional complex. Accordingly, the same nuclear extract was analyzed further via supershift assay using antibodies specific for BCL6, AP-2α, and MEF2B to determine authenticity of the transcriptional complex. EMSA analysis demonstrated that MEF2B protein is included in the protein complex binding to the consensus sequences of both AP-2α and BCL6 (Fig. 4c). However, given that AP-2α and BCL6 were known members of the BCL6 transcription complex [39], the detection of MEF2B along the AP-2α and BCL6 proteins in one complex indicates MEF2B essential role in activating the transcriptional complex of BCL6.
MEF2B expression is associated with BCL6 and CD10 expression and strongly correlated to GC-DLBCL by IHC and dataset analysis
Of the 111 DLBCL tissue samples included in the study, 34 samples (30.6%) were classified as GC type according to the IHC Visco–Young algorithm. Compared to the original COO classification at the time of diagnosis among in-house cases, the percentage of concordance was 74%. However, it should be noted that the original classification at the time of diagnosis had been performed using the Hans algorithm rather than the Visco–Young algorithm used in this study [23, 40]. Interestingly, we are reporting a percentage of GC-DLBCL lower than general Western data, and very similar to data reported from Saudi Arabia and Brazil [41, 42]. Total H-scores ranged from 0 to 190 out of a maximum possible 200. Using the AUROC, we found that the most relevant H-score value for MEF2B cutoff in the diagnosis of GC-type DLCBL would be 105, reaching a sensitivity of 87.5% and a specificity of 59.8% (Fig. 5b). With the H-score of 105, MEF2B is positive in 54 out of 111 samples of all DLCBL cases in the study (48%). Of the 34 samples in the GC group, 30 (88%) were positive for MEF2B. Out of 77 samples in the non-GC group, 32 (42%) were positive for MEF2B. In general, MEF2B expression was positively associated with BCL6 expression (R value 0.43; p value <0.01). MEF2B was also associated with CD10 expression, but to a lesser extent (R value 0.22; p value 0.01). MEF2B expression in the IHC-classified GC group demonstrates strong statistical significance (p value <0.0001).

a Pattern of expression of BCL6, CD10, and MEF2B by IHC in a normal germinal center control (left-side pictures) and in a core of DLBCL expressing the three proteins (right-side pictures). MEF2B shows nuclear expression similar to BCL6. b Receiver operating characteristic (ROC) curve for determining the cutoff value of MEF2B for diagnosing GC-DLBCL. In the plot, the highest specificity is reached at a cutoff for MEF2B positivity at an H-score of 105. The area under the ROC value is 0.76, indicating a high predictive power
Of the 24 cases used for NanoString analysis, 14 were found to be GC-DLBCL cell lines and 7 were found to be ABC-GCDLC cell lines. Three cases were unclassified. The agreement between the NanoString and IHC classification was good (r = 0.69; p< 0.05), but the direct relationship between expression of MEF2B by IHC to the NanoString subgrouping was not statistically significant (p = 0.3) (Table 1).
Analysis of the differential expression of the DLBCL GEO public datasets showed that MEF2B expression was statistically significant in all datasets (Table 2).
Discussion
In the present study, we analyzed the function and expression of MEF2B using models of DLBCL cell lines and TMAs. We addressed the mechanistic role of MEF2B in the regulation of DLBCL cell lines, where we showed for the first time that MEF2B induces BCL6 expression by acting as a BCL6 gene transcriptional co-activator, along AP-2α and BCL6 proteins. Our results indicate that knocking down MEF2B suppresses BCL6 and CD10 expression, as well as ERK1/2 phosphorylation. We also confirmed previous findings wherein MEF2B promoted the growth and proliferation of GC-derived lymphomas. We further revealed that MEF2B is preferentially expressed in the GC-DLBCL cell lines using IHC. Our results support that MEF2B may act as an oncogene in GC-DLBCL cell lines.
GEP revealed that the DLBCL disease could be classified into at least two distinct subgroups: GCB type and ABC type [2]. In addition to their prognostic distinctions, these molecular subtypes possess important differences in both their biology and the intracellular oncogenic pathways that drive their lymphomagenesis. Accordingly, the GCB type is characterized by the expression of a number of GC markers such as BCL6, LMO2, and CD10. Meanwhile, the ABC type is characterized by suppression of BCL6 expression and activation of NF-κB pathway [43]. BCL6 is essential for the development of GCs, as knocking down of BCL6 leads to failure of their development [3, 4]. Several studies have shown a parallel regulation of both MEF2B and BCL6 proteins in a panel of DLBCL cell lines [5, 6, 10]. Also, MEF2B was found to regulate BCL6-mediated expression of GC markers such as MYBL1 and LMO2 [5]. In the present study, we suggest an essential role for MEF2B protein as a co-transcriptional activator of BCL6 promoter. MEF2B protein, together with the transcriptional factor AP-2α and BCL6 protein, form a transcriptional complex that can bind to the BCL6 promoter and subsequently enhance the expression of BCL6. Consequently, the continuous expression of BCL6 accelerates the growth rate of GC-DLBCL-derived cell lines. In agreement with Ying et al. [6], our data revealed that the DLBCL cells harboring MEF2B protein show growth advantage over the MEF2B-deficient cells. Also, the knockdown of MEF2B by its specific siRNA or with a defective mutant resulted in BCL6 suppression and cell growth inhibition. This suggests the importance of MEF2B-induced BCL6 expression in the modulation of GC-DLBCL cell growth and maintenance.
Hideshima et al. [44] demonstrated that BCL6-expressing cell lines display phosphorylation of ERK1. Given that ERK is activated in the majority of B-cell lymphomas [45], it is most likely that the ERK pathway is a common pathway in lymphomagenesis. Accordingly, the phosphorylation of ERK1/2 in MEF2B-expressing cell lines and the suppression of ERK phosphorylation by MEF2B knockdown implicate that MEF2B-induced BCL6 expression may activate the ERK pathway.
In this study, IHC data revealed that the majority of GC-DLBCL cell lines were positive for MEF2B (88%) and that the association between MEF2B expression and GC phenotype using IHC was statistically significant. However, we were unable to show such significance when executing the DLBCL subgrouping using NanoString analysis. That may be attributed to the small number of cases included in the NanoString analysis (24 cases). We also showed a positive correlation between MEF2B and BCL6 expression, and a positive (yet weaker) correlation between MEF2B and CD10 expression. While Krenács et al. [12] previously reported the correlation between MEF2B and BCL6 in cases of DLBCL by IHC, they did not report a correlation between MEF2B and COO in DLBCL. Likewise, Moore et al. [11] indicated that MEF2B was not sensitive or specific for GC-DLBCL. However, it should be noted that this may be attributed to the fact that, in our study, we used an ROC-determined cutoff point, whereas Krenács et al. [12] and Moore et al. [11] used cutoff points of 50 and 20%, respectively, with no indication of the methodology that was used in choosing the cutoff percentages. The relationship between MEF2B and the GC B cells increases the possibility of MEF2B utilization as a GC marker, and may be useful as an add-on marker in the IHC algorithms developed to differentiate cases of GC-DLBCL from non-GC-DLBCL [46].
In summary, we showed that MEF2B is a member of BCL6 transcriptional complex and is likely essential for the regulation of BCL6-mediated DLBCL lymphomagenesis via the promotion of both BCL6 expression and activation of the ERK1/2 pathway. These findings indicate that MEF2B-induced BCL6 expression is essential for GC-DLBCL development. In addition to its mechanistic role in the regulation of DLBCL growth, we also confirmed that MEF2B expression is correlated with the expression of BCL6 and CD10 proteins, as well as being a potential marker for GC-DLBCL.
References
Swerdlow S, Campo E, Lee Harris N, et al. WHO classification of tumors of haematopoietic and lymphoid tissues. Lyon: IARC Press; 2008.
Alizadeh AA, Eisen MB, Davis RE, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–11.
Cattoretti G, Chang CC, Cechova K, et al. BCL-6 protein is expressed in germinal-center B cells. Blood. 1995;86:45–53.
Ye BH, Cattoretti G, Shen Q, et al. The BCL-6 proto-oncogene controls germinal-centre formation and Th2-type inflammation. Nat Genet. 1997;16:161–70.
Ding J, Dirks WG, Ehrentraut S, et al. BCL6—regulated by AhR/ARNT and wild-type MEF2B—drives expression of germinal center markers MYBL1 and LMO2. Haematologica. 2015;100:801–9.
Ying CY, Dominguez-Sola D, Fabi M, et al. MEF2B mutations lead to deregulated expression of the oncogene BCL6 in diffuse large B cell lymphoma. Nat Immunol. 2013;14:1084–92.
Basso K, Saito M, Sumazin P, et al. Integrated biochemical and computational approach identifies BCL6 direct target genes controlling multiple pathways in normal germinal center B cells. Blood. 2010;115:975–84.
Pon JR, Marra MA. MEF2 transcription factors: developmental regulators and emerging cancer genes. Oncotarget. 2016;7:2297–312.
Basso K, Brescia P, Schneider C, et al. MEF2B instructs germinal center development and acts as an oncogene in B cell lymphomagenesis. Chantilly, VA: ASH Meeting on Lymphoma Biology; 2018.
Pon JR, Wong J, Saberi S, et al. MEF2B mutations in non-Hodgkin lymphoma dysregulate cell migration by decreasing MEF2B target gene activation. Nat Commun. 2015;6:7953.
Moore EM, Swerdlow SH, Gibson SE. Comparison of myocyte enhancer factor 2B versus other germinal center-associated antigens in the differential diagnosis of B-cell non-Hodgkin lymphomas. Am J Surg Pathol. 2018;42:342–50.
Krenács D, Borbényi Z, Bedekovics J, et al. Pattern of MEF2B expression in lymphoid tissues and in malignant lymphomas. Virchows Arch. 2015;467:345–55.
Schuetz JM, Johnson NA, Morin RD, et al. BCL2 mutations in diffuse large B-cell lymphoma. Leukemia. 2012;26:1383–90.
Yang Y, Shaffer AL, Emre NC, et al. Exploiting synthetic lethality for the therapy of ABC diffuse large B cell lymphoma. Cancer Cell. 2012;21:723–37.
Camicia R, Bachmann SB, Winkler HC, et al. BAL1/ARTD9 represses the anti-proliferative and pro-apoptotic IFNÂ-STAT1-IRF1-p53 axis in diffuse large B-cell lymphoma. J Cell Sci. 2013;126:1969–80.
Hassan M, Mirmohammadsadegh A, Selimovic D, et al. Identification of functional genes during Fas-mediated apoptosis using a randomly fragmented cDNA library. Cell Mol Life Sci. 2005;62:2015–26.
Negi AK, Kansal S, Bhatnagar A, et al. Alteration in apoptosis and cell cycle by celecoxib and/or fish oil in 7,12-dimethyl benzene (α) anthracene-induced mammary carcinogenesis. Tumour Biol. 2013;34:3753–64.
El Jamal SM, Taylor EB, Abd Elmageed ZY, et al. Interferon gamma-induced apoptosis of head and neck squamous cell carcinoma is connected to indoleamine-2,3-dioxygenase via mitochondrial and ER stress-associated pathways. Cell Div. 2016;11:11.
Selimovic D, Porzig BB, El-Khattouti A, et al. Bortezomib/proteasome inhibitor triggers both apoptosis and autophagy-dependent pathways in melanoma cells. Cell Signal. 2013;25:308–18.
Selimovic D, Sprenger A, Hannig M, et al. Apoptosis related protein-1 triggers melanoma cell death via interaction with the juxtamembrane region of p75 neurotrophin receptor. J Cell Mol Med. 2012;16:349–61.
El-Khattouti A, Selimovic D, Hannig M, et al. Imiquimod-induced apoptosis of melanoma cells is mediated by ER stress-dependent Noxa induction and enhanced by NF-κB inhibition. J Cell Mol Med. 2016;20:266–86.
Hassan M, Selimovic D, Ghozlan H, et al. Induction of high-molecular-weight (HMW) tumor necrosis factor(TNF) alpha by hepatitis C virus (HCV) non-structural protein 3 (NS3) in liver cells is AP-1 and NF-kappaB-dependent activation. Cell Signal. 2007;19:301–11.
Visco C, Li Y, Xu-Monette ZY, et al. Comprehensive gene expression profiling and immunohistochemical studies support application of immunophenotypic algorithm for molecular subtype classification in diffuse large B-cell lymphoma: a report from the International DLBCL Rituximab-CHOP Consortium Program Study. Leukemia. 2012;26:2103–13.
Scott DW, Wright GW, Williams PM, et al. Determining cell-of-origin subtypes of diffuse large B-cell lymphoma using gene expression in formalin-fixed paraffin-embedded tissue. Blood. 2014;123:1214–7.
Saad AG, Grada Z, Bishop B, et al. nCounter NanoString Assay shows variable concordance With Immunohistochemistry-based Algorithms in Classifying Cases of Diffuse Large B-Cell Lymphoma According to the Cell-of-Origin. Appl Immunohistochem Mol Morphol 2018.
El Jamal SM, Yaseen AA, Alatassi H, et al. Strong NFκB expression is associated with high-grade dysplasia in barrett's esophagus. Appl Immunohistochem Mol Morphol. 2016;2:329–33.
Daems C, Martin LJ, Brousseau C, et al. MEF2 is restricted to the male gonad and regulates expression of the orphan nuclear receptor NR4A1. Mol Endocrinol. 2014;28:886–98.
McCarty KS, Miller LS, Cox EB, et al. Estrogen receptor analyses. Correlation of biochemical and immunohistochemical methods using monoclonal antireceptor antibodies. Arch Pathol Lab Med. 1985;109:716–21.
Hirsch FR, Varella-Garcia M, Bunn PA, et al. Epidermal growth factor receptor in non-small-cell lung carcinomas: correlation between gene copy number and protein expression and impact on prognosis. J Clin Oncol. 2003;21:3798–807.
Budczies J, Klauschen F, Sinn BV, et al. Cutoff Finder: a comprehensive and straightforward Web application enabling rapid biomarker cutoff optimization. PLoS ONE. 2012;7:e51862.
Tzankov A, Zlobec I, Went P, et al. Prognostic immunophenotypic biomarker studies in diffuse large B cell lymphoma with special emphasis on rational determination of cut-off scores. Leuk Lymphoma. 2010;51:199–212.
Williams PM, Li R, Johnson NA, et al. A novel method of amplification of FFPET-derived RNA enables accurate disease classification with microarrays. J Mol Diagn. 2010;12:680–6.
Shaknovich R, Geng H, Johnson NA, et al. DNA methylation signatures define molecular subtypes of diffuse large B-cell lymphoma. Blood. 2010;116:e81–89.
Hummel M, Bentink S, Berger H, et al. A biologic definition of Burkitt's lymphoma from transcriptional and genomic profiling. N Engl J Med. 2006;354:2419–30.
Salaverria I, Philipp C, Oschlies I, et al. Translocations activating IRF4 identify a subtype of germinal center-derived B-cell lymphoma affecting predominantly children and young adults. Blood. 2011;118:139–47.
Kikuchi M, Miki T, Kumagai T, et al. Identification of negative regulatory regions within the first exon and intron of the BCL6 gene. Oncogene. 2000;19:4941–5.
Pasqualucci L, Migliazza A, Basso K, et al. Mutations of the BCL6 proto-oncogene disrupt its negative autoregulation in diffuse large B-cell lymphoma. Blood. 2003;101:2914–23.
Wang X, Li Z, Naganuma A, et al. Negative autoregulation of BCL-6 is bypassed by genetic alterations in diffuse large B cell lymphomas. Proc Natl Acad Sci USA. 2002;99:15018–23.
Ohashi K, Miki T, Hirosawa S, et al. Characterization of the promoter region of human BCL-6 gene. Biochem Biophys Res Commun. 1995;214:461–7.
Hans CP, Weisenburger DD, Greiner TC, et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood. 2004;103:275–82.
Bavi P, Uddin S, Ahmed M, et al. Bortezomib stabilizes mitotic cyclins and prevents cell cycle progression via inhibition of UBE2C in colorectal carcinoma. Am J Pathol. 2011;178:2109–20.
Gualco G, Bacchi LM, Domeny-Duarte P, et al. The contribution of HGAL/GCET2 in immunohistological algorithms: a comparative study in 424 cases of nodal diffuse large B-cell lymphoma. Mod Pathol. 2012;25:1439–45.
Jovanovic MP, Mihaljevic B, Jakovic L, et al. BCL2 positive and BCL6 negative diffuse large B cell lymphoma patients benefit from R-CHOP therapy irrespective of germinal and non germinal center B cell like subtypes. J BUON. 2015;20:820–8.
Hideshima T, Mitsiades C, Ikeda H, et al. A proto-oncogene BCL6 is up-regulated in the bone marrow microenvironment in multiple myeloma cells. Blood. 2010;115:3772–5.
Bhalla S, Evens AM, Dai B, et al. The novel anti-MEK small molecule AZD6244 induces BIM-dependent and AKT-independent apoptosis in diffuse large B-cell lymphoma. Blood. 2011;118:1052–61.
Bernieh A, El Dinali M, Abulsayen H, et al. The incorporation of MEF2B in DLBCL immunostains algorithms: a tissue microarray comparative study. In Laboratory Investigation (Vol. 97, pp. 339A–339A) New York: Nature Publishing Group; 2017.
Acknowledgements
This work was supported by grants from the German Research Foundation (HA 5081/3-1) and German Cancer Research Foundation (10-2202-Ha1) to M Hassan. We would like to thank Dr. Carols Cordon-Cardo at the Icahn School of Medicine at Mount Sinai for his support to this study. We would like to thank Dr. Francisco Vega at the University of Miami and Dr. Katia Basso at the Columbia University for providing some of the reagents, nuclear extracts, and MEF2B vectors required for this study. We would also like to thank Ms. Barbara Bishop, Ms. Denise Kelley, and Ms. Melissa Peak at Special Procedures Lab at the University of Louisville for performing the IHC stains.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
El Jamal, S.M., Grada, Z., El Dinali, M.H. et al. MEF2B is a member of the BCL6 gene transcriptional complex and induces its expression in diffuse large B-cell lymphoma of the germinal center B-cell-like type. Lab Invest 99, 539–550 (2019). https://doi.org/10.1038/s41374-018-0152-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-018-0152-2
This article is cited by
-
Bob1 maintains T follicular helper cells for long-term humoral immunity
Communications Biology (2024)
-
In-depth molecular analysis of lymphomas with lymphoplasmacytic differentiation may provide more precise diagnosis and rational treatment allocation
Annals of Hematology (2024)
