
Abstract
Micrognathia is a common craniofacial deformity which represents hypoplastic development of the mandible, accompanied by retrognathia and consequent airway problems. Usually, micrognathia is accompanied by multiple systematic defects, known as syndromic micrognathia, and is in close association with genetic factors. Now, large quantities of pathogenic genes of syndromic micrognathia have been revealed. However, how these different pathogenic genes could lead to similar phenotypes, and whether there are some common characteristics among these pathogenic genes are still unknown. In this study, we proposed a genetic-phenotypic classification of syndromic micrognathia based on pathogenic genes information obtained from Phenolyzer, DAVID, OMIM, and PubMed database. Pathogenic genes of syndromic micrognathia could be divided into four groups based on gene function, including cellular processes and structures, cell metabolism, cartilage and bone development, and neuromuscular function. In addition, these four groups exhibited various clinical characteristics, and the affected systems, such as central nervous system, skeletal system, cardiovascular system, oral and dental system, respiratory system and muscle, were different in these four groups. This classification could provide meaningful insights into the pathogenesis of syndromic micrognathia, and offer some clues for understanding the molecular mechanism, as well as guiding precise clinical diagnosis and treatment for syndromic micrognathia.
Similar content being viewed by others

Introduction
As one of the most common craniofacial deformities, micrognathia represents hypoplastic development of the mandible, accompanied by retrognathia and consequent airway problems, which will greatly damage patients’ appearance and even bring great risk for children survival. Micrognathia could occur independently, but usually is associated with different syndromes, such as Cerebro-oculofacio-skeletal syndrome, Meier–Gorlin syndrome and Pierre Robin syndrome [1]. Previous researchers have made meaningful attempts at classifying this usual but etiologically complex malformation. Pruzansky proposed the first nosology of mandibular anomaly in patients with hemifacial microsomia based on morphologic changes [2]. Davinder et al. summarized that mandibular hypoplasia can be classified into three groups: congenital, developmental and acquired [1]. However, there was no systematic and comprehensive classification of syndromic micrognathia based on genetic mechanisms. Many mutant genes of syndromic micrognathia, such as SOX9, PIEZO2, TCOF1 and EZH2, have been found in recent years [3,4,5,6], which provides potential chances to gain deeper insights into syndromic micrognathia. As we know, these different pathogenic genes have different functions, and different syndromic micrognathia is accompanied by different affected systems. Whether there are some common characteristics among these genes, and the exact differences of phenotypes among different syndromic micrognathia need further investigation. In this study, we propose a comprehensive and flexible classification of syndromic micrognathia based on different gene functions and phenotypic characteristics, which would provide useful insights for genetic aspects of syndromic micrognathia and could offer helpful guidance in clinical diagnosis and treatment.
Materials and methods
Study design
Firstly, collection of pathogenic genes of micrognathia was performed. Then functional enrichment analysis of these genes was performed to identify significant gene functional classification. A genetic classification of syndromic micrognathia was then proposed based on different functions of pathogenic genes. Finally, the phenotypic characteristics of syndromic micrognathia among different classification groups were analyzed (Fig. 1).

Study outline. Pathogenic genes of syndromic micrognathia were firstly collected. Then functional enrichment analysis was performed to identify significant biological processes and molecular functions of these genes. A genetic classification of syndromic micrognathia was then proposed based on different functions of pathogenic genes. Finally, the phenotypic characteristics of syndromic micrognathia were analyzed
Identification of pathogenic genes of syndromic micrognathia
Possible pathogenic genes of syndromic micrognathia were retrieved online using Phenolyzer (http://phenolyzer.usc.edu), a tool to prioritize human disease genes based on disease or phenotype information provided by users as free text [7]. We queried for possible pathogenic genes of syndromic micrognathia with the term “micrognathia”. By 8 September 2018, a total of 582 seed genes were found by Phenolyzer. Phenolyzer also adapts a machine learning model that integrates multiple features to score and prioritize all candidate genes.
As Phenolyzer uses an algorithm to predict previously unknown disease genes, we further evaluated these seed genes of syndromic micrognathia through references and Online Mendelian Inheritance in Man (OMIM, http://www.ncbi.nlm.nih.gov/omim), which is a comprehensive, authoritative and timely knowledgebase of human genes and genetic disorders [8]. To exclude the unconfirmed genes, an exclusion criteria was adopted. Genes responsible for diseases which do not have the typical manifestation of micrognathia, or syndromes without clear genetic pathogenesis, or chromosomal abnormalities without definite pathogenic genes were excluded. Predicted genes without reference or solid evidence support were also excluded. The rest of seed genes were designated as pathogenic genes of syndromic micrognathia, and were used for further analysis.
Functional enrichment analysis of pathogenic genes of syndromic micrognathia
The database for annotation, visualization and integrated discovery (DAVID) bioinformatics resources integrate useful analytic tools for functional enrichment analysis, such as Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases. DAVID could systematically extract biological meaning from large gene/protein lists. The submitted gene list could also be divided into several groups by DAVID based on their enriched and pertinent biological features [9]. We performed a systematic analysis of the syndromic micrognathia related genes using DAVID. Firstly, we went to the DAVID website (http://david.abcc.ncifcrf.gov/home.jsp) and copied pathogenic genes of syndromic micrognathia as input gene list. The official gene symbol was selected as the gene identifier type and the annotations were limited to Homo sapiens. Default setting was used as gene population background. The gene list was then submitted and we used DAVID’s analytic modules to perform a systematic evaluation.
Classification of pathogenic genes of syndromic micrognathia and phenotypic analysis
Each pathogenic gene of syndromic micrognathia was classified into different groups according to the major function of the gene and its possible role in the pathogenesis of micrognathia, which was retrieved on PubMed (http://www.ncbi.nlm.nih.gov/PubMed). The results of functional enrichment analysis were also used as references.
The phenotypic characteristics of each syndrome in association with micrognathia were obtained by collecting information from the “Clinical Synopsis” part of each disease on OMIM or references. Statistical analyses on phenotypic differences among groups were then performed. The number of times of a certain type of systematic defects, such as skeletal defects, cardiovascular defects and pulmonary defects, was calculated in different groups. The rates of these systematic defects were determined in terms of patients with a certain systematic defect compared to total syndromic micrognathia patients. The rates of these systematic defects in different groups were also calculated, and statistical analysis was carried using Pearson’s chi-squared test. P values were adjusted with FDR method. All statistical analyses were performed with SPSS software (version 24.0 for Windows, IBM Corp., Armonk, NY, USA).
Results
Identification of pathogenic genes of syndromic micrognathia
Phenolyzer provided a list of 582 seed genes, as well as their corresponding diseases according to our query term “micrognathia”. A thorough evaluation of the candidate gene list was then performed through searching function of each gene and their corresponding diseases on OMIM. Genes that were unrelated to micrognathia, or genes related to syndromes without a definite pathogenesis and chromosomal abnormalities were excluded. If the gene’s function was not confirmed, more information from published reference for further evaluation will be performed. Totally, 325 pathogenic genes and 172 syndromes were confirmed in association with syndromic micrognathia, and were included in this study for further analysis (Supplementary Table 1).
Functional enrichment analysis of pathogenic genes of syndromic micrognathia
The list of pathogenic genes of syndromic micrognathia was input to DAVID, which then generated comprehensive enrichment analysis results of these genes, including their enrichment in GO terms, pathways, and protein domains. As we focused on the different functional groups of these genes, we used the Gene Functional Classification module to analyze these results. Input genes are classified into 18 groups based on functional similarity, with the highest enrichment score of 20.31. These groups of genes show several different biological functions, including peroxisomal biogenesis, DNA replication, ciliary function (Table 1), which indicates that syndromic micrognathia could possibly be related with abnormalities of many different biological processes.
Classification of pathogenic genes of syndromic micrognathia
The functional classification from DAVID analysis is instructive in guiding the classification of pathogenic genes of syndromic micrognathia. However, only a part of the input genes could be classified into the 18 functional groups. Genes that are not in the output fail to map to any of the functional groups. This could be possibly due to the high threshold of forming a functional group according to the algorithm of DAVID. In order to conduct a comprehensive classification of pathogenic genes of syndromic micrognathia, we then carefully searched the function of each gene and their possible role in the occurrence of micrognathia on OMIM and PubMed. Finally, a functional classification of pathogenic genes of syndromic micrognathia was proposed, which included four groups: cellular processes and structures, cell metabolism, cartilage and bone development, and neuromuscular function (Table 2), according to their functional clustering and their role in the pathogenesis of micrognathia. A detailed list of these genes, their related syndromes, their possible role in the occurrence of micrognathia and references is shown in Supplementary Table 1.
Phenotypic analysis of syndromic micrognathia
The phenotypic characteristics of each syndrome in association with micrognathia were obtained by collecting information from the “Clinical Synopsis” part of each disease on OMIM, genetics home reference (https://ghr.nlm.nih.gov/) and published references, and some examples of patient images could also be obtained from it. A total of 172 syndromes were analyzed. We focused on the common clinical manifestations of these syndromes other than micrognathia, such as skeletal system, respiratory system, cardiovascular system disease and so on (Supplementary Table 2).
The distributions of affected systems in syndromic micrognathia among different groups are shown in Table 3. General occurrence rate of systematic defects in all syndromic micrognathia were calculated, and skeletal system defects have the highest occurrence rate among all syndromic micrognathia diseases (88.37%, 152/172), followed by central nervous system defects, facial dysmorphism (including auricular, ocular, nasal, oral and dental defects) and general growth problems. Defects of these systems are accompanied with micrognathia in nearly at least 70% of cases. Approximately half of the 172 syndromes also have defects in genitourinary, digestive, cardiovascular and respiratory systems. Muscular and soft tissue defects (31.40%, 54/172), as well as endocrine and immunologic problems (20.35%, 35/172) have relative lower occurrence rates, whereas cancer and hematic problems are rare, accounting for less than 10% of the cases.
Different patterns of the affected systems are prominent among the four groups, and each group exhibited phenotypic similarities or overlaps but showed different appearance rate of multiple systematic defects among different groups. Group I has relatively higher rates of multiple systematic defects, including central nervous system, cardiovascular, genitourinary, dermal, as well as endocrine and immunologic defects, and mental retardation, ventricular septal defect, atrial septal defect, cutis laxa, as well as diabetes may be phenotypic similarities in this group. The distribution of systematic defects of Group II was similar to the overall level of the other three groups, as no statistical significance could be determined in all 16 systems. However, the appearance rate of digestive system defects such as hepatomegaly, a possible phenotypic similarity in this group, seemingly much higher than other three groups, and the raw P value was 0.001 according to the Chi-square value. The phenomenon of reduced rates in multiple systematic defects is striking in Group III. The rates for defects in central nervous system, cardiovascular system, digestive system, genitourinary system, endocrine and immunologic system, as well as muscle and soft tissues were lower with statistical significance. However, the incidence rate for skeletal system defects in Group III is 93.94%, which is the highest incidence rate among four groups, and defects of pelvis, limbs and hands, such as scoliosis, arthropathy and brachydactyly, may be the characteristic clinical similarities in this group. For Group IV, there was an increased involvement of muscle and soft tissue defects. Muscle weakness, muscle atrophy or increased muscle tone, are typically seen in conditions of Group IV.
Discussion
Genetic classification of syndromic micrognathia and its relationship with mandibular development
In this article, we propose a novel classification method of syndromic micrognathia based on analysis of pathogenic gene function using DAVID, OMIM, and PubMed. This classification organizes syndromic micrognathia into a logical and comprehensive system based on known functions of the pathogenic genes. The four groups of genes could correspond to the key steps of mandibular development, and may shed light on the complicated pathogenesis behind syndromic micrognathia.
Genes in Group I are involved in several fundamental cellular processes and structures, including DNA, RNA and protein biosynthesis, epigenetic modifications, cell signaling pathways, as well as integrity of cytoskeleton, cell membrane and organelle [10,11,12,13,14]. These basic cellular processes and structures are essential in the development of multiple systems. In particular, mutations of these genes are associated with micrognathia possibly through influencing the proliferation, migration and differentiation of cranial neural crest cells (CNCCs), a group of multipotential stem cells which contribute to the formation of most craniofacial structures [15].
Group II is composed of genes responsible for cell metabolism of several biologically important substances, such as cholesterol, carbon hydrates, and trace elements [16,17,18]. Although the exact mechanism remains unknown, it has been proven true that metabolic disorders could have negative effects on mandibular development, and such an example is reduced mandibular growth in streptozotocin-induced diabetic mice, and diabetic pregnancy is also a risk factor for hemifacial microsomia [19, 20].
Group III represents genes essential in the development of cartilage and bone, comprising mainly of genes responsible for the regulation of chondrocyte and osteocyte proliferation and differentiation, such as TGF-β superfamily, FGF family, RUNX2, SOX9, growth hormone, parathyroid hormone, and other factors [3, 21, 22]. Genes associated with cartilage extracellular matrix (ECM) function are also included in this group, as ECM has a regulatory effect on chondrocyte behavior and function by providing the structural support for chondrocytes, and also serves as a reservoir for growth factors [23]. Another subgroup in Group III include genes regulating bone remodeling of the mandible, which is active both in prenatal and postnatal life [24, 25], and is controlled by the relative activities of osteoblasts and osteoclasts [26].
Group IV include genes responsible for neuromuscular function, as abnormalities in the muscle or tendon itself, or the conduction of motor nerve impulse, or the central nervous system, could all affect the normal muscular function and thus show a negative effect on mandibular development [27,28,29]. Skeletal muscles play important roles in mandibular skeletogenesis, either through mechanical ways, as muscle contraction provides functional loading onto mandible, or through secretory ways on skeleton to stimulate bone development [30]. Previous animal model has also demonstrated that decrease in masticatory demand during the growth period leads to insufficient mandibular development [31].
Phenotypic analysis of syndromic micrognathia and clinical meanings
The results from our phenotypic analysis of the total 172 syndromic micrognathia and the four different groups showed instructive clues in clinic. In general, skeletal defects, central nervous system defects, facial dysmorphism and general growth problems are most commonly involved in patients with syndromic micrognathia. This may be due to these structures and systems share some common developmental pathways. For example, Clim-2, which is pronounced in both facial and limb bud mesenchyme, plays crucial roles in mandibular arch and limb development under the regulation of Fgf-8 [32]. In addition, Pitx1 is a Bicoid-related homeodomain factor which is expressed in the hindlimb, the developing anterior pituitary gland and first branchial arch, and Pitx1 gene-deleted mice showed significant structural defects of tibia and fibula, as well as abnormalities in pituitary cell types and the first branchial arch [33]. In this sense, the combined manifestations of micrognathia with one or more of these defects may be instructive to the diagnosis of a certain craniofacial syndrome. On the other hand, this could also give some clues to get clinical practitioners to pay attention to those often overlooked defects, such as genitourinary, digestive, cardiovascular and respiratory system defects and even cancer susceptibility, and avoid missed diagnosis.
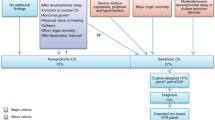
The phenotypic differences among the four groups of syndromic micrognathia may also provide interesting insights. Group I has a generally greater involvement in multiple systems, which may be due to genes in Group I are involved in fundamental cellular structures, processes and functions, and are important in development of multiple systems. For those with multiple systematic defects in accompany with micrognathia, it implied that doctors need to carefully examine the patients to avoid missed diagnosis, and a multidisciplinary treatment based on the diagnosis is also necessary. In particular, Group I has a relative higher rate of cardiovascular defects, which may call for extra attention in clinic practice. The high rate of digestive defects in Group II may be a characteristic of this group. The rates for systematic defects in Group III are reduced in multiple systems, including central nervous system, cardiovascular system, digestive system, genitourinary system, endocrine and immunologic system, as well as muscle and soft tissues. However, it showed the highest rate of skeletal defects among these four groups. This high rate of skeletal defects and relative low rates of defects of many other systems is a typical characteristic of syndromes in Group III, which is due to that these genes, or their regulatory elements such as tissue specific enhancers, exhibited relative specific function in skeleton tissues or craniofacial tissues, and thus treatment may mainly focus on skeleton tissues. Finally, conditions of Group IV have characteristic high rates of muscle and soft tissue defects, which revealed that force generated by muscles or tendons according to the neural activation is important for skeletal development, and treatment for syndromic micrognathia should also focus on muscle and tendon diseases, such as restoring the muscle and tendon function through gene manipulation. Based on the different phenotypic distributions of these four conditions, a possible flow path to help surgeons to relatively quickly focus on accurate diagnosis of syndromic micrognathia is illustrated in Fig. 2, which could also help surgeons to decrease the number of genes that need to be detected to confirm the diagnosis.

A possible diagnostic flow of syndromic micrognathia. The accompanying skeletal, CNS, growth defects and facial dysmorphism are indicative of syndromic micrognathia, which suggests clinicians to examine other less common defects. Different groups of syndromic micrognathia have their own characteristic involvement of one of more certain systems, which provide possible diagnostic clues as indicated by the dashed lines
In summary, there were some shortcomings for this classification method. For example, not all syndromic micrognathia have a definite pathogenic gene, and some of them are also influenced by environmental factors. A good example of one such disease is hemifacial microsomia, which is believed to be caused by environmental, maternal and genetic factors, while not only by genetic factors [34], and also exhibited difference with syndromic micrognathia that mentioned in this study (Fig. 3). In addition, many pathogenic genes may exhibit multiple functions, and this classification just focus on the main function that contribute to syndromic micrognathia. Also, it should be noted that the mandibles are not frequently and severely affected in all included syndromes. Some syndromes only have mild or less frequent involvement of micrognathia. These syndromes include myopathy, lactic acidosis, and sideroblastic anemia, Barber-Say syndrome, nemaline myopathy 9 and so forth [35,36,37]. Treatment for these syndromes may thus focus on defects of many other systems, as the clinical consequences of micrognathia in theses conditions may be mild and do not require surgical correction. Furthermore, with the growing application of high-throughput sequencing, more congenital disorders with micrognathia and their pathogenic genes may also need to be added to update the classification. However, this classification of syndromic micrognathia based on pathogenic gene function and phenotype is a novel method with potential instructive clinical significance to help surgeons to better understand the mechanism of syndromic micrognathia, and make accurate diagnosis and treatment plan. Precise classification diagnosis based on genomic and phenotypic information is also a future research hotpot in craniofacial deformity related syndromes. Besides, application of artificial intelligence or big data database, such as Face2gene [38], might also be useful in guiding clinical diagnosis and treatment.

Three dimension reconstruction images of skull showed difference of syndromic micrognathia among different disorders. Treacher Collins syndrome (Group I) showed bilateral hypoplastic mandible. Hemifacial microsomia (did not included in any group in this study) showed unilateral hypoplastic mandible
References
Singh DJ, Bartlett SP. Congenital mandibular hypoplasia: analysis and classification. J Craniofacial Surg. 2005;16:291.
Gundlach KK, Holtje WJ. The isolated mandibular ramus - a hitherto rarely described anomaly of the mandible. Pathog Treat J Cranio-Maxillo-Facial Surg. 2013;41:450–6.
Benko S, Fantes JA, Amiel J, Kleinjan DJ, Thomas S, Ramsay J, et al. Highly conserved non-coding elements on either side of SOX9 associated with Pierre Robin sequence. Nat Genet. 2009;41:359–64.
Cohen ASA, Yap DB, Suzanne LME, Chieko C, Ramos‐Arroyo MA, Natália T, et al. Weaver syndrome‐associated EZH2 protein variants show impaired histone methyltransferase function in vitro. Hum Mutat. 2016;37:301–7.
Dai J, Si J, Wang M, Huang L, Fang B, Shi J, et al. Tcof1-related molecular networks in Treacher Collins syndrome. J Craniofacial Surg. 2016;27:1420.
McMillin MJ, Beck AE, Chong JX, Shively KM, Buckingham KJ, Gildersleeve HI, et al. Mutations in PIEZO2 cause Gordon syndrome, Marden-Walker syndrome, and distal arthrogryposis type 5. Am J Hum Genet. 2014;94:734–44.
Yang H, Robinson PN, Wang K. Phenolyzer: phenotype-based prioritization of candidate genes for human diseases. Nat Methods. 2015;12:841.
Hamosh A, Scott AF, Amberger JS, Bocchini CA, Mckusick VA. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005;33:514–7.
Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57.
Bicknell LS, Bongers EMHF, Leitch A, Brown S, Schoots J, Harley ME, et al. Mutations in the pre-replication complex cause Meier-Gorlin syndrome. Nat Genet. 2011;43:356–9.
Bacrot S, Doyard M, Huber C, Alibeu O, Feldhahn N, Lehalle D, et al. Mutations in SNRPB, encoding components of the core splicing machinery, cause cerebro-costo-mandibular syndrome. Hum Mutat. 2015;36:187–90.
Hess D, Keusch JJ, Oberstein SA, Hennekam RC, Hofsteenge J. Peters Plus syndrome is a new congenital disorder of glycosylation and involves defective Omicron-glycosylation of thrombospondin type 1 repeats. J Biol Chem. 2008;283:7354–60.
Robertson SP. Otopalatodigital syndrome spectrum disorders: otopalatodigital syndrome types 1 and 2, frontometaphyseal dysplasia and Melnick-Needles syndrome. Eur J Hum Genet Ejhg. 2007;15:3–9.
Schindeler A, Little DG. Ras‐MAPK Signaling in osteogenic differentiation: friend or foe? J Bone Miner Res. 2010;21:1331–8.
Chai Y, Jiang X, Ito Y, Jr BP, Han J, Rowitch DH, et al. Fate of the mammalian cranial neural crest during tooth and mandibular morphogenesis. Development . 2000;127:1671–9.
De BP, Muller P, Wijmenga C, Klomp LW. Molecular pathogenesis of Wilson and Menkes disease: correlation of mutations with molecular defects and disease phenotypes. J Med Genet. 2007;44:673.
Grubenmann CE, Frank CG, Hülsmeier AJ, Schollen E, Matthijs G, Mayatepek E, et al. Deficiency of the first mannosylation step in the N-glycosylation pathway causes congenital disorder of glycosylation type Ik. Hum Mol Genet. 2017;13:535.
Waterham HR, Koster J, Romeijn GJ, Hennekam RC, Vreken P, Andersson HC, et al. Mutations in the 3beta-hydroxysterol Delta24-reductase gene cause desmosterolosis, an autosomal recessive disorder of cholesterol biosynthesis. Am J Hum Genet. 2001;69:685–94.
Giglio MJ, Lama MA. Effect of experimental diabetes on mandible growth in rats. Eur J Oral Sci. 2001;109:193–7.
Wang R, Martinez-Frias ML, Graham JM Jr. Infants of diabetic mothers are at increased risk for the oculo-auriculo-vertebral sequence: a case-based and case-control approach. J Pedia. 2002;141:611–7.
Cancedda R, Descalzi CF, Castagnola P. Chondrocyte differentiation. Int Rev Cytol. 1995;159:265–358.
Yoshida CA, Furuichi T, Fujita T, Fukuyama R, Kanatani N, Kobayashi S, et al. Core-binding factor beta interacts with Runx2 and is required for skeletal development. Nat Genet. 2002;32:633–8.
Pm VDK, Buma P, Van KT, Wb VDB. Interaction of chondrocytes, extracellular matrix and growth factors: relevance for articular cartilage tissue engineering. Osteoarthr Cartil. 2002;10:631–7.
Radlanski RJ, Klarkowski MC. Bone remodeling of the human mandible during prenatal development. J Orofac Orthop. 2001;62:191–201.
Yaeger JA. Principles of bone remodeling; an account of post-natal growth and remodeling processes in long bones and the mandible. J Am Med Assoc. 1963;184:906.
Motyckova G, Fisher DE. Pycnodysostosis: role and regulation of cathepsin K in osteoclast function and human disease. Curr Mol Med. 2002;2:407.
Robinson P, Lipscomb S, Preston LC, Altin E, Watkins H, Ashley CC, et al. Mutations in fast skeletal troponin I, troponin T, and β-tropomyosin that cause distal arthrogryposis all increase contractile function. FASEB J. 2007;21:896–905.
Salinas S, Proukakis C, Crosby A, Warner TT. Hereditary spastic paraplegia: clinical features and pathogenetic mechanisms. Lancet Neurol. 2008;7:1127–38.
Benomran T, Fahiminiya S, Sorfazlian N, Almuriekhi M, Nawaz Z, Nadaf J, et al. Nonsense mutation in the WDR73 gene is associated with Galloway-Mowat syndrome. J Med Genet. 2015;52:381–90.
Rot I, Mardesicbrakus S, Costain WJ, Saragababic M, Kablar B. Role of skeletal muscle in mandible development. Histol Histopathol. 2014;29:1377–94.
Hichijo N, Kawai N, Mori H, Sano R, Ohnuki Y, Okumura S, et al. Effects of the masticatory demand on the rat mandibular development. J Oral Rehabil. 2014;41:581–7.
Tucker AS, Al KA, Ferguson CA, Bach I, Rosenfeld MG, Sharpe PT. Conserved regulation of mesenchymal gene expression by Fgf-8 in face and limb development. Development. 1999;126:221–8.
Szeto DP, Rodriguez-Esteban C, Ryan AK, O’Connell SM, Liu F, Kioussi C, et al. Role of the Bicoid-related homeodomain factor Pitx1 in specifying hindlimb morphogenesis and pituitary development. Genes Dev. 1999;13:484–94.
Chen Q, Zhao Y, Shen G, Dai J. Etiology and Pathogenesis of Hemifacial Microsomia. J Dental Res. 2018:0022034518795609.
Roche N, Houtmeyers P, Janssens S, Blondeel P. Barber-Say syndrome in a father and daughter. Am J Med Genet A. 2010;152a:2563–8.
Gupta VA, Ravenscroft G, Shaheen R, Todd EJ, Swanson LC, Shiina M, et al. Identification of KLHL41 mutations implicates BTB-Kelch-mediated ubiquitination as an alternate pathway to myofibrillar disruption in Nemaline Myopathy. Am J Hum Genet. 2013;93:1108–17.
Inbal A, Avissar N, Shaklai M, Kuritzky A, Schejter A, Ben-David E, et al. Myopathy, lactic acidosis, and sideroblastic anemia: a new syndrome. Am J Med Genet. 1995;55:372–8.
Basel‐Vanagaite L, Wolf L, Orin M, Larizza L, Gervasini C, Krantz ID, et al. Recognition of the Cornelia de Lange syndrome phenotype with facial dysmorphology novel analysis. Clin Genet. 2016;89:557–63.
Acknowledgements
The study is supported by The National Natural Science Foundation of China (No. 81741028), Shanghai Jiaotong University Joint Foundation of Medicine and Engineering (No. YG2016MS08).
Author information
Authors and Affiliations
Contributions
QC, YZ, YQ, CL, GS, and JD contributed to conception and design, drafted and critically revised the manuscript. GS and JD gave final approval and ensured that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Chen, Q., Zhao, Y., Qian, Y. et al. A genetic-phenotypic classification for syndromic micrognathia. J Hum Genet 64, 875–883 (2019). https://doi.org/10.1038/s10038-019-0630-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-019-0630-4
