
Abstract
Dysregulated autophagy may be a central player in trehe pathogenesis of primary biliary cholangitis (PBC) by inducing autoimmune processes via abnormal expression of mitochondrial antigens such as pyruvate dehydrogenase complex, E2 component (PDC-E2) and also by inducing cellular senescence in biliary epithelial cells (BECs) in bile duct lesions in PBC. We examined the association of an impaired “biliary bicarbonate umbrella” due to dysfunction of anion exchanger 2 (AE2) with dysregulated autophagy and cellular senescence in PBC. The expression of AE2 was examined in cultured BECs treated with bile acids such as glycochenodeoxycholic acid (GCDC) and tauro-ursodeoxycholic acid (TUDCA), various cytokines (IL-4, IL-13, IFNγ, TNFα, TGFβ), and serum deprivation. The effect of AE2 knockdown using siRNA on autophagy, cell surface expression of PDC-E2, and cellular senescence was also examined. The expression of AE2 and its association with autophagy-related markers and senescent markers p16INK4a and p21WAF1/Cip1 were immunohistochemically determined in livers taken from the patients with PBC (n = 50) and 69 control diseased and normal livers. The expression of AE2 was significantly induced in the cultured BECs shortly treated with GCDC and other stresses, whereas it was significantly decreased in senescent BECs induced by GCDC and other stresses (p < 0.05). Dysregulated autophagy, cell surface expression of PDC-E2, and cellular senescence were significantly increased by knockdown of AE2 (p < 0.05). The expression of AE2 was significantly decreased in cholangitis in PBC, compared to control livers (p < 0.05). The decreased expression of AE2 was correlated with dysregulated autophagy, abnormal expression of PDC-E2, and cellular senescence in bile duct lesions in PBC. In conclusion, an impaired biliary bicarbonate umbrella may be involved in the pathogenesis of PBC by inducing dysregulated autophagy.
Similar content being viewed by others


Introduction
Primary biliary cholangitis (PBC) is an autoimmune cholestatic liver disease presenting with chronic, progressive cholestasis, biliary fibrosis, and liver failure [1,2,3]. PBC is characterized by a high frequency of serum anti-mitochondrial antibodies (AMAs) and unique cholangitis in small bile ducts referred as chronic non-suppurative destructive cholangitis (CNSDC) [1, 4]. Several genome-wide association studies (GWAS) revealed that PBC is associated with specific HLA and non-HLA genes relating immune systems, such as T cell activation [5, 6]. However, there are still many enigmas concerning the pathogenesis of PBC. For example, how do AMAs play a role in the pathogenesis of bile duct lesions [1,2,3,4, 7,8,9]? We previously reported that dysregulated autophagy might be a central player in the pathogenesis of bile duct lesions in PBC by inducing autoimmune process via abnormal expression of mitochondrial antigens and also cellular senescence in biliary epithelial cells (BECs) in bile duct lesions in PBC [10,11,12,13,14,15,16,17,18]. However, the mechanisms of dysregulated autophagy were not explored in our previous studies.
Accumulating evidence suggests that anion exchanger 2 (AE2) plays a central role in maintaining a “biliary bicarbonate umbrella” for protecting biliary mucosa and that dysfunctional AE2 may be related to the pathogenesis of PBC [19,20,21,22]. An impaired “biliary bicarbonate umbrella” due to dysfunction of AE2 is supposed to be critical for the toxicity of hydrophobic bile acids, such as glycochenodeoxycholic acid (GCDC), and other stresses to BECs [19, 23]. For example, Ae2a,b-deficient mouse is reportedly PBC model mice presenting serum AMA, increase of IgM, progressive cholestasis, and portal inflammation [22]. Upregulation of miR-506 is reportedly related to the decreased expression of AE2 in PBC [24].
We hypothesized that an impaired “biliary bicarbonate umbrella” may be related to dysregulated autophagy, followed by abnormal expression of mitochondrial antigens and cellular senescence in biliary epithelial lesions in PBC. In this study, we examined the association of the expression of AE2 and dysregulated autophagy, abnormal expression of mitochondrial antigens, and cellular senescence in PBC.
Materials and methods
Culture study
Cell culture and treatments
Mouse intrahepatic BECs were isolated from 8-week-old female BALB/c mice and were purified and cultured as described previously [25]. The cell density of the cells was less than 80% during experiments. In several experiments, BECs were treated with GCDC, deoxycholic acid (DCA), ursodeoxycholic acid (UDCA), tauro-UDCA, Etoposid (Sigma-Aldrich, St Louis, MO), and various cytokines (IL-4, IL-13, IFNγ, TNFα, TGFβ) (R&D, Minneapolis, MN). In some experiments, BECs were treated with serum deprivation for induction of autophagy, as described previously [10].
Knockdown of AE2 by small interfering RNA (siRNA)
Validated siRNA for AE2 and negative control siRNA were purchased from Santa-Cruz biotech (Santa-Cruz, CA, USA) and QIAGEN (Hilden, Germany), respectively. One day before transfection, BECs were plated in 35-mm dishes (5 × 105 cells) or Lab-Tek chambers (5 × 104 cells/well), and then the cells were transiently transfected with either AE2 or control siRNA (100 nM) using Lipofectamine 3000 (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol. Bafilomycin A1 (Baf, 5 nM) was used to assess autophagy flux.
Real-time quantitative reverse transcriptase-polymerase chain reaction (RT-PCR)
Total RNA was extracted from the cells with a QIAGEN RNeasy Mini kit (QIAGEN) according to the manufacturer’s protocol. After cDNA was synthesized, quantitative real-time PCR was performed to measure AE2 and β-actin mRNA according to a standard protocol using the SYBR Green PCR Master Mix (Toyobo, Tokyo, Japan). Primers used are shown in Table 1. Each experiment was performed twice in triplicate, and the mean was adopted in each experiment.
Immunoblotting
The cell lysate samples (10 μg) were resolved by SDS-PAGE and transferred to a nitrocellulose membrane as described previously. [26] After transfer, the membranes were processed for immunoblotting as described previously [26]. The primary antibodies used are shown in Table 2. Densitometry of the resulting bands was performed using Image-J software and normalized to the loading control.
Immunofluorescence staining for cultured cells
The BECs growing in a Lab-Tek chamber were fixed and immunostained using the primary antibodies for AE2, PDC-E2, LC3, or p62 (Table 2), as described previously [10]. A similar dilution of the control mouse or rabbit Immunoglobulin G (IgG) (Dako) was applied instead of the primary antibody as a negative control. The cells were then incubated with Alexa-488-labeled or Alexa-592-labeled anti-mouse or rabbit IgG (Molecular probes, Eugene, OR) for 30 min, counterstained with 10 µg/ml 4′,6-diamidino-2-phenylindole (DAPI) and evaluated under a conventional fluorescence microscope (Olympus, Tokyo, Japan).
Assessment of cell-surface expression of mitochondrial antigen
The cell-surface expression of PDC-E2 was detected in the absence of membrane-perforating agent. Cultured BECs treated with AE2 siRNA or control siRNA were fixed in buffered 10% formalin, then immunostained with anti-PDC-E2, as described previously [10]. The level of cell surface mitochondrial antigen expression was evaluated semiquantitatively by Image J software.
Assay for cellular senescence
SA-β-gal activity was detected by using the senescence detection kit (Bio Vision, Mountain View, CA) according to manufacturer’s protocol [27]. The proportion of senescent cells in each condition was assessed at Day 4 by counting the percentage of SA-β-gal-positive cells in at least 1 × 103 total cells using light microscopy. In addition, cellular senescence was assessed by SenTraGorTM system (Arriani pharmaceuticals, Attica, Greece) according to manufacturer’s protocol [28]. This is a sensitive method for identification of senescent cells using an analog of Sudan Black B histochemical dye coupled with biotin (GL13) [28].
Human study
Classification of intrahepatic biliary tree
The intrahepatic biliary tree is classified into intrahepatic large and small bile ducts (septal and interlobular bile ducts) by their size and distributions in the portal tracts [29]. Bile ductules, which are characterized by tubular or glandular structures with a poorly defined lumen and located at the periphery of the portal tracts [29, 30], are not included in the small bile ducts.
Liver tissue preparation
A total of 119 liver tissue specimens (all were biopsied or surgically resected) were collected from the liver disease file of our laboratory and affiliated hospitals. The Ethics Committee of Kanazawa University approved this study. The liver specimens in this study were 50 PBC, 8 primary sclerosing cholangitis (PSC), 38 chronic viral hepatitis (CVH), 10 livers with extrahepatic biliary obstruction (EBO), and 13 “histologically normal” livers. All PBC were from patients fulfilling the clinical, serological, and histological characteristics consistent with the diagnosis of PBC [1]. PBC livers were staged histologically [1], and 25 and 15 of PBC were stages 1, 2 (early PBC) and stages 3, 4 (advanced PBC), respectively. Fifty PBC livers were divided into 38 CA3 (presence of CNSDC) and 12 CA0-2 (absence of CNSDC), respectively, according to Nakanuma classification [31]. Nineteen and 19 CVH were regarded as F0-2 and as F3, 4, respectively [32]. Eight and 30 of CVH cases were serologically positive for hepatitis B surface antigen and anti-hepatitis C viral antibody, respectively. Causes of EBO were obstruction of the bile duct at the hepatic hilum or the extrahepatic bile ducts, because of stone or carcinoma, and the duration of jaundice was less than 1 month. “Histologically normal” livers were obtained from surgically resected livers for traumatic hepatic rupture or metastatic liver tumor. Normal liver tissues were obtained from an area apart from the tumor, and carcinoma tissues were not evaluated. Chemotherapy was not performed before liver resection in all patients with histologically normal liver.
Liver tissue samples were fixed in 10% neutral-buffered formalin, and embedded in paraffin. More than 20 serial sections, 4 µm thick, were cut from each block. Several sections were processed routinely for histologic study, and the remainder was processed for the subsequent immunohistochemistry.
Immunohistochemistry
We examined immunohistochemically the expression of AE2 using the primary antibody shown in Table 2, as described previously [14]. A similar dilution of the control mouse or rabbit IgG (Dako) was applied instead of the primary antibody as a negative control. Positive and negative controls were routinely included. Histological analysis was performed in a blinded manner. For the assessment of AE2 expression in small bile ducts, the extent of expression was evaluated as increase, no change, and decrease, compared with small bile ducts in normal liver.
Double immunofluorescence
We also performed double immunofluorescence for AE2 with autophagy related markers (LC3 and p62), senescent markers (p16INK4a and p21WAF1/Cip1), and mitochondrial antigen PDC-E2. Antibodies used are shown in Table 2. In brief, either LC3, p62, PDC-E2, p16INK4a, or p21WAF1/Cip1 was detected using Vector Red Alkaline Phosphatase Substrate Kit (Vector Lab, Burlingame, CA), followed by second staining for AE2 using Alexa-488-labeled anti-rabbit IgG. The sections were counterstained with DAPI and evaluated under a conventional fluorescence microscope.
Statistical analysis
Statistical analysis of differences was performed using the Kruskal–Wallis test with Dunn’s posttest. When the number of groups is 2, statistical analysis of difference was performed using the Mann–Whitney test. When the p value was less than 0.05, the difference was regarded as significant.
Results
Culture study
Increased expression of AE2 mRNA by GCDC, TUDCA, and cytokines in BECs
We examined the mRNA expression level of AE2, in cultured BECs treated with bile acids (GCDC, DCA, UDCA, tauro-UDCA) and various cytokines (IL-4, IL-13, IFNγ, TNFα, TGFβ) for 4 h. Supplemental-Fig. 1 summarizes the data. The expression of AE2 was significantly increased in cultured BECs treated with GCDC, TUDCA, IL-4, and IL-13, compared with control (p < 0.05).

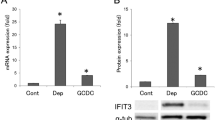
Decreased expression of AE2 in senescent BECs. a Cellular senescence was assessed by senescence-associated β-galactosidase activity (SA-β-gal) at day 4 after treatment with Etoposid (Etop, 100 μM), serum deprivation (Dep), GCDC (0.5 μM), or DCA (25 μM); n = 10 for each group, *p < 0.05 compared to control (Cont). b The expression of AE2 mRNA was significantly decreased in senescent BECs induced by GCDC and other stresses (p < 0.05); *p < 0.05 compared to control. c The expression of AE2 protein was significantly decreased in senescent BECs induced by GCDC and other stresses (p < 0.05); *p < 0.05 compared to control
Decreased expression of AE2 mRNA in senescent BECs induced by various stressors
Cellular senescence was induced by the treatment with Etoposid, serum deprivation, GCDC, and DCA, as reported previously (Fig. 1a). The expression of AE2 was significantly decreased in senescent BECs induced by GCDC and other stresses in mRNA and protein levels (p < 0.05)(Fig. 1b, c).
AE2 knockdown induced dysregulated autophagy, cell surface expression of PDC-E2, and cellular senescence
Effective knockdown of AE2
We examined the effect of AE2 knockdown on autophagy, cell surface expression of PDC-E2, and cellular senescence using AE2 siRNA. An effective knockdown of AE2 using siRNA was confirmed in mRNA and protein levels (Fig. 2a, b)

Induction of dysregulated autophagy, abnormal cell surface expression of mitochondrial antigen PDC-E2 and cellular senescence by AE2 knockdown. a The expression of AE2 mRNA in BECs treated with AE2 small interfering RNA (siRNA) or control siRNA for 1 day. AE2 mRNA was quantified with real-time PCR and normalized as a ratio using b-actin as the housekeeping gene. Data are expressed as the means ± SD; *p < 0.01 compared to the control, n = 3 for each group. b The protein level expression of AE2 assessed by immunoblotting in BECs treated with AE2 siRNA orcontrol siRNA for 2 days; *p < 0.05 vs. control siRNA, n = 3 for each group. c Expression of autophagy marker, LC3-II, was greater in BECs treated with AE2 siRNA compared to BECs with control siRNA by representative western blots and quantitative analysis of LC3-II band intensity. The degree of increase in LC3-II level by treatment with Bafilomycin A1 (100 nM) was less in BECs with AE2 siRNA compared to BECs with control siRNA, demonstrating decreased autophagic flux; *p < 0.05 compared to control siRNA, #p < 0.05 compared to AE2 siRNA. d Expression of p62 was greater in BECs treated with AE2 siRNA compared to BECs with control siRNA by representative western blots and quantitative analysis of LC3-II band intensity; *p < 0.05 compared to control siRNA. e Double Immunofluorescent staining for LC3 and p62 in BECs treated with AE2 siRNA (AE2si) or control siRNA (Contsi) for 2 days. The expression of LC3 and p62 was absent in BECs treated with control siRNA, whereas a granular expression of LC3 and p62 was increased in the cytoplasm of BECs treated by AE2 siRNA. f Cell-surface expression of PDC-E2 in BECs treated with AE2 siRNA (AE2si) or control siRNA (Contsi) with or without glycochenodeoxycholate (GCDC, 0.5 μM) for 2 days. Left, the expression of PDC-E2 was absent on the surface of BECs treated with control siRNA, whereas the cell-surface expression of PDC-E2 was increased on the surface of BECs treated with AE2 siRNA. Immunofluorescent staining for PDC-E2 in the absence of a membrane-permeabilizing agent. Original magnification, ×400. Right, semiquantitative analysis of cell-surface expression of PDC-E2 in BECs. The cell-surface signal for PDC-E2 was significantly increased in cells treated with AE2 siRNA (15.3 ± 6.1), AE2 siRNA + GCDC (10.1 ± 3.7), and control siRNA + GCDC (8.5 ± 3.6), when compared with control siRNA BECs (1.0 ± 0.4). Data are expressed as the mean ±SD (n = 5); *p < 0.01 and #p < 0.05, compared to the control siRNA. g Cellular senescence in BECs treated with AE2 siRNA (AE2si) or control siRNA (Contsi) with or without GCDC for 4 days. Cellular senescence was assessed by senescence-associated β-galactosidase activity (SA-β-gal). Percentage of cells positive for SA-β-gal was significantly increased in cells treated with AE2 siRNA (SA-β-gal labeling index, 30.3±5.6), compared to control siRNA (4.8±1.9) and treatment with GCDC tended to further accelerate cellular senescence (control siRNA + GCDC, 26.2±3.0; AE2 siRNA + GCDC, 29.6±5.6). Data were expressed as mean ± SD; *p < 0.05 compared to control. h Cellular senescence was assessed by SenTraGorTM system in BECs treated with AE2 siRNA (AE2si) or control siRNA (Contsi) for 4 days. Percentage of cells positive for GL13 was significantly increased in cells treated with AE2 siRNA (GL13 labeling index, 18.7±6.4), compared to control siRNA (4.4±2.8). Data was expressed as mean ± SD; *p < 0.01 compared to control
AE2 knockdown induced dysregulated autophagy
Immunoblotting showed that autophagy was dysregulated in BECs treated with AE2 siRNA, compared to control siRNA. Expression of the autophagy marker, LC3-II, in BECs with AE2 siRNA was greater than in BECs with control siRNA (Fig. 2c). The expression of LC3-II was further increased in the presence of Bafilomycin A1 in both BECs with AE2 siRNA and control siRNA (Fig. 2c). The degree of increase was less in BECs with control siRNA, compared to BECs with AE2 siRNA, indicating decreased autophagic flux by AE2 knockdown (Fig. 2c). The expression of autophagy-related p62 was increased in BECs with AE2 siRNA, compared to control siRNA, indicating dysregulated autophagy (Fig. 2d).
Immunofluorescent staining disclosed that a granular expression of LC3 and p62 was observed as dots in BECs treated with AE2 siRNA, which suggests dysregulated autophagy (Fig. 3e).

Expression of AE2 in small bile ducts in primary biliary cholangitis (PBC) and control livers. a Top left, AE2 was mainly expressed in the basolateral side in BECs in small bile ducts (arrow) in normal liver. Top right, Luminal expression of AE2 was increased in small bile ducts in CVH (arrow). Bottom, the expression of AE2 was decreased in the damaged small bile ducts (arrow) in PBC. Whereas, it was increased in some bile ductules (thin long arrow). PBC cases #1 and #3 (stage 2). Immunostaining for AE2. Original magnification, ×400. b Evaluation of the expression of AE2 in small bile ducts in PBC and control livers. The expression of AE2 was significantly decreased in small bile ducts in PBC with CNSDC (PBC, CA3), compared with other groups (p < 0.01). In contrast, the expression of AE2 was significantly increased in small bile ducts in chronic viral hepatitis (CVH), compared with other groups (p < 0.01). Gray column, decrease; white column, no change; and stripe column, increase. Decrease: *p < 0.01 compared to other groups; Increase #p < 0.01 compared to PBC, EBO, and NL. NL normal liver, PSC primary sclerosing cholangitis
AE2 knockdown increased a cell-surface expression of PDC-E2
The expression of mitochondrial antigen PDC-E2 was absent on the surface of control BECs (Fig. 2f), whereas, the cell-surface expression of PDC-E2 was increased on the surface of BECs treated with AE2 siRNA (Fig. 2f). Semi-quantitative analysis disclosed that the cell-surface expression of PDC-E2 was significantly greater in BECs by knockdown of AE2 using siRNA than BECs with control siRNA (p < 0.01) (Fig. 2f). The cell-surface expression of PDC-E2 was significantly increased by the treatment with GCDC (p < 0.05) (Fig. 2f), as reported previously [12]. These findings suggest that dysregulated autophagy caused by AE2siRNA may induce the increased expression of mitochondrial antigen PDC-E2 on the cell surface of BECs
AE2 knockdown induced cellular senescence
Cellular senescence assessed by SA-β-gal activity was significantly increased by the knockdown of AE2 using siRNA even in control culture condition (p < 0.01) (Fig. 2g). The treatment with GCDC tended to further accelerate cellular senescence, although there was no significant difference. Cellular senescence assessed by SenTraGorTM system was also significantly increased by knockdown of AE2 using siRNA even in control culture condition (p < 0.01) (Fig. 2h).
Human studies
Decreased expression of AE2 in damaged BECs in small bile ducts in PBC
AE2 was mainly expressed in the basolateral side in BECs in small bile ducts in normal liver (Fig. 3a). Luminal expression of AE2 was increased in small bile ducts in some patients with CVH. In contrast, the expression of AE2 was decreased in the damaged small bile ducts (Fig. 3a). AE2 expression was increased in non-inflamed bile ducts and bile ductules (Fig. 3a).
The expression of AE2 was significantly decreased in PBC with CNSDC (PBC, CA3), compared with PBC without CNSDC (PBC, CA0-2), CVH, PSC, EBO, and normal livers (p < 0.05) (Fig. 3b). The expression of AE2 was significantly increased in small bile ducts in CVH, compared with PBC, EBO, and normal livers (p < 0.01) (Fig. 3b).
Decreased expression of AE2 correlated with dysregulated autophagy, increased expression of mitochondrial antigen PDC-E2, and cellular senescence
Double immunostaining revealed that the expression of AE2 was frequently decreased in the damaged bile ducts in which autophagy markers (LC3 and p62), PDC-E2, senescent markers (p16INK4a and p21WAF1/Cip1) were expressed in PBC (Fig. 4, Supplemental Fig. 2). Whereas, the expression of AE2 was seen in small bile ducts in control livers in which autophagy markers (LC3 and p62), PDC-E2, senescent markers (p16INK4a and p21WAF1/Cip1) were not expressed (Fig. 4, Supplemental Fig. 2).

Association of AE2 expression with PDC-E2 or senescent markers p16INK4a in small bile ducts in primary biliary cholangitis (PBC) and chronic viral hepatitis (CVH). a Increased expression of PDC-E2 (top middle) was seen in a damaged small bile duct (arrow) showing decreased expression of AE2 (top left) in PBC. In contrast, the expression of PDC-E2 (bottom middle) was not seen in a small bile duct (arrow) with AE2 expression in CVH (bottom left). b Increased expression of p16INK4a (top middle) was seen in the nuclei of BECs in a damaged small bile duct (arrow) showing decreased expression of AE2 (top left) in PBC. In contrast, the expression of p16INK4a (bottom middle) was not seen in a small bile duct (arrow) with AE2 expression in CVH (bottom left). Double immunostaining for AE2 (green) and PDC-E2 or p16INK4a (red). Original magnification, ×400
Discussion
The data obtained in this study are summarized as follows: (1) the expression of AE2 was significantly increased in cultured BECs treated with GCDC, TUDCA, IL-4, and IL-13 for 4 h and it was significantly decreased in senescent BECs induced by GCDC and other stresses; (2) AE2 knockdown using siRNA induced dysregulated autophagy, cell surface expression of PDC-E2, and cellular senescence in cultured BECs; (3) the expression of AE2 was significantly decreased in small bile ducts involved in CNSDC in PBC, compared to control livers. In contrast, it was significantly increased in small bile ducts in CVH, compared to PBC and other livers. (4) The decreased expression of AE2 was correlated with abnormal expression of PDC-E2, dysregulated autophagy, and cellular senescence in PBC.
Several lines of studies indicated that AE2, the principal bicarbonate secretor in the human biliary tree, is downregulated in PBC [19,20,21,22]. A recent study demonstrated that downregulation of AE2 may sensitize BECs to apoptotic insults activating soluble adenylyl cyclase [23]. However, the mechanisms describing how downregulated AE2 participates in the pathogenesis of PBC have not been fully clarified. In this study, we disclosed for the first time that the decreased expression of AE2 might be involved in dysregulated autophagy, followed by abnormal expression of mitochondrial antigens and cellular senescence in BECs in PBC (Fig. 5). We have previously reported that dysregulated autophagy may be a central player in the pathogenesis of PBC by inducing autoimmune process via abnormal expression of mitochondrial antigens such as PDC-E2 and also induction of cellular senescence in BECs in bile duct lesions in PBC [10,11,12,13,14,15,16,17,18, 33]. In fact, abnormal cell surface expression of mitochondrial antigen PDC-E2 was observed by dysregulated autophagy caused by GCDC with and without Bafilomycin A [12]. Hydrophobic bile acid such as GCDC, endoplasmic reticulum (ER) stress, and oxidative stress are candidate triggers of dysregulated autophagy; however, the exact mechanism of dysregulated autophagy was not elucidated in our previous studies [10,11,12, 33].

Working hypothesis on the association of AE2 downregulation with dysregulated autophagy and cellular senescence in PBC. AE2 anion exchanger 2, GCDC glycochenodeoxycholic acid, Mit-Ag mitochondrial antigen, SASP senescence-associated secretory phenotype
The expression of AE2 was significantly reduced in PBC, especially in the damaged small bile ducts involved in CNSDC, in agreement with previous studies [21, 34]. Furthermore, this study disclosed for the first time that the decreased expression of AE2 was closely correlated with abnormal expression of PDC-E2 and autophagy-related markers LC3 and p62. The present study also demonstrated that AE2 knockdown effectively induced dysregulated autophagy and cell surface expression of PDC-E2 in BECs. These findings suggest that an impaired bicarbonate umbrella caused by AE2 dysfunction may be involved in dysregulated autophagy in the pathogenesis of PBC (Fig. 5).
It is conceivable that bile acid signaling may also participate in dysregulated autophagy and other pathophysiology of BECs in PBC. Bile acid may regulate BECs through bile acid nuclear receptor, farnesoid X receptor (FXR), and plasma membrane receptor TGR5 [35]. FXR suppressed autophagy in hepatocytes, whereas nuclear receptor peroxisome proliferator activated receptor α (PPARα) induced autophagy [36]. In cholestatic livers, the accumulation of bile acids caused the accumulation of autophagosomes via suppressed autophagy flux and accumulation of p62 in hepatocytes, although the signaling pathway including FXR and TGR5 was undetermined [37]. It remains to be seen how bile acids regulate autophagy and cause dysregulated autophagy and which bile acid receptors participate in the process in BECs in PBC.
Furthermore, this study demonstrates that AE2 knockdown effectively induced cellular senescence, in agreement with a recent study [34]. Our group previously reported that cellular senescence was caused via dysregulated autophagy [10]; therefore, it is likely that cellular senescence may be induced via dysregulated autophagy due to AE2 downregulation. Conversely, AE2 expression was decreased in senescent BECs induced by GCDC, serum deprivation, and other stresses. Since a short-term treatment with GCDC and other several factors rather increased AE2 mRNA expression, chronic exposure to GCDC seems to be critical to decrease AE2 expression together with the induction of cellular senescence. It is conceivable that impairment of bicarbonate umbrella, due to senescent BECs, further exacerbates injuries of BECs (Fig. 5). It has not yet been determined whether AE2 overexpression could prevent dysregulated autophagy and cellular senescence and other injuries in BECs caused by GCDC and other toxic agents.
In addition, it is now well known that senescent BECs express various chemokines and cytokines as senescence-associated secretory phenotypes (SASPs) in PBC and PSC, which modulate inflammatory cell infiltration and fibrosis in cholangiopathies, as our group reported for the first time [17, 18, 38, 39]. Taken together, these studies indicate that it is plausible that impairment of AE2 in parallel with cellular senescence may participate in the exacerbated inflammation and other pathologic events via SASPs in PBC.
It is of interest that allelic variations in AE2 gene have a significant association with the progression rate of liver disease in PBC under UDCA therapy in a previous study [40]. Recent GWAS studies revealed several candidates of susceptible genes/loci in PBC [5, 6]; however, AE2 has not been identified as a susceptible gene, so far. Generally, most susceptible genes/loci are HLA and non-HLA genes relating adaptive and innate immunity such as cytokines, chemokines, and their receptors in PBC [5, 6]. There were few genes/loci relating to the susceptibility/vulnerability of BECs in PBC [5, 6]. Some ER stress-related genes, such as ORM1-like protein 3 (ORMDL3), which are included in susceptible genes/loci in PBC [5, 6], might be related to autophagy of epithelial cells as suggested in inflammatory bowel disease. Our previous study showed that ER stress might be involved in dysregulated autophagy and cellular senescence in BECs. Therefore, ER stress may be associated with an impairment of AE2 in BECs in PBC.
In conclusion, an impaired biliary bicarbonate umbrella may be involved in dysregulated autophagy followed by abnormal expression of PDC-E2 and cellular senescence in BECs in PBC. Therapeutic strategies to relieve the “impaired biliary bicarbonate umbrella” may be effective for PBC.
References
Portmann B, Nakanuma Y. Diseases of the bile ducts. In: Burt A, Portmann B, Ferrell L, editors. Pathology of the liver. 6th ed. London: Churchill Livingstone; 2011. p. 491–562.
Kaplan MM, Gershwin ME. Primary biliary cirrhosis. N Engl J Med. 2005;353:1261–73.
Lindor KD, Gershwin ME, Poupon R, et al. Primary biliary cirrhosis. Hepatology. 2009;50:291–308.
Nakanuma Y, Ohta G. Histometric and serial section observations of the intrahepatic bile ducts in primary biliary cirrhosis. Gastroenterology. 1979;76:1326–32.
Nakamura M, Nishida N, Kawashima M, et al. Genome-wide association study identifies TNFSF15 and POU2AF1 as susceptibility loci for primary biliary cirrhosis in the Japanese population. Am J Hum Genet. 2012;91:721–8.
Mells GF, Floyd JA, Morley KI, et al. Genome-wide association study identifies 12 new susceptibility loci for primary biliary cirrhosis. Nat Genet. 2011;43:329–32.
Fussey SP, Guest JR, James OF, et al. Identification and analysis of the major M2 autoantigens in primary biliary cirrhosis. Proc Natl Acad Sci USA. 1988;85:8654–8.
Kita H, Matsumura S, He XS, et al. Quantitative and functional analysis of PDC-E2-specific autoreactive cytotoxic T lymphocytes in primary biliary cirrhosis. J Clin Invest. 2002;109:1231–40.
Shimoda S, Van de Water J, Ansari A, et al. Identification and precursor frequency analysis of a common T cell epitope motif in mitochondrial autoantigens in primary biliary cirrhosis. J Clin Invest. 1998;102:1831–40.
Sasaki M, Miyakoshi M, Sato Y, et al. Autophagy mediates the process of cellular senescence characterizing bile duct damages in primary biliary cirrhosis. Lab Invest. 2010;90:835–43.
Sasaki M, Miyakoshi M, Sato Y, et al. A possible involvement of p62/sequestosome-1 in the process of biliary epithelial autophagy and senescence in primary biliary cirrhosis. Liver Int. 2012;32:487–99.
Sasaki M, Miyakoshi M, Sato Y, et al. Increased expression of mitochondrial proteins associated with autophagy in biliary epithelial lesions in primary biliary cirrhosis. Liver Int. 2013;33:312–20.
Sasaki M, Ikeda H, Haga H, et al. Frequent cellular senescence in small bile ducts in primary biliary cirrhosis: a possible role in bile duct loss. J Pathol. 2005;205:451–9.
Sasaki M, Ikeda H, Sato Y, et al. Decreased expression of Bmi1 is closely associated with cellular senescence in small bile ducts in primary biliary cirrhosis. Am J Pathol. 2006;169:831–45.
Sasaki M, Ikeda H, Yamaguchi J, et al. Telomere shortening in the damaged small bile ducts in primary biliary cirrhosis reflects ongoing cellular senescence. Hepatology. 2008;48:186–95.
Sasaki M, Ikeda H, Nakanuma Y. Activation of ATM signaling pathway is involved in oxidative stress-induced expression of mito-inhibitoryp21(WAF1/Cip1) in chronic non-suppurative destructive cholangitis in primary biliary cirrhosis: An immunohistochemical study. J Autoimmun. 2008;31:73–78.
Sasaki M, Nakanuma Y. Bile acids and deregulated cholangiocyte autophagy in primary biliary cholangitis. Dig Dis. 2017;35:210–6.
Sasaki M, Miyakoshi M, Sato Y, et al. Modulation of the microenvironment by senescent biliary epithelial cells may be involved in the pathogenesis of primary biliary cirrhosis. J Hepatol. 2010;53:318–25.
Beuers U, Hohenester S, de Buy Wenniger LJ, et al. The biliary HCO(3)(-) umbrella: a unifying hypothesis on pathogenetic and therapeutic aspects of fibrosing cholangiopathies. Hepatology. 2010;52:1489–96.
Hohenester S, Wenniger LM, Paulusma CC, et al. A biliary HCO3- umbrella constitutes a protective mechanism against bile acid-induced injury in human cholangiocytes. Hepatology. 2012;55:173–83.
Prieto J, Qian C, Garcia N, et al. Abnormal expression of anion exchanger genes in primary biliary cirrhosis. Gastroenterology. 1993;105:572–8.
Salas JT, Banales JM, Sarvide S, et al. Ae2a,b-deficient mice develop antimitochondrial antibodies and other features resembling primary biliary cirrhosis. Gastroenterology. 2008;134:1482–93.
Chang JC, Go S, de Waart DR, et al. Soluble adenylyl cyclase regulates bile salt-induced apoptosis in human cholangiocytes. Hepatology. 2016;64:522–34.
Banales JM, Saez E, Uriz M, et al. Up-regulation of microRNA 506 leads to decreased Cl-/HCO3- anion exchanger 2 expression in biliary epithelium of patients with primary biliary cirrhosis. Hepatology. 2012;56:687–97.
Katayanagi K, Kono N, Nakanuma Y. Isolation, culture and characterization of biliary epithelial cells from different anatomical levels of the intrahepatic and extrahepatic biliary tree from a mouse. Liver. 1998;18:90–98.
Sasaki M, Van De Water J, Kenny TP, et al. Immunoglobulin gene usage and immunohistochemical characteristics of human monoclonal antibodies to the mitochondrial autoantigens of primary biliary cirrhosis induced in the XenoMouse. Hepatology. 2001;34:631–7.
Dimri GP, Lee X, Basile G, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92:9363–7.
Evangelou K, Lougiakis N, Rizou SV, et al. Robust, universal biomarker assay to detect senescent cells in biological specimens. Aging Cell. 2017;16:192–7.
Nakanuma Y, Sasaki M. Expression of blood-group-related antigens in the intrahepatic biliary tree and hepatocytes in normal livers and various hepatobiliary diseases. Hepatology. 1989;10:174–8.
Roskams TA, Theise ND, Balabaud C, et al. Nomenclature of the finer branches of the biliary tree: canals, ductules, and ductular reactions in human livers. Hepatology. 2004;39:1739–45.
Nakanuma Y, Zen Y, Harada K, et al. Application of a new histological staging and grading system for primary biliary cirrhosis to liver biopsy specimens: interobserver agreement. Pathol Int. 2010;60:167–74.
Desmet V, Gerber M, Hoofnagle J, et al. Classification of chronic hepatitis: diagnosis, grading and staging. Hepatology. 1994;19:1513–20.
Sasaki M, Yoshimura-Miyakoshi M, Sato Y, et al. A possible involvement of endoplasmic reticulum stress in biliary epithelial autophagy and senescence in primary biliary cirrhosis. J Gastroenterol. 2015;50:984–95.
Hisamoto S, Shimoda S, Harada K, et al. Hydrophobic bile acids suppress expression of AE2 in biliary epithelial cells and induce bile duct inflammation in primary biliary cholangitis. J Autoimmun. 2016;75:150–60.
Marin JJ, Macias RI, Briz O, et al. Bile acids in physiology, pathology and pharmacology. Curr Drug Metab. 2015;17:4–29.
Preidis GA, Kim KH, Moore DD. Nutrient-sensing nuclear receptors PPARalpha and FXR control liver energy balance. J Clin Invest. 2017;127:1193–201.
Kim S, Han SY, Yu KS, et al. Impaired autophagy promotes bile acid-induced hepatic injury and accumulation of ubiquitinated proteins. Biochem Biophys Res Commun. 2018;495:1541–7.
Tabibian JH, O’Hara SP, Splinter PL, et al. Cholangiocyte senescence by way of N-ras activation is a characteristic of primary sclerosing cholangitis. Hepatology. 2014;59:2263–75.
Sasaki M, Miyakoshi M, Sato Y, et al. Chemokine-chemokine receptor CCL2-CCR2 and CX3CL1-CX3CR1 axis may play a role in the aggravated inflammation in primary biliary cirrhosis. Dig Dis Sci. 2014;59:358–64.
Poupon R, Ping C, Chretien Y, et al. Genetic factors of susceptibility and of severity in primary biliary cirrhosis. J Hepatol. 2008;49:1038–45.
Acknowledgements
This study was supported in part by a Grant-in-Aid for Scientific Research (C) from the Ministry of Education, Culture, Sports and Science and Technology of Japan (15K08341).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Sasaki, M., Sato, Y. & Nakanuma, Y. An impaired biliary bicarbonate umbrella may be involved in dysregulated autophagy in primary biliary cholangitis. Lab Invest 98, 745–754 (2018). https://doi.org/10.1038/s41374-018-0045-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-018-0045-4
This article is cited by
-
Knockout of secretin ameliorates biliary and liver phenotypes during alcohol-induced hepatotoxicity
Cell & Bioscience (2023)
-
Algorithmic assessment of cellular senescence in experimental and clinical specimens
Nature Protocols (2021)
-
Interferon-induced protein with tetratricopeptide repeats 3 may be a key factor in primary biliary cholangitis
Scientific Reports (2021)
