
Abstract
During the past few years, considerable evidence has uncovered a strong relationship between fat and bone metabolism. Consequently, alterations in plasma lipid metabolic pathways strongly affect bone mass and quality. We recently showed that the deficiency of apolipoprotein A-1 (APOA1), a central regulator of high-density lipoprotein cholesterol (HDL-C) metabolism, results in reduced bone mass in C57BL/6 mice. It is documented that apolipoprotein E (APOE), a lipoprotein know for its atheroprotective functions and de novo biogenesis of HDL-C, is associated with the accumulation of fat in the liver and other organs and regulates bone mass in mice. We further studied the mechanism of APOE in bone metabolism using well-characterized APOE knockout mice. We found that bone mass was remarkably reduced in APOE deficient mice fed Western-type diet (WTD) compared to wild type counterparts. Static (microCT-based) and dynamic histomorphometry showed that the reduced bone mass in APOΕ−/− mice is attributed to both decreased osteoblastic bone synthesis and elevated osteoclastic bone resorption. Interestingly, histologic analysis of femoral sections revealed a significant reduction in the number of bone marrow lipoblasts in APOΕ−/− compared to wild type mice under WTD. Analyses of whole bone marrow cells obtained from femora of both animal groups showed that APOE null mice had significantly reduced levels of the osteoblastic (RUNX2 and Osterix) and lipoblastic (PPARγ and CEBPα) cardinal regulators. Additionally, the modulators of bone remodeling RANK, RANKL, and cathepsin K were greatly increased, while OPG and the OPG/RANKL ratio were remarkably decreased in APOΕ−/− mice fed WTD, compared to their wild-type counterparts. These findings suggest that APOE deficiency challenged with WTD reduces osteoblastic and lipoblastic differentiation and activity, whereas it enhances osteoclastic function, ultimately resulting in reduced bone mass, in mice.
Similar content being viewed by others

Introduction
Osteoporosis (OP) is a metabolic condition characterized by reduced bone mass and disturbances in bone architecture that eventually lead to bone fragility and thus susceptibility for fractures [1, 2]. Since the common form of OP is the consequence of bone cell aging, OP constitutes a significant public health problem worldwide. Indeed, it is estimated that more than 44 million people suffer from OP or low bone mass in the United States alone [3]. Not surprisingly several studies have focused on the clarification of the mechanism that govern the pathogenesis of OP. Notably, the past few years several epidemiological and animal model studies have documented a tight link between lipid metabolism and bone homeostasis [4,5,6]. Towards this direction it has been proposed that perturbation of high-density lipoprotein cholesterol (HDL) metabolic pathway are associated with reduced bone in mice [7,8,9]. Indeed, we recently demonstrated that the deficiency of apolipoprotein A-1 (APOA1) the principal component of HDL biogenesis, results in great reduction of bone mass in mice; importantly, this effect is primarily associated with reduced osteoblastic activity and not with elevated osteoclastic function [10]. Moreover, APOA1 knockout (KO) mice are characterized by augmented bone marrow adiposity, a finding that highlights the importance of bone marrow white fat in the regulation osteoblastic function and thus bone mass maintenance [10]. Other components of the HDL metabolic cassette, namely the lipid transporter, ATP-binding cassette 1 (ABCA1) and the major HDL receptor scavenger receptor class B type I (SRB1) have also been found to be differentially involved in the pathobiology of OP [11].
In addition to APOA1, apolipoprotein E (APOE) also participates in the de novo biogenesis of HDL [12]. APOE is a 34.5 kDa glycoprotein produced by the liver and other peripheral organs. It mediates the clearance of triglyceride rich lipoproteins from circulation via the LDL receptors, and serves as a significant structural element of HDL. Therefore, its main functions are atheroprotection and maintenance of plasma lipid homeostasis, since APOE mediates the cellular uptake of chylomicron remnants, very low-density lipoprotein and low-density lipoprotein and their clearance from the circulation [13, 14]. Notably, Ackert-Bicknell et al. [15] recently reported that APOE, together with PPARγ, ESR1, and IL6, can regulate both HDL and bone mineral density. Nonetheless, the role of APOE in bone pathophysiology remains enigmatic. Histomorphometrical, biomechanical, and in vitro approaches on mouse models by Schilling et al. [16] showed that the APOE-deficient mice display increased osteoblastic function and augmented bone formation rate. Surprisingly, however, a more recent study by the same research group showed that bone mass of APOE KO mice is remarkably reduced following the consumption of high fat (diabetogenic) diet for 16 weeks [17], adding to the complexity of the relationship between APOE and bone metabolism.
Triggered by the current knowledge and recent data from our research team, in the present study we aimed at further exploring the puzzling mechanism of APOE in the regulation of bone metabolism. For this reason we chose to use APOE-/- and wild-type (WT) C57BL/6 mice fed with Western-type diet (WTD) that resembles the dietary habits and corresponds to the average daily energy consumption seen in western societies. Applying histological, histomorphometrical, biochemical, and molecular methods we examined whether APOE deficiency affects the expression of factors that are involved in the regulation of osteoblasts, lipoblasts, and osteoclasts under the influence of WTD. We uncovered that following WTD, the APOE null mice display remarkably reduced bone mass and bone marrow adiposity compared to their WT litermates. Moreover, the expression levels of factors that regulate osteoblastogenesis, osteoclastogenesis, and lipogenesis were greatly, but differentially, affected, indicating that APOE is requirement for normal skeletal homeostasis and development.
Materials and methods
Animal studies
We employed APOE deficient (APOΕ−/−) mice backcrossed on the C57BL/6 mice for ten generations and their legitimates as control (Jackson Labs, Bar Harbor, Maine, USA). Mice were genotyped with PCR of tail DNA samples. Animals were kept on a 12 h light cycle in a pathogen-free environment, caged individually (one mouse per cage) and allowed unrestricted access to food and water. At 6 weeks of age animals were switched to WTD (17.3% protein, 48.5% carbohydrates, 21.2% fat, 0.2% cholesterol, and 4.5 Kcal g−1), ad libitum in a 12 h dark/light cycle (7 am − 7 pm light) for 24 weeks. The age and starting body-weights of the tested animals were similar. Body weight measurements were performed every 6 weeks. Αt the end of the 24th week, the animals were sacrificed by CO2 inhalation, followed by cervical dislocation and femora and lumbar vertebrae were surgical isolated for histological and μCT analyses. Additionally, bone marrow was flashed and cells were isolated for molecular analyses. All animal studies were performed according to the EU guidelines for the Protocol for the Protection and Welfare of Animals. The estimated sample size was determined by power analysis, using the tool (http://www.stat.ubc.ca/~rollin/stats/ssize/n2.html). The work was approved by the committee of the Laboratory Animal Center of The University of Patras Medical School and the Veterinary Authority of the Prefecture of Western Greece. All animal experiments comply with the ARRIVE guidelines and have been carried out in accordance with the U.K. Animals (Scientific Procedures) Act, 1986 and associated guidelines, EU Directive 2010/63/EU for animal experiments, or the National Institutes of Health guide for the care and use of Laboratory animals (NIH Publications No. 8023, revised 1978).
Histological and histomorphometrical analyses
Mice were injected with calcein (25 μg g−1 of animal weight) according to a standard double-labeling protocol, including injection at day 7 and 2 before sacrifice [10] for the determination of new bone formation rate. Fluorochrome measurements were made using Fluorescence Microscope (Nikon TE2000 inverted phase-fluorescence microscope with a 12 bit 1600 × 1200 pixel CCD). Tartrate-resistant acid phosphatase (TRAP) was labeled using 0.01% naphthol phosphate substrate (Sigma Chemical Company, St Louis, MO, USA) in N-N-dimethylformamide (Sigma Chemical Company), followed by the addition of 5 mg ml−1 fast red violet in 50 mM sodium tartrate and 90 mM sodium acetate at pH 5.0.
Following sacrifice femora and lumbar vertebrae were collected and stored either at −80 °C or fixed in 4% formalin (Merck, NJ, USA) overnight at 4 °C and stored at 70% ethanol at −20 °C until used. Fixed sections were decalcified with ethylenediaminetetraacetic acid (EDTA), embedded in paraffin and sectioned at 4 μm for histological evaluation. Conventional hematoxylin and eosin (H&E) stain was applied for the assessment of bone marrow histology.
Fixed samples were mounted on the CT specimen tube for scanning at a resolution of 12 μm, with a slice increment of 10 μm, using a micro-CT Scanner (Scanco vivaCT 40 in vivo μCT scanner, Scanco Medical AG). For dynamic histomorphometry, undecalcified frozen bone tissues were sectioned at 4 μm on a Microm HM 505E cryostat using tape transfer (CryoJane Instrumetics, St. Luis, MO).
Whole bone marrow cell isolation
Femora was removed under aseptic conditions from C57BL/6 and APOΕ−/− mice, and whole bone marrow cells (WBMCs) were flushed using a 26G syringe filled with cell isolation media (CIM) (RPMI-1640 with 10% FBS, 1% Pen/Strep (Gibco, UK). Following red blood cell lysis in ammonium chloride (BD Pharm LyseTM Lysing Buffer, cat no. 555899, BD Biosciences Pharmingen) and centrifugation at 20 °C for 5 min, WBMCs were further assessed for molecular analysis.
Spectrometric analysis
Right femora from four WT and four APOΕ−/− animals were used for the Raman analysis. Bone samples were rinsed with ultra pure water (MilliQ, Millipore) for formalin solution cleaning. Bone samples were maintained at −20 °C until use.
Raman analysis acquired focusing a 785 nm laser through an objective lens (20×) onto femora using an in Via Raman microscope (Renishaw, UK). The laser power was 35 mW on the edge of the objective lens. Raman emission from 700–1800 cm−1 was scanned. The time required was about 2 min and the resolution was 2 cm−1. Spectra were collected from the periosteal surface of the entire diaphysis. The amide I (1590–1710 cm−1) region in the Raman spectrum was isolated for further analysis. Sub-bands were derived by the second-derivative method. Further analysis included deconvolution and curve fitting of the sub-bands to unconstrained Gaussian-Lorentzian profiles used Peakfit software (Jandel Scientific, San Rafael, CA). Raman intensities (areas) were measured. The bands at 1668 and 1690 cm−1 were to evaluate the cross-linking and the quality of collagen [18].
Protein extraction and western blot analysis
For the protein extraction, WBMCs were isolated from the bone marrow of WT and APOΕ−/− mice and washed with sterile, cold PBS and then lysed with RIPA buffer (0.1% SDS, 1% octylphenoxypolyethoxyethanol, 1% sodium deoxycholate, 10 mM Tris-HCL, pH 7.5, 150 mM NaCl, and 0.5 mM EDTA) in the presence of a protease and phosphatase inhibitor cocktail (Sigma-Aldrich Corp., St. Louis, MO, USA). Protein extracts were mixed with 2× Laemmli buffer (1 M Tris-HCl, pH 6.8, 10% 2-mercaptoethanol, 10% SDS, 50% glycerol, and 0.01% bromophenol blue) and boiled for 5 min. The protein content of cell homogenates was determined using NanoDrop, and equal amounts of protein were subjected to sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis. After electrophoresis, proteins were transferred to immobilon fluoride (PVDF) membranes (Millipore Corporation, Billerica, MA, USA) at constant current of 200 mA at 4 °C for 1 h in tris-glycine. The membranes were blocked with 5% dry skimmed milk in phosphate buffer saline-Tween 20 and incubated overnight with the appropriate primary antibodies, including PPARγ (sc7273, mouse monoclonal antibody, Santa Cruz Biotechnology, Santa Cruz, CA, USA) and RUNX2 (sc10758 (M-70), rabbit polyclonal, Santa Cruz Biotechnology, Santa Cruz, CA, USA). Incubation with secondary antibody (conjugated to horseradish peroxidase goat anti-mouse IgG-HRP, sc-2005, Santa Cruz or goat anti-rabbit IgG-HRP, AP132P, Millipore, Billerica, MA, USA) was 1 h at room temperature. Antibody binding was visualized using enhanced chemiluminescence detection system (SuperSignal, ThermoScientific, Rockford, IL, USA). The results are expressed in relation to actin mouse monoclonal antibody MAB1501 (Millipore).
Flow cytometric analysis
For flow cytometric analysis, the cells washed with phosphate-buffered saline, fixed and permeabilized using Cytofix-Cytoperm kit (cat. no. 554714, BD Biosciences, NJ, USA) for 20 min at 4 °C). Cells were labeled with the primary antibodies [for PPARγ, cat no. sc7273 mouse monoclonal, RUNX2, cat no. sc-10758 (M-70) rabbit polyclonal, both from Santa Cruz Biotechnology, Santa Cruz, CA, USA)] for 1 h at 4 °C. Cells were then incubated with secondary antibodies conjugated with Alexa Fluor 488 (goat anti-mouse Alexa Fluor 488, cat no. A11001 and goat anti-rabbit Alexa Fluor 488, cat no. A11008, Molecular Probes, LifeTechnologies, MA, USA) for 1 h at 4 °C and then subjected to flow cytometric analysis. The samples (at least 100,000 cells) were analyzed by EPICS-XL/ Coulter flow cytometer. Data were analyzed using FlowJo software (Tree Star Inc., Ashland, OR). Values are expressed as arbitrary units of mean fluorescence intensity (MFI) relateve to the isotype control of every single experiment (AU = MFIH sample-MFIisot.ct/ MFIHisot.ct).
RNA extraction, cDNA synthesis, and real-time-PCR
Total RNA was extracted from WBMCs obtained from WT and APOΕ−/− mice using silica membrane spin columns, NucleoSpin RNA (MACHEREY-NAGEL, Duren, Germany), as described by the manufacturer. Extracted RNA was treated with RNase-free DNase (MACHEREY-NAGEL, Duren, Germany) to remove contaminating genomic DNA. The concentration of total RNA was determined using a nanodrop spectrophotometer (TECAN, Switzerland). First-strand cDNA was synthesized from total RNA using PrimeScript reverse transcriptase (PrimeScript 1st strand cDNA Synthesis kit, 6110 A, TaKaRa Biotechnology, Japan). Real time PCR was performed using the MX3000P apparatus (Stratagene, La Jolla, CA). Primer sets of target and housekeeping genes (Table 1) were from VBC BIOTECH (Austria). Polymerase chain reaction amplification was carried out in a final volume of 20 μl including 10 µl of premixed SYBR green, NTPs, buffer, and polymerase (KAPA SYBR® FAST qPCR Kit, KAPA Biosystems, Boston, US), plus 1 µl of each primer (20 pmol μl−1) and 1 μl of first strand cDNA (5 ng). PCR was performed according to the following protocol: 95 °C for 5 min 1 cycle, followed by 35 cycles of 95 °C for 10 sec and 60 °C for 30 sec, and 1 cycle of 95 °C 10 sec, 60 °C 2 min, 95 °C 0 sec continuous, and finally 1 cycle cooling at 40 °C for 30 sec. Each reaction was run in duplicate. The obtained CT (cycle number at which amplification threshold of detection was reached) values were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The mean ΔΔCT was converted to relative expression value by the equation 2–ΔΔCt, were ΔΔCt = ΔCt (sample) – ΔCt (calibrator) and ΔCt is the Ct of the housekeeping gene (GAPDH) subtracted from the Ct of the target gene. As assessed by melting curve analysis, no primer-dimer was obtained for any primer sets, and the specificity of the products was also confirmed by agarose electrophoresis. The primer sets are shown in Table 1.
Statistical analysis
Statistical analysis and comparison performed using Student’s t test. Data are reported as mean ± standard deviation (mean ± SD). Differences were considered statistically significant at p ≤ 0.05. All analyses were performed using GraphPad Prism (version 5.03, GraphPad Software, San Diego, CA, USA).
Results
Body weight measurements
Consistent with our published results [19], APOE deficient mice were less sensitive to the development of diet-induced obesity than WT litermates [20]. Indeed, during the course of the experiment, APOE KO challenged with WTD mice showed only a modest increase in body weight (Fig. 1a, b). More specifically, at week 6 of the experiment, the APOΕ−/− mouse group had an average body weight of 26.7 ± 0.6 g (5.52 ± 1.45% increase relative to their starting weight of 25.7 ± 0.2 g at week 0, p < 0.05). At week 12, their average body weight was 30.7 ± 1.1 g and, at week 24, it showed a further slight increase to 31.6 ± 1.7 g (19.7 ± 7.3% increase relative to their starting weight at week 0, p < 0.05) (Fig. 1a). In contrast, WT mice showed a much more remarkable elevation in their body weight as a function of time on WTD. In fact, at week 6, they had an average body weight of 31.8 ± 1.7 g (23.5 ± 3.9% increase relative to their starting weight of 25.8 ± 1 g at week 0, p < 0.05). At week 12, their body weight reached 35.3 ± 0.6 g and, at week 24, their body weight was 42.8 ± 1.7 g (66.7 ± 5.6% increase relative to their starting weight at week 0, p < 0.05) (Fig. 1a). The other body characteristics and dimensions were not affected.

Effect of western type died on body weight and bone composition in APOE deficient mice and wild type controls. In Fig.1, mean ± SD, N = 5. a, b APOE deficient mice fed WTD mice display significantly reduced body weight in comparison to C57BL/6 WTD mice at week 6, 12, 18, and 24 of the experiment (a). Note the remarkable differenced in the physical appearance between mice that belong to the two experimental groups (b). c Hematoxylin and eosin stain on sections obtained from mouse femora showed that the APOE deficient mice consumed WTD for 24 weeks had virtually no BM adipocytes, in sharp contrast to their WT legitimates (bone marrow adipocytes are pointed by yellow arrows) (original magnification 4×). d Collagen cross-linking study for control WTD and knockout WTD mice, determined by Raman spectroscopy, revealed that the APOE deficient mice fed WTD had a substantial increase (41%) in the collagen cross-links, compared to the C57BL/6 WTD mice. Statistical significance was considered at p value less than 0.05
APOE deficiency results in reduced bone mass and bone marrow adiposity after the consumption of WTD
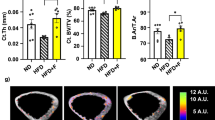
Micro computed analyses on undecalcified sections of mice lumbar vertebrae and femora (Fig. 2a–c) showed a significant reduction in the bone mass of the APOΕ−/− as compared to the C57BL/6 mice fed WTD. Indeed, static histomorphometry parameters such as bone volume/total volume, trabecular thickness, and trabecular number were greatly decreased, while trabecular separation was significantly increased in APOΕ−/− mice (Fig. 2b–d). Dynamic histomorphometry on trabecular bone unveiled significant differences in the bone formation rate between the two groups. Particularly, calcein labeled and double-labeled surfaces were greatly reduced in the APOΕ−/− compared to the C57BL/6 mice on WTD (Fig. 3 a, b). On the contrary, the number of the TRAP positive cells on bone surfaces was significantly augmented in the APOΕ−/−as compared to the WT mice fed WTD (Fig. 3c, d). The biochemical composition of mice femora was evaluated by Raman spectroscopy. We found that the collagen cross-linking was substantially increased (41%) in the KO compared to the WT mice (p < 0.05) (Fig. 1d).

Effect of APOE deficiency on bone mass and architecture by micro computerized tomography. Static histomorphometric analyses with the use of microCT on mice lumbar vertebrae (a, b) and femora (c, d) revealed significant differences between the APOE deficient and the C57BL/6 mice fed WTD in all the static parameters examined [BV/TV: bone volume/total volume, thickness of bone trabeculae, number of bone trabeculae/spicules, distance between separate bone trabeculae (trabecular separation)]. Bone quality and architecture were remarkably impaired in the APOE knock-out mice (*p < 0.05, **p < 0.01)

Effect of APOE deficiency on bone formation and resorption by dynamic histomorphometry. Reduced bone formation and increased osteoclast resorption in the lumbar vertebrae of APOE deficient mice that were fed WTD mice relatively to C57BL/6 WTD. a, b Calcein labeled surfaces (green lines) and calcein double-labeling (yellow arrows) are more intense in the vertebrae of the C57BL/6 WTD compared to the APOE knockout mice (original magnification 20×). c, d TRAP stain showed increased TRAP activity in the APOE deficient compared to C57BL/6 WTD mice (the TRAP positive cells/areas are colored red) (original magnification 10×)
APOE deficiency differentially affects factors that regulate both the osteoblastic and the osteoclastic differentiation following exposure to WTD
To further explore the impact of APOE on bone mass, we tested the expression of a set of molecules that are known to modulate bone remodeling. First, we examined the expression levels of RUNX2, the master regulator of osteoblastic differentiation, in WBMCs isolated from bone marrow cells obtained from mice femora. In parallel with the histomorphometrical results, we found that runt-related transcription factor 2 (RUNX2) was significantly reduced in the APOΕ−/− compared to their control counterparts fed WTD at both protein [Western-blotting analysis (p = 0.048), flow cytometry (p = 0.015)], and mRNA [RT-PCR (p = 0.0006)] levels (Figs. 4 and 5).

Comparison of osteoblast- and osteoclast-related mRNA expression in APOE deficient and wild type animals. All graphs indicate mean ± SD, N = 5. Each real time PCR result is expressed as fraction of the wild type normalized to unity. The expression levels of the osteoblast-related genes RUNX2, Osterix, and COL1α1, the RANK/RANKL/OPG complexes, the osteoclast-related genes TRAP and cathepsin K, as well as the lipoblastic regulators, PPARγ and CEBPα, are graphically demonstrated. Detail analysis is presented in the “results” section

Western blots of key bone and lipoblast regulatory proteins in APOE deficient and wild type mice. a Western blot results from RUNX2 and PPARγ analyses, normalized to actin expression. The protein levels of both the osteoblastic (RUNX2) and the lipoblastic regulator (PPARγ) were significantly reduced to the APOE deficient mice challenged with WTD, in comparison to the control mice. b Determination of intracellular RUNX2 and PPARγ protein levels in WΒΜCs by flow cytometry presented as median fluorescent intensities (MFI). Histograms of count and intensity are shown on the left; summaries of quantitative data on mean PPARγ and RUNX2 antibody labeling are depicted on the right
In line with the aforementioned results, we uncovered that the expression levels of Osterix, another major regulator of osteoblastic function, were considerably elevated “touching” the threshold of statistical significance (p = 0.05). In addition, we assessed the the mRNA levels of receptor activator of nuclear factor kappa-B (RANK), a receptor that is expressed in the surface of osteoclasts, as well as the expression levels of its ligand (RANKL) and osteoprotegerin (OPG), a decoy receptor for RANKL, molecules that are are produced by bone marrow osteoblasts/stromal cells and are involved in the regulation of osteoclast differentiation. We found that the expression levels of RANK and its ligand RANKL were significantly increased in the APOΕ−/− mice compared to their WT counterparts (Fig. 4). On the contrary, OPG mRNA levels as well as the RANKL/OPG ratio were significantly decreased in the APOΕ−/− in comparison to C57BL/6 mice (Fig. 4). The mRNA levels of TRAP (Acp5 gene) were increased (13.3%) in the APOΕ−/− group, even though this reduction did not reach significance (p = 0.2). Notably, however, the expression levels of Cathepsin K (Ctsk gene), a central factor of osteoclastic-bone resorbing function, were greatly augmented (p = 0.009) (Fig. 4a). In line with the results from Raman studies, the expression levels of collagen type 1a were increased (38.5%) in the APOΕ−/− in comparison to the WT mice. Overall, these findings suggest APOE deficiency results in alterations of bone remodeling that differentially affect both the osteoblastic and the osteoclastic functions, eventually leading to bone mass reduction in mice challenged with WTD.
APOE deficiency restrains lipogenesis in the BM of mice following consumption of WTD
Histological analysis of sections obtained from the femora of C57BL/6 mice fed WTD revealed the presence of numerous adipocytes (Fig.1c). Surprisingly, however, BM from femora obtained from the APOΕ−/− had virtually no adipocytes, even though these animals were also challenged with WTD for the same period of time (Fig.1c). In line with this histological finding, molecular analyses on WBMCs revealed that the master regulator of lipoblastogenesis PPARγ and CCAAT/enhancer-binding protein (CEBPA), a molecule involved in adipocyte differentiation, were greatly reduced in the APOE null compared to their WT counterparts, at both mRNA and protein level (Figs. 4a and 5a, b).
Discussion
The role of APOE in bone metabolism is puzzling, yet intriguing [11]. Indeed, morphologic and histomorphometric experiments on KO mice have shown that APOE deficiency is associated with elevated bone mass and increased osteoblastic function [16]. The authors suggested that this stems from the fact that APOE deficiency is associated with reduced uptake of lipoproteins that contain of vitamin K by osteoblasts [21]. Since vitamin K is crucial for osteocalcin carboxylation, it is most plausible that decreased APOE levels result in bone mass elevation, a phenotype which is similar to that encountered in osteocalcin null mice [21, 22]. Interestingly, however, APOE deficient mice that were stressed with very high-fat diabetogenic diet for a period of 16 weeks displayed substantially reduced bone mass followed by normal weight and lowered levels of serum insulin, glucose, and leptin [17].
The effects of APOE on metabolism have also attracted our interest. In fact, we recently demonstrated that APOE serves as a key peripheral contributor to hepatic lipid homeostasis and has a protective role against the accumulation of fat in the liver, thus against the development of diet-induced nonalcoholic fatty liver disease [19, 23].
Triggered by the aforementioned findings, in the present study we aimed at further exploring the role of APOE on bone homeostasis. In contrast to previous studies that used diabetogenic very high-fat diets [17], in the present project we chose to use WTD for 24 weeks, which is very close to the daily dietary habits of Western world. Applying static, microCT-based histomorphometry we found that under the influence of WTD, the bone mass of the APOE deficient mice is greatly reduced compared to the WT mice. Dynamic histomorphometry showed that the APOE-/- mice had substantially reduced bone formation rate compared to WT counterparts. This finding is in line with the study of Bartet et al. [17], having shown that the consumption of diabetogenic diet also results in reduced bone mass in APOE null mice. We next examined whether this effect on bone mass was associated with alterations in the biochemical characteristics of mouse femora. Notably, Raman analysis showed that collagen cross-linking was significantly increased (41%) in the knock-out compared to the WT mice. This may arise from an increase in the amount of the non-reducible (trivalent) cross-links due to transformation of the reducible (divalent), cross-links. Therefore, fibers get more efficiently interconnected and collagen network becomes stiffer and more rigid, thus losing its elasticity. A similar behaviour (44% increase of collagen cross-linking ratio) has been reported for female wistar rats due to severe OP induced through ovariectomy [18]. In symphony with Raman results, the expression levels of collagen type 1a were increased in the APOE knock out compared to WT groups.
Further to investigate the impact of APOE deficiency on bone, we studied the molecular signature of WBMC from APOΕ−/− and WT animals fed WTD. It is well established that bone mass and quality are determined by the balance between osteoblastic bone formation and osteoclastic bone resorption, a finely-tuned process that is called the “coupling phenomenon” [24, 25]. Osteoblasts secrete RANKL a molecule that binds RANK a receptor that resides on osteoclast cell membrane. The formation of the RANK-RANKL complexes promote osteoclast differentiation and activation, and hence bone resorption [26, 27]. Except from RANKL, osteoblast also produce and secrete osteoprotegerin (OPG) a soluble RANKL decoy receptor, which prevents RANKL from acting at distant sites as a scavenger, inhibiting osteoclast formation and maturation [26, 28]. Obviously, increased OPG/RANKL ratio prevents osteoclast formation and consequently bone resorption. In the present study, we found that the osteoblast master regulators, RUNX2 and Osterix, as well as the OPG/RANKL ratio were greatly reduced in the APOΕ−/− mice fed WTD in comparison to their WT counterparts, consistent with impaired osteoblastic differentiation and augmented osteoclastic activation.
TRAP stain on undecalcified mice femora revealed that TRAP positive surface was significantly increased in the APOE-/- mice compared to the WT, a finding that is compatible with enhanced osteoclastic activity. In line with the aforementioned findings, our molecular analysis on WBMC unveiled that the mRNA levels of TRAP and Cathepsin K, were also increased in the APOE KO group stressed with WTD, suggesting elevated osteoclastic maturation and function.
Taken together, our findings, provide evidence that further supports the observation that APOE deficiency under the influence of WTD has a strong impact on bone mass. Moreover, our data, in combination with the findings of Schilling et al. showing that ApoE deficient mice fed chow diet develop a high-bone mass phenotype, introduces the intriguing possibility that dietary habits, and in particular increased fat intake (WTD), differentially affect both bone synthesis and degradation, leading to the reduction of bone mass.
Under the influence of specific modulators mesenchymal stem cells follow different lines of differentiation giving genesis to osteoblasts, lipoblasts, fibroblasts, chondroblasts, and myoblasts. It is widely accepted that osteoblast and lipoblasts represent “the two different sides of the same coin” [29, 30]. For this reason in the present study in addition to the osteoblast-related molecules we also examined the expression levels of PPARγ, the master regulator of lipoblastic differentiation [31, 32]. Interestingly, our results showed that the lipoblastic modulators, PPARγ and CEBPA, were greatly reduced in the APOΕ−/− mice fed WTD, compared to the control group at both mRNA and protein level. This novel finding implies that following stress with WTD the APOE-dependent effects are mediated, at least in part, through PPARγ-mediated pathways. Furthermore, since our results showed that APOE deficiency is also associated with reduced osteoblastic differentiation and activation, we suggest that lack of APOE curbs the differentiation of mesenchymal stem cells at early stages, before the phase of committed osteoblasts or lipoblast. This explains the histological finding on femora sections that the bone marrow of the APOE null mice has virtually no osteoblasts. Consistent with decreased committed adipoblasts and BM adiposity the weight of the APOE KO mice fed WTD was significantly reduced in comparison to the weight of the C57BL/6 WTD mice fed chow diet.
A rational question that stems from our study is what component of WTD is most effective on bones, since the composition of WTD is quite complex. A recent metanalysis of data from 18 countries from five continents, suggested that high carbohydrate intake was associated with higher risk of total mortality, whereas total fat and individual types of fat were related to lower total mortality [33]. Total fat and types of fat were not associated with cardiovascular disease, myocardial infarction, or cardiovascular disease mortality, whereas saturated fat had an inverse association with stroke. In light of this information, in our study we compared the composition of the two diets used. Both chow and western type diets were commercially available diets. The content of the main ingredients of the chow diet used in our studies was 24% protein, 55% carbohydrate from crude fibers, 3.5% oils and fats, 7.5% crude ash. On the other hand the composition of the western type diet fed to the mice is 17.3% protein, 48.5% carbohydrate, 21.2% fat, 0.2% cholesterol. Based on this rough comparison, it appears that in our study fat and cholesterol are the contributing diet factors to the modulation of osteoblast, osteoclast and lipoblast differentiation and activation under the impact of APOE deficiency. Future studies with custom diets should be performed in order to dissect the precise role of each component on the observed phenotype.
Collectively the present study provides additional evidence that dietary fat modulates osteoblastic, osteoclastic and lipoblastic differentiation, and activity in a background of ΑPOE deficiency. The precise molecular mechanisms that govern this effect are still vague and warrant further exploration. It is possible that APOE exerts a direct effect on bone marrow and/or an indirect effect through regulation of triglyceride-rich lipoprotein deposition to bone marrow. Apparently, however, APOE deficiency challenged with WTD leads to alterations in signaling pathways that converge on mesenchymal stem cells affecting both osteoblastic and lipoblastic differentiation and activation. On the other other hand, it may fire signal transduction cascades that promote hematopoietic stem cells progression towards functional osteoclasts (Fig. 6).

Schematic illustration of our hypothesis as regards the effect of APOE deficiency on mice stressed with WTD. At the upper part of the figure are depicted the effects of WTD on wild-type mice. Note that in the bone marrow there is a considerable number of lipoblasts and that the bone spicules are lined by both osteoblasts and osteoclasts. The lower half of the figure displays the significant reduction in the number of osteoblasts, lipoblasts and bone spicules in the bone marrow of APOE KO mice fed WTD for 24 weeks. The increased or decreased effect on the distinct types of bone marrow cells is represented by thicker or thinner arrows and increased or decreased number of cells (osteoclasts, osteoblasts, lipoblasts) and bone spicules. HSC hematopoietic stem cells, LBL lipoblast, MSC mesenchymal stem cell, OBL osteoblast, WTD Western-type diet
Additional in vitro and biomechanical studies will be required to demonstrate the complete effect of WTD on osteoblastogenesis/osteoclastogenesis and bone strength of mice lacking APOE. Nonetheless, by expanding the current knowledge on bone and fat connection, our findings raise the intriguing possibility that APOE may serve as a potential target for the treatment of OP, obesity, and related pathologies.
References
Marini F, Brandi ML. Pharmacogenetics of osteoporosis: what is the evidence? Curr Osteoporos Rep. 2012;10:221–7.
Sobieszczańska M, Jonkisz J, Tabin M, et al. Osteoporosis: genetic determinants and relationship with cardiovascular disease. Adv Clin Exp Med. 2013;22:119–24.
Dempster DW. Osteoporosis and the burden of osteoporosis-related fractures. Am J Manag Care. 2011;17:S164–S169.
Papachristou DJ, Blair HC. Bone and high-density lipoprotein: the beginning of a beautiful friendship. World J Orthop. 2016;7:74–77.
Zhao LJ, Jiang H, Papasian CJ, et al. Correlation of obesity and osteoporosis: effect of fat mass on the determination of osteoporosis. J Bone Miner Res. 2008;23:17–29.
Triantaphyllidou I-E, Kalyvioti E, Karavia E, et al. Perturbations in the HDL metabolic pathway predispose to the development of osteoarthritis in mice following long-term exposure to western-type diet. Osteoarthr Cartil. 2013;21:322–30.
Colaianni G, Brunetti G, Faienza MF, et al. Osteoporosis and obesity: role of Wnt pathway in human and murine models. World J Orthop. 2014;5:242–6.
Yamauchi M, Yamaguchi T, Nawata K, et al. Increased low-density lipoprotein cholesterol level is associated with non-vertebral fractures in postmenopausal women. Endocrine. 2015;48:279–86.
Lin YY, Chen CY, Chuang TY, et al. Adiponectin receptor 1 regulates bone formation and osteoblast differentiation by GSK-3β/β-catenin signaling in mice. Bone. 2014;64:147–54.
Blair HC, Kalyvioti E, Papachristou NI, et al. Apolipoprotein A-1 regulates osteoblast and lipoblast precursor cells in mice. Lab Invest. 2016;96:763–72.
Papachristou NI, Blair HC, Kypreos KE, et al. High-density lipoprotein (HDL) metabolism and bone mass. J Endocrinol. 2017;233:R95–R107.
Kypreos KE, Zannis V. Pathway of biogenesis of apolipoprotein E-containing HDL in vivo with the participation of ABCA1 and LCAT. Biochem J. 2007;403:359–67.
Kypreos KE, Karagiannides I, Fotiadou EH, et al. Mechanisms of obesity and related pathologies: role of apolipoprotein E in the development of obesity. FEBS J. 2009;276:5720–8.
Kypreos KE, Teusink B, Van Dijk KW, et al. Analysis of the structure and function relationship of the human apolipoprotein E in vivo, using adenovirus-mediated gene transfer. FASEB J. 2001;15:1598–1600.
Ackert-Bichnell CL. HDL cholesterol and bone mineral density: is there a genetic link? Bone. 2012;50:525–33.
Schilling AF, Schinke T, Münch C, et al. Increased bone formation in mice lacking apolipoprotein E. J Bone Miner Res. 2005;20:274–82.
Bartelt A, Beil T, Schinke T, et al. Apolipoprotein E-dependent inverse regulation of vertebral bone and adipose tissue mass in C57Bl/6 mice: modulation by diet-induced obesity. Bone. 2010;47:736–45.
Orkoula MG, Vardaki MZ, Kontoyannis CG. Study of bone matrix changes induced by osteoporosis in rat tibia using Raman spectroscopy. Vib Spectrosc. 2012;63:404–8.
Karavia E, Papachristou DJ, Kotsikogianni I, et al. Deficiency in apolipoprotein E has a protective effect on diet-induced nonalcoholic fatty liver disease in mice. FEBS J. 2011;278:3119–29.
Lecka-Czernik B. Marrow fat metabolism is linked to the systemic energy metabolism. Bone. 2012;50:534–9.
Niemeier A, Schinke T, Heeren J, et al. The role of apolipoprotein E in bone metabolism. Bone. 2012;50:518–24.
Newman P, Bonello F, Wierzbicki AS, et al. The uptake of lipoprotein-bome phylloquinone (vitamin K1) by osteoblasts and osteoblast-like cells. Role of heparan sulfate proteoglycans and apolipoprotein E. J Bone Miner Res. 2002;17:426–33.
Karavia EA, Papachristou NI, Sakellaropoulos GC, et al. Scavenger receptor class B type I regulates plasma apolipoprotein E levels and dietary lipid deposition to the liver. Biochemistry. 2015;54:5605–516.
Suda T, Takahashi N, Udagawa N. Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr Rev. 1999;20:345–57.
Walsh MC, Choi Y. Biology of the RANKL-RANK-OPG system in immunity, bone, and beyond. Front Immunol. 2014;5:511.
Martin TJ, Sims NA. RANKL/OPG; Critical role in bone physiology. Rev Endocr Metab Disord. 2015;16:131–9.
Kobayashi Y, Udagawa N, Takahashi N. Action of RANKL and OPG for osteoclastogenesis. Crit Rev Eukaryot Gene Expr. 2009;19:61–72.
Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337–42.
Takada I, Kouzmenko AP, Kato S. Molecular switching of osteoblastogenesis versus adipogenesis: implications for targeted therapies. Expert Opin Ther Targets. 2009;13:593–603.
Harada S, Rodan GA. Control of osteoblast function and regulation of bone mass. Nature. 2003;423:349–55.
Takada I, Kouzmenko AP, Kato S. Wnt and PPARγ signaling in osteoblastogenesis and adipogenesis. Nat Rev Rheumatol. 2009;5:442–7.
Zhang Y, Shao J, Wang Z, et al. Growth differentiation factor 11 is a protective factor for osteoblastogenesis by targeting PPARgamma. Gene. 2015;557:209–14.
Dehghan M, Mente A, Zhang X, et al. Associations of fats and carbohydrate intake with cardiovascular disease and mortality in 18 countries from five continents (PURE): a prospective cohort study. Lancet. 2017;390:2050–62.
Acknowledgements
This work was supported in part by the Marie Curie Reintegration Grant (IRG), FP7-PEOPLE-RG-2009, #256402-«apoeostearthritis»; the University of Patras, Faculty of Medicine KARATHODORI Research Grants D.155 and E.073; the “ARISTIA I” of Hellenic GSRT Grant 248; the Department of Veteran’s Affairs Grant BX002490 and by National Institutes of Health (USA) grants AR055208, and AR065407. EAK is a fellow of the “Hellenic Scholarship Foundation” (IKY). This research study is an activity of the research network OsteoNet, of the University of Patras. We thank Dr. C.G. Kontoyannis and Dr. M.G. Orkoula (School of Pharmacy, University of Patras, Greece) for assistance with Raman spectometry.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Papachristou, N.I., Blair, H.C., Kalyvioti, E.S. et al. Western-type diet differentially modulates osteoblast, osteoclast, and lipoblast differentiation and activation in a background of APOE deficiency. Lab Invest 98, 1516–1526 (2018). https://doi.org/10.1038/s41374-018-0107-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-018-0107-7
This article is cited by
-
Lipid metabolism within the bone micro-environment is closely associated with bone metabolism in physiological and pathophysiological stages
Lipids in Health and Disease (2022)
-
High-density Lipoprotein Cholesterol Is Negatively Correlated with Bone Mineral Density and Has Potential Predictive Value for Bone Loss
Lipids in Health and Disease (2021)
-
The high-density lipoprotein receptor Scarb1 is required for normal bone differentiation in vivo and in vitro
Laboratory Investigation (2019)
