
Abstract
Aspirin has been found to diminish hypertriglyceridemia and hyperglycemia in both obese rodents and patients with type 2 diabetes mellitus. We aimed to test whether low-dose aspirin can prevent obesity and the progression of non-alcoholic fatty liver disease (NAFLD) in high-risk subjects. We used offspring mice with maternal over-nutrition as a high-risk model of obesity and NAFLD. The offspring were given postnatal HF-diet and diethylnitrosamine (DEN) to induce obesity and NAFLD, and were treated with or without a low dose of aspirin for 12 weeks (ASP or CTL groups). Aspirin treatment reduced body weight gain, reversed glucose intolerance, and depressed hepatic lipid accumulation in female, but not in male mice. Female mice displayed re-sensitized insulin/Akt signaling and overactivated AMPK signaling, with enhanced level of hepatic PPAR-γ, Glut4, and Glut2, while male mice only enhanced hepatic PPAR-α and PPAR-γ levels. The female ASP mice had inhibited p44/42 MAPK activity and enhanced Pten expression, while male displayed activated p38 MAPK signaling. Furthermore, the female but not the male ASP mice reduced Wnt-signaling activity via both the epigenetic regulation of Apc expression and the post-transcriptional regulation of β-catenin degradation. In summary, our study demonstrates a sex-associated effect of low-dose aspirin on obesity and NAFLD prevention in female but not in male mice.
Similar content being viewed by others


Introduction
The rapid rise in obesity and associated diseases throughout the world now has a major negative impact on human health and healthcare resources. Regarded as the hepatic manifestation of metabolic syndrome associated with obesity, hyperinsulinemia and peripheral insulin resistance, non-alcoholic fatty liver disease (NAFLD) affects 10–24% of the general population in various countries and the prevalence is up to 75 percent in obese people [1]. NAFLD encompasses a spectrum of diseases from simple steatosis to non-alcoholic steatohepatitis (NASH), which can progress to cirrhosis and hepatocellular carcinoma (HCC). In recent years, with the changes in diet and decrease in physical activity, the population of NAFLD patients is increasing and becoming younger [2].
Recent studies have put more effort into investigating maternal over-nutrition, which reflects the dietary habits of the Western society and supports the hypothesis that maternal over-nutrition is associated with increased obesity and NAFLD. In humans, maternal obesity markedly influenced the risk of NAFLD in adolescents [3]. It was recently reported that maternal pre-pregnancy obesity and early introduction of supplementary milk is associated with a higher risk for adolescent NAFLD [3]. In a mouse model, peri-conceptional obesogenic exposure induces sex-specific programming of fatty liver in offspring [3,4,5,6,7,8]. In male offspring with maternal high-fat (HF) diet, simple steatosis was presented at the age of 30 weeks with normal chow diet; while NASH was developed as early as week 15 if exposed to postnatal HF-diet [4]. These evidence support the theory that the next generation with over-nutrition during early-development is a high-risk population for NAFLD. Thus, there is an urgent need for an effective early prevention strategy for adolescent fatty liver disease, for which current recommendations provide no guidelines.
Aspirin has been used to prevent the development of atherosclerosis due to its anti-inflammatory role and inhibition of the enzyme clyclooxygenase [9]. High-dose aspirin treatment diminishes hypertriglyceridemia in both obese rodents [10] and patients with type 2 diabetes mellitus [11]. Similarly, low-dose aspirin ameliorated hyperlipidemia induced by HF-diet in rodent models and the mechanisms involve decreased adhesion molecule and chemokine production in platelets and lymphocytes [12], and reduced VLDL-triglyceride production [13]. Regardless of the cumulative-dose response or duration-response of aspirin, a large population-based study has shown that aspirin intake is associated with a 41% lower risk of hepatocellular carcinoma [14], for which NASH has been identified as an important risk factor. These findings suggest a preventive effect of aspirin on liver injury.
In this study, we aimed to evaluate whether aspirin is effective in preventing juvenile liver injury in a NAFLD high-risk population. We used mice (offspring) from a dam of maternal HF-diet and the liver injury was induced by postnatal HF-diet and diethylnitrosamine (DEN) for 12 weeks. With this model, we provide evidence-based data to support a preventive strategy for adolescent fatty liver diseases for those who have high risk due to early exposure to over-nutrition. The effect of low-dose aspirin on preventing liver injury, glucose intolerance, and the associated molecular and cellular alterations were investigated.
Materials and methods
Chemicals, antibodies, and reagents
Diethylnitrosamine (DEN) and aspirin were purchased from Sigma Chemical Co. (St. Louis, MO, USA). Antibodies against IRS-1, phospho- IRS-1(Ser1101), NF-kappaB, phospho-NF-kappaB, AKT, phospho-AKT(Ser473), phosphor-AKT (Thr308), JNK, phospho-SAPK/JNK (Thr183/Tyr185), phospho-p38MARK (Thr180/Tyr182), phospho-p44/42 MARK (Erk1/2) (Thr202/Tyr204), AMPK-α, phospho-AMPK-α(Thr172),ACC, phospho-ACC(Ser79),β-Catenin, phospho-β-Catenin (Ser33/37/Thr41), and GAPDH were purchased from Cell Signaling Technology (USA). Antibodies against Srebp-1c, Glut4, Glut2 were from Santa Cruz Biotechnology Inc. (USA). The antibody against PPAR-α was acquired from Millipore Sigma (USA). The antibody against PPAR-γ was obtained from Abcam Co. (USA). The bicinchonic acid (BCA) protein assay kit and the phosphatase inhibitor tablets were purchased from Thermo Fisher Scientific (USA). Concentrations of the primary and secondary antibodies are described in the supplemental table.
Study design
Three-month-old mice of mixed background (B6/129/SvEv) were fed a HF-diet (60% fat) for 9 weeks before conception. The breeding pairs were given the HF-diet throughout gestation and lactation. The offspring were divided into two groups: the ASP group and the CTL group. After weaning, all offspring were given DEN and the HF-diet for 12 weeks, while the ASP group were treated with aspirin at the same time. The REF group, which were fed the control diet and were born to breeding pairs given the control diet, served as the reference. Then, they were euthanized by CO2 with the flow rate 3.1–4 LPM, and cervical dislocations were placed to ensure mice were completely dead. After that, the blood and the liver were collected for additional experiments. Mouse experiments were completed according to a protocol reviewed and approved by the Institutional Animal Care and Use Committee of Texas A&M University, in compliance with the USA Public Health Service Policy on Humane Care and Use of Laboratory Animals.
Diet and treatments
Diet was purchased from Research Diets, LLC (New Brunswick, NJ, USA). The HF-diet (cat. no. D12492) had an energy density of 5.157 kcal/g (60% fat energy, 20% carbohydrate energy and 20% protein energy). The reference diet (cat. no. D12450B) had an energy density of 3.771 kcal /g (10% fat energy, 70% carbohydrate energy, and 20% protein energy). The fat source is composed of 92% lard and 8% soybean oil. The concentration of vitamins, minerals, and proteins were modified to ensure that these nutrients in the HF-diet were equivalent to those in the NF diet on a per kilocalorie basis.
DEN treatments
DEN was injected intraperitoneally (i.p.) with 20–25 µg/g and 50 µg/l in their drink water at 21 days of age.
Aspirin treatments
Aspirin (30 mg/l in drinking water, pH 6.4) was treated in their drinking water, which was replaced with fresh water every other day. Considering that each animal drinks in average 3 to 4 ml of water per day, this would be equal to 90 µg to 120 µg aspirin per day for a mouse of 30 g weight.
Intraperitoneal-injected glucose tolerance test (IPGTT)
At the end of week 12, offspring mice from each experimental group fasted overnight and were subsequently subjected to IPGTT early the next morning. Glucose tolerance tests were conducted with 20% d-glucose in 0.9% saline.The final concentration of the administered dose was 2 g/kg body weight.Tail vein blood glucose level was measured with an automated glucometer (Bayer, Elkhart, IN) at baseline and 30, 60, and 120 min after the injection.
Analysis of serum alanine aminotransferase (ALT) activity and insulin concentration
Serums were collected and stored in deep freeze −80 °C until measured. Serum ALT activity was measured using an ALT Activity Assay kit (MAK052, Sigma-Aldrich,USA) according to the manufacturer’s instructions. The plasma concentrations of insulin were determined using an insulin enzyme-linked immunosorbent assay (ELISA) commercial kits (RayBiotech, USA) according to the manufacturer’s instructions.
Histological analysis
Liver tissues were fixed in 4% paraformaldehyde overnight at 4 °C, dehydrated with FLEX 100 (70, 80, 90, 95, and 100%), and embedded in paraffin. Specimens were sliced into sections of 5 µm thickness using a Leica RM2235 microtome. Hematoxylin and eosin (H&E) staining was performed according to a standard method. All H&E slides were blindly examined according to the Kleiner scoring system.
Protein extraction and western blot
100 mg Frozen tissue was homogenized in 1 ml of lysis buffer (RIPA10ml-Add 1 tablet of cOmplete ULTRA Tablet, 1 tablet of Phosphatase inhibitor Tablet, and 200 mM PMSF 100 µl). The lysate was then centrifuged at 12,000 rmp for 20 min, and the supernatant fraction was collected. The protein concentration was determined using a BCA protein assay. Aliquots of 150 µg were diluted in SDS sample buffer, boiled and run immediately on 7% acrylamide SDS–PAGE gels. Proteins were transferred electrophoretically to nitrocellulose membranes, and the membranes were blocked in 5% Bovine Serum Albumin (BSA)/Tris-buffered saline and reacted with primary antibody to each specific protein overnight in Tris-buffered saline plus 5% BSA. After washing three times with Tris-buffered saline containing 0.1% Tween-20, the membranes were treated with peroxidase-conjugated secondary antibodies and visualized using ECL Ultra western HRP Substrate (Millipore Sigma,USA).
RNA extraction and RT PCR
Total RNA was extracted using TRIzol reagent (Thermo Fisher Scientific, USA), and concentration was determined in triplicate using a NanoDrop ND-1000 spectrophotometer (Thermo Scientific,USA). Total mRNA (1 µg) was amplified and reversed transcribed using ReadyScript®cDNA Synthesis Mix (Sigma-Aldrich, USA), and qPCR was performed using CFX384TM Real-Time System (BIO-RAD, USA). Each reaction consisted of 0.2 µl amplified cDNA, 2 μl primers, 5 μl All-in-One qPCR Mix (Gene Copoeia Co.,USA), and nuclease-free water (final volume of 10 μl). All reactions were performed in triplicate on a Bio-Rad real-time PCR machine with the CFX Manager 3.1 software. Primers used for RT PCR analysis are shown in Table 1.
DNA extraction, bisulfite conversion, and methylation-specific PCR (MSP)
To extract genomic DNA, 20 mg frozen liver tissue was used and followed the work flow of solid tissues, as instructed by the Quick-DNATM Universal Kit (Zymo Research,USA). One microgram of genomic DNA was bisulfite converted using EZ DNA Methylation-GoldTM Kit (Zymo Research, USA) according to the manufacture’s instruction. Bisulfite converts unmethylated cytosine into uracil, whereas methylated cytosine is left unchanged. The sequences of the Apc promoter region of mouse was retrieved from (http://genome.ucsc.edu). The tested GC-rich regions is located 185 bp upstream of the Apc start codon, and appropriate primers were designed in the website http://www.urogene.org/cgi-bin/methprimer/methprimer.cgi (shown in Table 2). MSPs were performed using CFX384TM Real-Time System (BIO-RAD, USA). All methylation-specific PCRs (MSPs) were performed under the following conditions: 95 °C for 10 min, then 40 cycles of 95 °C for 10 s, 58 °C for 20 s, and 72 °C for 15 s. The CFX Manager 3.1 software (Bio-Rad) was used to measure threshold cycle (Ct) values.
Statistical analysis
Differences among the control and ASP groups across different genders were analyzed by one-way ANOVA. For the longitudinal data such as body weight and food consumption, a linear mixed model was used for the analysis of repeated measures with each individual mouse as a random effect. All analyses were carried out by using SAS JMP software (SAS Institute Inc., Cary, NC, USA) and R statistical programming language. P < 0.05 is considered significant different.
Results
Aspirin treatment prevented excess body weight gain and the glucose intolerance in female but not in the male mice
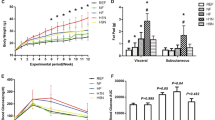
We monitored the body weight gain during the 12-week experimental period after weaning. Both male and female mice were weaned at similar body weight and significantly gained body weight by a HF-diet through the experimental period. However, low-dose aspirin treatment reduced the amount of body weight gain in female mice, but not in the male mice (Fig. 1a, b). At the end of week 12, the female mice treated with aspirin weighed significantly less than the control females (Fig. 1a).

Aspirin treatment prevented excess body weight gain and glucose intolerance in female but not male mice. a, b. Body weight of the female (a) and male (b) offspring were recorded weekly after weaning for 12 weeks. c, d An intraperitoneal injected glucose tolerance test (IPGTT) was performed at the end of week 12 in female (c) and male (d) mice. e, f Area under the curve (AUC) was calculated for the results of IPGTT in female (e) and male (f) mice. g, h Fasting insulin levels were measured at the end of week 12 in female (g) and male (h) mice. Data are presented as mean ± SD, n = 4–6. Significance (P < 0.05) is presented between two groups. *P < 0.05 vs. CTL/REF group
IPGTT was performed at the end of week 12. The fasting glucose levels of the CTL group and the ASP group were similar to that of REF mice (offspring from the dam fed normally and with postnatal chow diet), regardless of sex (Fig. 1c, d). Both the male and female offspring from overfed dams displayed glucose intolerance after a 12-week exposure to postnatal HF-diet and DEN (Fig. 1c–f, P = 0.03 vs. REF for female; P = 0.035 vs. REF for male). However, aspirin treatment prevented glucose intolerance in the female ASP group, while the male ASP mice remained glucose intolerant (Fig. 1c–f, P = 0.793 vs. REF for female; P = 0.044 vs. REF for male). In addition, we measured the fasting insulin levels at the end of week 12. ASP male mice had significantly higher serum insulin concentration vs. either the REF or the CTL mice (Fig. 1h), while the female mice’s insulin levels were not different among the REF, CTL, and ASP groups (Fig. 1g).
Aspirin treatment reduced fatty acid accumulation in female hepatocytes, but did not ameliorate NASH in male mice
Consistent with previous reports [15,16,17], we observed significant different histological and pathological changes in the female and the male liver at the end of week 12 in this study. Clearly, the male mice displayed NASH with severe hepatic steatosis and liver injury featured with balloon-like hepatocytes (Fig. 2b), while the female mice only had simple fatty acid accumulation in hepatocytes (Fig. 2a). ASP treatment reduced the amount of fatty acid accumulation in female hepatocytes (Fig. 2a); however, liver injury and hepatosteatosis in ASP male mice was not ameliorated compared to the CTL male mice (Fig. 2b). We further measured serum ALT levels which were unchanged under ASP treatment in either female or male offspring, compared to the CTL group (Fig. 2c, d).

Aspirin treatment reduced fatty acid accumulation in female hepatocytes, but did not ameliorate NASH in males. a Hematoxylin and eosin (H&E) staining shows that ASP treatment reduced the amount of fatty acid accumulation in female hepatocytes. b. H&E staining shows that liver injury and hepatosteatosis in ASP male mice were not ameliorated compared to CTL male mice. The blue arrow indicates inflammatory cell infiltration. c, d Serum ALT levels were not altered by ASP treatment in either female or male offspring compared to the CTL group in female (c) and male (d) mice. Data are presented as mean ± SD, n = 4–6. Significance (P < 0.05) is presented between two groups. *P < 0.05 vs. CTL group
Aspirin treatment re-sensitized hepatic insulin-Akt signaling in female but not male mice
The reversal of glucose intolerance in ASP female offspring drove us to measure insulin signaling activity in liver. In female livers, IRS-1 level was unchanged in the ASP group vs. the CTL group (Fig. 3a, c). However, the phosphorylation at Ser1101 of IRS-1, one of the well-known negative phosphor-regulatory sites, was depressed in the ASP group (Fig. 3a, c), suggesting re-sensitiziation of insulin signaling by aspirin treatment. Unlike the female mice, insulin signaling in male livers was not altered by aspirin treatment (Fig. 3b, d).

Aspirin treatment re-sensitized hepatic insulin-AKT signaling in female but not male mice. a, b IRS-1, phosphorylation of IRS-1 at Ser1101, Glut2 and Glut4 in liver tissue were detected by Western blotting in female (a) and male (b) mice. c, d Relative protein quantities are expressed as ratios of IRS-1/GAPDH, Phospho-IRS-1-Ser[1101]/GAPDH, Glut2/ GAPDH, and Glut4/ GAPDH in female (c) and male (d) mice. e, f Akt, phosphorylation of Akt at Thr308, phosphorylation of Akt at Ser473, in liver tissue were detected by western blotting in female (e) and male (f) mice. g, h Relative protein quantities are expressed as ratios of Akt/GAPDH, Phospho-Akt (Thr308)/GAPDH, Phospho-Akt (Ser473)/GAPDH in female (g) and male (h) mice. i Glut4 expression levels in liver was determined by real-time PCR in female and male mice. Data are presented as mean ± SD, n = 4–6. Significance (P < 0.05) is presented between two groups. *P < 0.05 vs. CTL group
We next examined whether insulin-dependent Akt signaling also displayed a higher basal activity after aspirin treatment. Consistently, the ASP female group showed hyper-phosphorylation at Thr308 of the Akt protein (Fig. 3e, g), suggesting a higher basal level of Akt signaling. However, Akt activation was not observed in male liver treated with aspirin although the expression of Akt protein was higher (Fig. 3f, h).
We further measured the expression of Glut4 and Glut2 to better understand whether aspirin promoted glucose transport in liver. Glut4 expression was significantly higher in liver of ASP female but not ASP male (Fig. 3a–d). Glut2 is the major hepatic glucose transporter, but is not insulin-responsive. ASP treatment significantly increased the level of Glut2 in female livers but not in male livers (Fig. 3a–d).
Aspirin treatment activated the AMPK signaling in female, but not male livers
We examined AMPK pathway activity to address how aspirin intervention promoted cellular lipid homeostasis in liver of the female and male mice. The expression of AMPK-α and ACC was the same in both groups regardless of sex (Fig. 4a–c). However, the p-AMPK-α levels were significantly higher in ASP group than in the CTL group of female mice, suggesting over-activation of AMPK signaling. Consistently, p-ACC/ACC was increased in the ASP group vs. the CTL group of female mice. These data suggest enhanced β-oxidation in the female hepatocytes (Fig. 4a, c). However, p-AMPK-α and p-ACC/ACC levels were unchanged in ASP male nice, compared to CTL male mice (Fig. 4b, d).

Aspirin treatment activated AMPK signaling in female, but not male livers. a, b AMPK-α, phosphorylation of AMPK-α at Thr172, ACC, and phosphorylation of ACC at Ser79 in liver tissue were detected by Western blotting in female (a) and male (b) mice. c, d Relative protein quantities are expressed as the ratios of phospho-AMPK-α (Thr172)/AMPK-α /GAPDH, Phospho- ACC (Ser79)/ACC/GAPDH in female (c) and male (d) mice. e, f Srebp1-1c, PPAR-α, and PPAR-γ in liver tissue were detected by western blotting in female (e) and male mice (f). g, h Relative protein quantities are expressed as the ratios of Srebp1-1c/GAPDH, PPAR-α/GAPDH, and PPAR-γ/GAPDH in female (g) and male (h) mice. i PPAR-γ expression levels were measured in liver by real-time PCR in female and male mice. Data are presented as mean ± SD, n = 4–6, significance (P < 0.05). *P < 0.05 vs. CTL group
Next, we measured the expression levels of three key transcription factors for lipid metabolism, Srebp-1c, PPAR-γ, and PPAR-α. Srebp-1c expression was unchanged in ASP hepatocytes, regardless of sex. Unlike the significantly higher hepatic expression of PPAR-γ but not PPAR-α seen in ASP female mice (Fig. 4e, g); the male mice significantly showed enhanced PPAR-α and PPAR-γ expression in liver (Fig. 4f, h).
Aspirin treatment enhanced activity of JNK signaling in male livers
According to the “two-hit” hypothesis, hepatic inflammation triggers the “second hit” for NAFLD progression [18]. Consistent with the histological changes in female liver that revealed the lack of hepatic inflammation, aspirin treatment had no effect on either JNK or the NF-κB signaling in female livers (Fig. 5a, c). Neutrophil infiltration was observed in the CTL and ASP male livers, thus we then determined whether aspirin activated JNK signaling and NF-κB signaling in liver. Aspirin treatment significantly activated JNK signaling via enhanced expression of JNK protein and hyper-phosphorylation of p54 JNK in male livers (Fig. 5b, d). The ASP male mice also showed increased expression of hepatic NF-κB although the phosphorylation of NF-κB, similar to the CTL male mice (Fig. 5b, d). We further detected the expression of inflammatorry genes including Il-6, Il-1β, Il-10, Tnf-α, Ccl1, Ccl2, and Il-4 by real-time PCR. However, ASP treatment did not alter the expression of any of these genes (Fig. 5e).

Aspirin treatment enhanced activity of JNK signaling in male livers. a, b. JNK, phosphorylation of JNK, NF-κB, and phosphorylation of NF-κB in liver tissue were detected by western blots in female (a) and male (b) mice. c, d. Relative protein quantities are expressed as the ratios of JNK /GAPDH, Phospho-JNK /GAPDH, NF-κB /GAPDH, Phospho-NF-κB /GAPDH in female (c) and male (d) mice. 6E. IL-6, IL-1β, IL-10, TNF-α, CCL-1, CCL-2, and IL-4 expression levels in liver were determined by real-time PCR in male mice (e). Data are presented as mean ± SD, n = 4–6, significance (P < 0.05). *P < 0.05 vs. CTL group
Aspirin treatment depressed hepatic p44/42 MAPK signaling in female mice and enhanced p38 MAPK signaling in male mice
DEN has been widely used as a chemical carcinogen to cause HCC. A previous study reported that DEN leads to carcinogenesis and cancer progression at doses above 25–30 mg/kg of body weight [19]. Although exposure to DEN at a dose of 20–25 mg/kg of body weight for 12 weeks might not be toxic enough to cause HCC, the liver injury was obvious. Therefore, we measured mitogen-activated protein kinase (MAPK) pathways in liver in order to determine the mechanisms of proliferation, differentiation, and apoptosis.
In female mice, aspirin treatment resulted in hypo-phosphorylation of p44/42 MAPK in liver exposed to a HF-diet and DEN for 12 weeks (Fig. 6a, c). Unlike the changes seen in female mice, the ASP male group showed hyper-phosphorylation of p38 MAPK (Fig. 6b, d).

Aspirin treatment depressed hepatic p44/42 MAPK signaling in female mice and enhanced p38 MAPK signaling in male mice. a, b. Phosphorylation of p44/42 MAPK and phosphorylation of p38 MAPK in liver tissue were detected by western blotting in female (a) and male (b) mice. c, d Relative protein quantities are expressed as the ratios of Phospho-p44/42 MAPK /GAPDH and Phospho-p38 /GAPDH in female (c) and male (d) mice. 6E-F. Atm, Cdk2, Cdk4, Cdk6, Chk1, Pal, Pten, P21, CycB, and CycD2 expression levels in liver were measured by real-time PCR in female (e) and male (f) mice. Data are presented as mean ± SD, n = 4–6, significance (P < 0.05). *P < 0.05 vs. CTL group
Liver expression of several important cell-cycle genes, including Atm, Cdk2, Cdk4, Cdk6, Chk1, Pal, Pten, P21, CycB, and CycD2, were determined. Aspirin treatment enhanced hepatic expression of Pten in female livers (Fig. 6e), while expression levels of these genes in male liver were unchanged (Fig. 6f).
Aspirin treatment inhibited Wnt-signaling via both epigenetic regulation of the Apc promoter region and post-transcriptional phosphorylation of β-catenin
Wnt/β-catenin signaling is known to have an important role in liver development and liver regeneration via regulation of cell-cycle progression. We thus investigated whether aspirin treatment could affect Wnt signaling via key modulator genes. In female ASP mice vs. CTL mice, there was higher expression of hepatic Apc (Fig. 7a); however, none of the Wnt modulator genes were found to be upregulated or downregulated in male ASP mice (Fig. 7b). We hypothesized that Apc upregulation was due to hypo-methylation at its CpG islands and therefore performed methylation-specific PCR. As expected, hypo-methylation of a CpG island located 185 bp upstream of the Apc start codon was found in ASP female offspring livers (Fig. 7c). In contrast, the methylation status was unchanged at this site in ASP male vs. the CTL male mice (Fig. 7d).

Aspirin treatment enhanced Apc expression in female liver via decreased DNA methylation at CpG sitsa of the Apc promoter region. a, b Hepatic expression of Wnt signaling-related genes were measured by real-time PCR in female (a) and male (b) mice. c, d Methylation status of the two CpG sites within the regulatory region of the Apc gene was measured by MSP in female (c) and male (d) mice. 7E-F.β-Catenin and phospho-β-catenin (Ser33/37/Thr41) in liver tissue were detected by western blotting in female (e) mice. Relative quantities are expressed as the ratios of β-catenin /GAPDH, phospho-β-catenin (Ser33/37/Thr41) in female (f) mice. Data are presented as mean ± SD, n = 4–6, significance (P < 0.05) *P < 0.05 vs. CTL group
We then determined whether aspirin also effects Wnt-signaling via post-transcriptional regulation of β-catenin. Protein levels and the phosphorylation status of β-catenin were determined. We found significant decreases in β-catenin expression in ASP livers compared to CTL livers (Fig. 7e, f), suggesting a potential increased degradation of this protein. The ASP liver showed increased phosphorylation at Ser33/37 and Thr41 of β-catenin (Fig. 7e, f), the major phosphorylation sites whose activation is associated with its inactivation.
Discussion
There are widely reported gender differences of hepatic phenotypes in mouse models of post-weaning diet-induced obesity (DIO) [16, 20,21,22,23,24,25,26] Low dose of aspirin has been previously studied only in male rodents with a HF-diet and was reported to reduce serum lipid content and inhibit hepatic inflammation [12, 13]. There is little information about aspirin modulating lipid metabolism in female mice. For the first time, we report that the effects of aspirin for the prevention of obesity and its related complications are different in male vs. in female mice. A low-dose aspirin regimen not only reduced body weight gain but also reversed impaired glucose tolerance in female mice; while such effects were not observed in male mice. Instead, impaired insulin sensitivity may occur in male mice, as evidenced by higher fasting serum insulin levels. This sex-associated effect is also manifested by different metabolic responses in liver: the female ASP mice displayed less fatty acid accumulation, while hepatic steatosis and liver injury remained severe in male ASP mice. Thus, one hypothesis to explain the disparity between male and female is that the effects were attributable to the more advanced NASH in males than in females; low doses of aspirin may have prevented the progression of NAFLD only in the early but not in the later stages. However, this hypothesis needs to be tested in animal models of the same sex in at different stages of NAFLD. Interestingly, this is not the first study to report the differing effects of aspirin in males and females, which has been widely reported in both human and animal models, and especially regard to its atherothrombotic benefits [27,28,29,30,31,32]. Although most studies about the sex-related differences of aspirin effects are observational, the data suggest that the contribution of the interaction of aspirin and sex hormones [33,34,35,36,37,38]. As the major organ that metabolizes sex hormones, a dysfunctional liver can greatly affect the levels and the normal function of sex hormones (reviewed in reference 39). The interactive effects of aspirin, sex hormones, and fatty liver need to be systematically tested in the future studies. Nonetheless, our data suggest that a low-dose aspirin regimen might be efficient to prevent excessive body weight gain, glucose intolerance and an early progression of NAFLD.
According to the “two-hit” hypothesis, the “first hit” initiates hepatic lipid accumulation. During the “first hit”, diminished insulin signaling by serine phosphorylation of IRS-1 and IRS-2 contributes to fat accumulation [40,41,42,43,44]. Notably, previous human data are not consistent about the effect of aspirin on insulin sensitivity. Earlier studies on diabetic patients support the theory that aspirin is responsible for insulin sensitization [45,46,47], while some recent studies in healthy subjects found a detrimental effect [48,49,50]. This difference is attributable to a higher dose and a longer duration in the former studies (6–9 g/d vs. <3 g/d and 1–3 weeks vs. a few days). In our study, we aimed to investigate the preventive effect of aspirin, thus a very low dose of aspirin (3–4 mg/kg/d) but a long-term treatment (12 weeks) was given to ASP mice daily. We demonstrated that long-term, low-dose aspirin improved insulin sensitivity via activation of insulin-Akt signaling via modulation of glucose and lipid metabolism, as evidenced by increased hepatic expression of Glut2 and PPAR-γ. These results are consistent with more recent studies in human [11] and rodent models [10] that support a role of aspirin in maintaining glucose homeostasis, although with a higher dose and for a shorter term. We also provide evidence to show that aspirin helps to maintain lipid homeostasis via promotion of lipid catabolism, through activation of AMPK signaling and enhancement of PPAR-γ, which transcriptionally regulates β-oxidation [51].
Previous studies have shown that aspirin may be useful for the treatment or prevention of a variety of cancers including colorectal cancer [52,53,54], squamous cell esophageal cancer [55,56,57] and prostate cancer [58,59,60,61], suggesting that aspirin can prevent hyper-proliferation during tumorigenesis. In our study with liver injury induced by DEN and HFD, a low dose of aspirin depressed p42/44 MAPK signaling in females. The female mice also showed enhanced expression of hepatic Pten, a well-known tumor suppressor and a well-established negative regulator of cell-cycle progression [62]. One mechanism that may explain the effect of aspirin in cancer prevention is its cyclooxygenase-independent effect on Wnt. A previous report has shown that aspirin treatment results in the increased ubiquination of β-catenin though increased phosphorylation of protein phosphatase 2 A (PP2A), which further leads to a reduction in enzymatic PP2A-activated Wnt-signaling [63]. Here we demonstrate a novel role of aspirin in the epigenetic regulation of Wnt-signaling via decreased DNA methylation, and identified the methylation site within its transcriptional regulation region. Apc protein is known to inhibit Wnt signaling by interacting with β-catenin and axin to form an Apc/Axin/β-catenin complex. We showed that the phosphorylation at Ser33/37 and Thr41 site of β-catenin further leads to its degradation, which is consistent with previous reports [64]. Our results suggeste a novel mechanism for aspirin’s cyclooxygenase-independent role in regulating Wnt signaling. Although Apc knockout is associated with HCC [65], it is not clear whether Apc overexpression is able to inhibit DEN-induced hyper-proliferation. Here we were unable to detect transcriptional changes in known proliferation-related Wnt-pathway downstream targets due to aspirin treatment, but we also could not exclude changes in unknown Wnt-pathway targets contributing to anti-hyper-proliferation. Nevertheless, our data confirm a potential role of aspirin for preventing the progression of NAFLD via downregulation of Wnt-signaling, which subsequently blocks DEN-triggered hyper-proliferation.
Our results show that aspirin does not prevent obesity, insulin resistance and NASH in high-risk male offspring. Although we detected overexpression of hepatic PPAR-α and PPAR-γ, we also showed glucose intolerance and increased fasting insulin levels in male mice, suggesting the observations might be attributable to a compensatory effect. Previous studies have shown that a high dose of aspirin activates IKKβ signaling [10, 66] and p38 MAPK signaling [67], suggesting that activated IKKβ/NFκB and p38 MAPK mediate anti-inflammatory effects and reduction in insulin resistance during liver injury. In our study, we did observe activated p38 MAPK, NFκB signaling and JNK signaling in male ASP offspring mice. However, expression of inflammatory cytokine genes in liver were not responsible for aspirin treatment in our study. Thus, the role of aspirin in mediating insulin sensitivity and hepatic fat accumulation-induced injury requires more comprehensive studies.
In summary, we adapted a mouse model with high risk of NAFLD to examine the preventive effect of low-dose aspirin on obesity and NAFLD, which is different in male vs female mice. Aspirin prevented body weight gain, glucose intolerance and lipid accumulation in female mice via sensitization of insulin/Akt signaling, activation of AMPK signaling, inhibition of MAPK signaling and Wnt-signaling, through both epigenetic regulation and post-transcriptional modification (Fig. 8). However, these effects of low-dose aspirin were not observed in male mice with NASH.

Schematic diagram of the mechanism for prevention of NAFLD progression by a low-dose aspirin regimen. Long-term low-dose of aspirin treatment in female mice re-sensitized insulin/Akt signaling via reducing phosphorylation at Ser1101 of IRS-1 and hyper-phosphorylation at Thr308 of Akt, resulting in increased glucose homeostasis via enhanced expression of hepatic Glut4, Glut2, and hepatic PPAR-γ. In female mice, aspirin helped maintain lipid homeostasis via promotion of lipid catabolism through activation of AMPK signaling and enhancement of PPAR-γ. Aspirin treatment suppressed cell proliferation by inhibiting the MAPK signaling via hypo-phosphorylation of p44/42 MAPK in liver and upregulation of Apc gene expression via hypo-methylation of a CpG island located 185 bp upstream of the Apc start codon. Overexpression of Apc further inhibited Wnt signaling by hyper-phosphorylating β-catenin at Ser33/37/Thr41 to form an Apc/Axin/β-catenin complex, which leads to β-catenin degradation
References
Miele L, Forgione A, Hernandez AP, Gabrieli ML, Vero V, Di Rocco P, et al. The natural history and risk factors for progression of non-alcoholic fatty liver disease and steatohepatitis. Eur Rev Med Pharmacol Sci. 2005;9:273–7.
Charlton M. Nonalcoholic fatty liver disease: a review of current understanding and future impact. Clin Gastroenterol Hepatol. 2004;2:1048–58.
Ayonrinde OT, Oddy WH, Adams LA, Mori TA, Beilin LJ, de Klerk N, et al. Infant nutrition and maternal obesity influence the risk of non-alcoholic fatty liver disease in adolescents. J Hepatol. 2017;67:568–76.
Bruce KD, Cagampang FR, Argenton M, Zhang J, Ethirajan PL, Burdge GC, et al. Maternal high-fat feeding primes steatohepatitis in adult mice offspring, involving mitochondrial dysfunction and altered lipogenesis gene expression. Hepatology. 2009;50:1796–808.
Ashino NG, Saito KN, Souza FD, Nakutz FS, Roman EA, Velloso LA, et al. Maternal high-fat feeding through pregnancy and lactation predisposes mouse offspring to molecular insulin resistance and fatty liver. J Nutr Biochem. 2012;23:341–8.
Dahlhoff M, Pfister S, Blutke A, Rozman J, Klingenspor M, Deutsch MJ, et al. Peri-conceptional obesogenic exposure induces sex-specific programming of disease susceptibilities in adult mouse offspring. Biochim Biophys Acta. 2014;1842:304–17.
Kruse M, Seki Y, Vuguin PM, Du XQ, Fiallo A, Glenn AS, et al. High-fat intake during pregnancy and lactation exacerbates high-fat diet-induced complications in male offspring in mice. Endocrinology. 2013;154:3565–76.
Li J, Huang J, Li JS, Chen H, Huang K, Zheng L. Accumulation of endoplasmic reticulum stress and lipogenesis in the liver through generational effects of high fat diets. J Hepatol. 2012;56:900–7.
Maranhao RC, Leite AC Jr. Development of anti-atherosclerosis therapy based on the inflammatory and proliferative aspects of the disease. Curr Pharm Des. 2015;21:1196–204.
Yuan M, Konstantopoulos N, Lee J, Hansen L, Li ZW, Karin M, et al. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science. 2001;293:1673–7.
Hundal RS, Petersen KF, Mayerson AB, Randhawa PS, Inzucchi S, Shoelson SE, et al. Mechanism by which high-dose aspirin improves glucose metabolism in type 2 diabetes. J Clin Invest. 2002;109:1321–6.
Lin HL, Yen HW, Hsieh SL, An LM, Shen KP. Low-dose aspirin ameliorated hyperlipidemia, adhesion molecule, and chemokine production induced by high-fat diet in Sprague-Dawley rats. Drug Dev Res. 2014;75:97–106.
van Diepen JA, Vroegrijk IO, Berbee JF, Shoelson SE, Romijn JA, Havekes LM, et al. Aspirin reduces hypertriglyceridemia by lowering VLDL-triglyceride production in mice fed a high-fat diet. Am J Physiol Endocrinol Metab. 2011;301:E1099–107.
Sahasrabuddhe VV, Gunja MZ, Graubard BI, Trabert B, Schwartz LM, Park Y, et al. Nonsteroidal anti-inflammatory drug use, chronic liver disease, and hepatocellular carcinoma. J Natl Cancer Inst. 2012;104:1808–14.
Burcelin R, Dolci W, Thorens B. Glucose sensing by the hepatoportal sensor is GLUT2-dependent: in vivo analysis in GLUT2-null mice. Diabetes. 2000;49:1643–8.
Hwang LL, Wang CH, Li TL, Chang SD, Lin LC, Chen CP, et al. Sex differences in high-fat diet-induced obesity, metabolic alterations and learning, and synaptic plasticity deficits in mice. Obesity. 2010;18:463–9.
Fu Q, Olson P, Rasmussen D, Keith B, Williamson M, Zhang KK, et al. A short-term transition from a high-fat diet to a normal-fat diet before pregnancy exacerbates female mouse offspring obesity. Int J Obes. 2016;40:564–72.
Byrne CD, Olufadi R, Bruce KD, Cagampang FR, Ahmed MH. Metabolic disturbances in non-alcoholic fatty liver disease. Clin Sci. 2009;116:539–64.
Verna L, Whysner J, Williams GM. N-nitrosodiethylamine mechanistic data and risk assessment: bioactivation, DNA-adduct formation, mutagenicity, and tumor initiation. Pharmacol Ther. 1996;71:57–81.
Cecconello AL, Trapp M, Hoefel AL, Marques CV, Arbo BD, Osterkamp G, et al. Sex-related differences in the effects of high-fat diets on DHEA-treated rats. Endocrine. 2014;48:985–94.
Bellisario V, Berry A, Capoccia S, Raggi C, Panetta P, Branchi I, et al. Gender-dependent resiliency to stressful and metabolic challenges following prenatal exposure to high-fat diet in the p66(Shc-/-) mouse. Front Behav Neurosci. 2014;8:285.
Hariri N, Thibault L. High-fat diet-induced obesity in animal models. Nutr Res Rev. 2010;23:270–99.
Deeb SS, Zambon A, Carr MC, Ayyobi AF, Brunzell JD. Hepatic lipase and dyslipidemia: interactions among genetic variants, obesity, gender, and diet. J Lipid Res. 2003;44:1279–86.
Mischke M, Pruis MG, Boekschoten MV, Groen AK, Fitri AR, van de Heijning BJ, et al. Maternal Western-style high fat diet induces sex-specific physiological and molecular changes in two-week-old mouse offspring. PLoS ONE. 2013;8:e78623.
Cecconello AL, Trapp M, Hoefel AL, Marques CV, Arbo BD, Osterkamp G, et al. Sex-related differences in the effects of high-fat diets on DHEA-treated rats. Endocrine. 2015;48:985–94.
Hwang LL, Wang CH, Li TL, Chang SD, Lin LC, Chen CP, et al. Sex differences in high-fat diet-induced obesity, metabolic alterations and learning, and synaptic plasticity deficits in mice. Obesity. 2010;18:463–9.
Adelman EE, Lisabeth L, Brown DL. Gender differences in the primary prevention of stroke with aspirin. Women’s Health. 2011;7:341–52. quiz52-3
Amateau SK, Alt JJ, Stamps CL, McCarthy MM. Brain estradiol content in newborn rats: sex differences, regional heterogeneity, and possible de novo synthesis by the female telencephalon. Endocrinology. 2004;145:2906–17.
Ayala DE, Hermida RC. Sex differences in the administration-time-dependent effects of low-dose aspirin on ambulatory blood pressure in hypertensive subjects. Chronobiol Int. 2010;27:345–62.
Kelton JG, Hirsh J, Carter CJ, Buchanan MR. Sex differences in the antithrombotic effects of aspirin. Blood. 1978;52:1073–6.
Morikawa M, Kojima T, Inoue M, Tsuboi M. Sex difference in the inhibitory effect of aspirin on prostacyclin production of rat aortae. Jpn J Pharmacol. 1984;35:1–7.
Menguy R, Desbaillets L, Masters YF, Okabe S. Evidence for a sex-linked difference in aspirin metabolism. Nature. 1972;239:102–3.
Albert O, Desdoits-Lethimonier C, Lesne L, Legrand A, Guille F, Bensalah K, et al. Paracetamol, aspirin and indomethacin display endocrine disrupting properties in the adult human testis in vitro. Hum Reprod. 2013;28:1890–8.
Bauer SR, Fortner RT, Gates MA, Eliassen AH, Hankinson SE, Tworoger SS. Analgesic use in relation to sex hormone and prolactin concentrations in premenopausal women. Cancer Causes Control. 2013;24:1087–97.
Didolkar AK, Gurjar A, Joshi UM, Sheth AR, Roychowdhury D. Effects of aspirin on blood plasma levels of testosterone, LH and FSH in maturing male rats. Int J Androl. 1980;3:312–8.
Duggan C, Wang CY, Xiao L, McTiernan A. Aspirin and serum estrogens in postmenopausal women: a randomized controlled clinical trial. Cancer Prev Res. 2014;7:906–12.
Gates MA, Tworoger SS, Eliassen AH, Missmer SA, Hankinson SE. Analgesic use and sex steroid hormone concentrations in postmenopausal women. Cancer Epidemiol Biomark Prev. 2010;19:1033–41.
Kristensen DM, Lesne L, Le Fol V, Desdoits-Lethimonier C, Dejucq-Rainsford N, Leffers H, et al. Paracetamol (acetaminophen), aspirin (acetylsalicylic acid) and indomethacin are anti-androgenic in the rat foetal testis. Int J Androl. 2012;35:377–84.
Gawrieh S. Sex hormones, sex hormone-binding globulin, and liver fat: which came first, the chicken or the egg? Clin Gastroenterol Hepatol. 2015;13:1694–6.
Kodama Y, Brenner DA. c-Jun N-terminal kinase signaling in the pathogenesis of nonalcoholic fatty liver disease: Multiple roles in multiple steps. Hepatology. 2009;49:6–8.
Tarantino G, Caputi A. JNKs, insulin resistance and inflammation: A possible link between NAFLD and coronary artery disease. World J Gastroenterol. 2011;17:3785–94.
Singh R, Wang Y, Xiang Y, Tanaka KE, Gaarde WA, Czaja MJ. Differential effects of JNK1 and JNK2 inhibition on murine steatohepatitis and insulin resistance. Hepatology. 2009;49:87–96.
Solinas G, Vilcu C, Neels JG, Bandyopadhyay GK, Luo JL, Naugler W, et al. JNK1 in hematopoietically derived cells contributes to diet-induced inflammation and insulin resistance without affecting obesity. Cell Metab. 2007;6:386–97.
Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120:649–61.
Reid J, Lightbody TD. The insulin equivalence of salicylate. Br Med J. 1959;1:897–900.
Reid J, Macdougall AI, Andrews MM. Aspirin and diabetes mellitus. Br Med J. 1957;2:1071–4.
Williamson RT. On the treatment of glycosuria and diabetes mellitus with sodium salicylate. Br Med J. 1901;1:760–2.
Bratusch-Marrain PR, Vierhapper H, Komjati M, Waldhausl WK. Acetyl-salicylic acid impairs insulin-mediated glucose utilization and reduces insulin clearance in healthy and non-insulin-dependent diabetic man. Diabetologia. 1985;28:671–6.
Giugliano D, Sacca L, Scognamiglio G, Ungaro B, Torella R. Influence of acetylsalicylic acid on glucose turnover in normal man. Diabete Metab. 1982;8:279–82.
Newman WP, Brodows RG. Aspirin causes tissue insensitivity to insulin in normal man. J Clin Endocrinol Metab. 1983;57:1102–6.
Varga T, Czimmerer Z, Nagy L. PPARs are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation. Biochim Biophys Acta. 2011;1812:1007–22.
Cook NR, Lee IM, Zhang SM, Moorthy MV, Buring JE. Alternate-day, low-dose aspirin and cancer risk: long-term observational follow-up of a randomized trial. Ann Intern Med. 2013;159:77–85.
Friis S, Riis AH, Erichsen R, Baron JA, Sorensen HT. Low-dose aspirin or nonsteroidal anti-inflammatory drug use and colorectal cancer risk: a population-based, case-control study. Ann Intern Med. 2015;163:347–55.
Rothwell PM, Wilson M, Elwin CE, Norrving B, Algra A, Warlow CP, et al. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet. 2010;376:1741–50.
Corley DA, Kerlikowske K, Verma R, Buffler P. Protective association of aspirin/NSAIDs and esophageal cancer: a systematic review and meta-analysis. Gastroenterology. 2003;124:47–56.
Malik S, Ullah S, Afzal M, Lal K, Haque S. Clinical and descriptive genetic study of polydactyly: a Pakistani experience of 313 cases. Clin Genet. 2014;85:482–6.
Zhang L, Wu YD, Li P, Tu J, Niu YL, Xu CM, et al. Effects of cyclooxygenase-2 on human esophageal squamous cell carcinoma. World J Gastroenterol. 2011;17:4572–80.
Bosetti C, Rosato V, Gallus S, La Vecchia C. Aspirin and prostate cancer prevention. Recent Results Cancer Res. 2014;202:93–100.
Moyad MA, Vogelzang NJ. Heart healthy equals prostate healthy and statins, aspirin, and/or metformin (S.A.M.) are the ideal recommendations for prostate cancer prevention. Asian J Androl. 2015;17:783–91.
Vidal AC, Freedland SJ. Aspirin and prostate cancer prevention. Aging. 2015;7:292–3.
Vidal AC, Howard LE, Moreira DM, Castro-Santamaria R, Andriole GL, Freedland SJ. Aspirin, NSAIDs, and risk of prostate cancer: results from the REDUCE study. Clin Cancer Res. 2015;21:756–62.
Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, et al. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39.
Bos CL, Kodach LL, van den Brink GR, Diks SH, van Santen MM, Richel DJ, et al. Effect of aspirin on the Wnt/beta-catenin pathway is mediated via protein phosphatase 2A. Oncogene. 2006;25:6447–56.
Yost C, Torres M, Miller JR, Huang E, Kimelman D, Moon RT. The axis-inducing activity, stability, and subcellular distribution of beta-catenin is regulated in Xenopus embryos by glycogen synthase kinase 3. Genes Dev. 1996;10:1443–54.
Colnot S, Decaens T, Niwa-Kawakita M, Godard C, Hamard G, Kahn A, et al. Liver-targeted disruption of Apc in mice activates beta-catenin signaling and leads to hepatocellular carcinomas. Proc Natl Acad Sci USA. 2004;101:17216–21.
Yin MJ, Yamamoto Y, Gaynor RB. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature. 1998;396:77–80.
Oshima T, Miwa H, Joh T. Aspirin induces gastric epithelial barrier dysfunction by activating p38 MAPK via claudin-7. Am J Physiol Cell Physiol. 2008;295:C800–6.
Acknowledgements
This project is supported by grants from the National Institutes of Health (NIDDK 1R01DK112368-01 to LX and KZ). This work is supported by the USDA National Institute of Food and Agriculture, [Hatch] project [1010406] to LX.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Zhou, Y., Peng, H., Liu, Z. et al. Sex-associated preventive effects of low-dose aspirin on obesity and non-alcoholic fatty liver disease in mouse offspring with over-nutrition in utero. Lab Invest 99, 244–259 (2019). https://doi.org/10.1038/s41374-018-0144-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-018-0144-2
This article is cited by
-
BPDE-DNA adduct formation and alterations of mRNA, protein, and DNA methylation of CYP1A1, GSTP1, and GSTM1 induced by benzo[a]pyrene and the intervention of aspirin in mice
Environmental Science and Pollution Research (2023)
