
Abstract
Variants have been identified in the embryonic ectoderm development (EED) gene in seven patients with syndromic overgrowth similar to that observed in Weaver syndrome. Here, we present three additional patients with missense variants in the EED gene. All the missense variants reported to date (including the three presented here) have localized to one of seven WD40 domains of the EED protein, which are necessary for interaction with enhancer of zeste 2 polycomb repressive complex 2 subunit (EZH2). In addition, among the seven patients reported in the literature and the three new patients presented here, all of the reported pathogenic variants except one occurred at one of four amino acid residues in the EED protein. The recurrence of pathogenic variation at these loci suggests that these residues are functionally important (mutation hotspots). In silico modeling and calculations of the free energy changes resulting from these variants suggested that they not only destabilize the EED protein structure but also adversely affect interactions between EED, EZH2, and/or H3K27me3. These cases help demonstrate the mechanism(s) by which apparently deleterious variants in the EED gene might cause overgrowth and lend further support that amino acid residues in the WD40 domain region may be mutation hotspots.
Similar content being viewed by others

Introduction
Histone tail modifications are epigenetic alterations that mediate packaging and expression of the genome. Among these modifications, tri-methylation of lysine 27 on histone 3 (H3K27me3) is known to be associated with chromatin repression and gene silencing [1], and the highly conserved histone methyltransferase (HMT) polycomb repressive complex 2 (PRC2) is responsible for establishing, spreading, and maintaining this epigenetic mark through a positive feedback loop [2]. Dysregulation of this epigenetic mark can lead to abnormalities of development as well as cancer progression [3, 4]. The PRC2 complex contains four core proteins—embryonic ectoderm development (EED), enhancer of zeste 2 (EZH2), suppressor of zeste 12 (SUZ12), and RB-binding protein 4, chromatin remodeling factor [5, 6]. However, EZH2 is incapable of achieving methyltransferase activity on its own owing to the inherent disorganization of the active site in the absence of other cofactors; therefore, other proteins are required to form the functional PRC2 methyltransferase complex [7, 8]. The EED protein is one of these required subunits. Disruption of specific amino acid residues in EED or EZH2 can inhibit the activity of the PRC2 complex by (1) affecting the enzymatic activity of the complex, or (2) affecting the ability of the complex to bind H3K27me3. Pathogenic de novo variants in EZH2 (OMIM #601573) are associated with Weaver syndrome (OMIM #277590), a rare overgrowth syndrome that is characterized by advanced bone age, a characteristic craniofacial appearance, intellectual disability, and developmental delay.
In the literature, several de novo pathogenic variants in the EED gene (OMIM #605984) have recently been identified in four unrelated males (aged 5, 8, 22, and 27 years at the time of publication), one 16-year-old female, and two individuals of unknown sex, aged 20 and 25 years, with a Weaver-like overgrowth syndrome (OMIM #617561, Cohen–Gibson syndrome) [9,10,11,12,13]. Here, we report de novo and rare missense changes in the WD repeat domains (domains of approximately 40 amino acids that encode tryptophan and aspartic acid residues in a repeating motif) encoded by the EED gene in three additional unrelated patients with syndromic overgrowth. We also provide in silico modeling of all the reported EED pathogenic variants as a step towards understanding the molecular mechanism by which they might cause disease.
Materials and methods
Clinical evaluation, consent, and prior testing
Patients 1 and 2 were evaluated clinically at the Greenwood Genetic Center (Greenwood, SC), and Patient 3 was evaluated at Nationwide Children’s Hospital (Columbus, OH) for the indication of overgrowth. Informed consent was obtained from all individual participants included in the study and additional informed consent was obtained from all individual participants for whom identifying information is included in this article. Patient 1 had previous testing that included microarray, NSD1 (OMIM #606681) sequencing and MLPA, and endocrinology evaluation all with normal results. Informed consent was obtained to perform WES on the proband–mother duo as the father was unavailable. Patient 2 was referred for genetic consultation for evaluation of intellectual disability and atypical physical features including dysmorphic facies, decreased adipose tissue, and overgrowth of her hands and feet. Prior cytogenetic testing in Patient 2 revealed a maternally-inherited copy number variant of 1q25.2 measuring 52 kb, which was classified as a variant of uncertain clinical significance, and a karyotype demonstrated a pericentric inversion of 9p22q21.3, which is a commonly seen polymorphism. Prior testing on patient 3 included karyotype and BAC array, both of which were normal.
Whole-exome sequencing (WES) and analysis
Sequencing and data analysis
DNA libraries were prepared from the proband and parental samples, using either the Agilent SureSelectXT Human All Exon v5 or the Agilent SureSelectXT Clinical Research Exome (CRE) Reagent Kits (Agilent Technologies, Santa Clara, CA) and were sequenced using an Illumina NextSeq 500® Sequencing System (Illumina Inc. San Diego, CA). Sequences were processed using NextGENe® software (SoftGenetics, LLC, State College, PA), mapped to the February 2009 human reference assembly (GRCh37/hg19), and analyzed using the Cartagenia Bench Lab NGS software (Agilent Technologies, Santa Clara, CA).
Sequencing for Patient 3 was performed using the CRE backbone as outlined above, but variants were subsequently filtered to focus on variation in genes associated with overgrowth and/or macrocephaly (specifically, BRWD3(OMIM #300553), EED (OMIM #605984), MED12 (OMIM #300188), PTCH1(OMIM #601309), CDKN1C(OMIM #600856), EZH2 (OMIM #601573), NFIX(OMIM #164005), PTEN(OMIM #601728), CUL4B(OMIM #300304), GLI3(OMIM #165240), NSD1(OMIM #606681), RNF135(OMIM #611358), DNMT3A(OMIM #602769), GPC3(OMIM #300037), PHF6(OMIM #300414), and UPF3B(OMIM #300298)).
Structure preparation for in silico analysis
To analyze the effect of apparently pathogenic EED variants on the functionality of the PRC2/EED–EZH2 complex, we compared two recently deposited Cryo–EM structures of the PRC2 complex bound to cofactors in two active states: compact and extended (PDB: 6C23 and 6C24) [14]. Supplementary Figure 1 shows the structural alignment of two conformational states, where visible differences between the compact and extended conformations can be appreciated. One of the most significant structural differences is the stimulation-responsive motif (SRM) of EZH2, which is disordered in the extended active conformation (PDB: 6C24) but stably bound with EED in the compact active conformation (PDB: 6C23). Among the four mutations sites, three (Arg236, His258, and Arg302) were located at the binding interface of the EED/EZH2–SRM. Here, the binding interface residues are quantitively defined as residues at which the solvent accessible surface area changes upon the binding of EED/EZH2–SRM. Mutations at these sites are expected to affect EED/EZH2–SRM binding. This is the reasoning behind selecting the compact active conformation structure (PDB: 6C23) to analyze each mutation’s effects not only of stability but on the EED-SRM interactions, as well.
Molecular dynamics (MD) simulations
The structure of the PRC2 compact active state (PDB: 6C23) was downloaded from the Protein Data Bank (PDB) for the MD simulation. As the focus of the analysis was on the effects of the mutations on the EED–EZH2 interactions, we did not include the cofactors located far from the mutation sites or other regions that were not relevant for this study. Thus, the final model was comprised of EED, EZH2, and SUZ12 (chain L, C, K, and M). The structure was then subjected to Profix to correct missing heavy atoms and loops [15]. MD simulations were performed by NAMD 2.11 with Charmm36 force field [16]. The parameter files were prepared with VMD psfgen plugin [17]. Proteins were solvated with 0.15 m NaCl in cubic water box with at least 10 Å from the protein to the edge of box and the systems contains total 117,620 atoms. Langevin dynamics with periodic boundary conditions were applied in the simulation. Van der Waals and electrostatic interactions were truncated at 12 Å with a switching function from 10 Å. Particle Mesh Ewald was applied for long-range electrostatic interaction calculations. First, the system underwent a 5000-step minimization with a fixed backbone, and then a subsequent 5000-step minimization without constraint. Then, all atoms in the protein were fixed for 100 ps equilibration of the water and ions. Harmonic constraint of 1 kcal mol/1Å2 was applied to the protein alpha carbon atoms (CA), and the system was then gradually heated from 0 K to 310 K with 1000-step/K in the fixed atom number, volume, and temperature (NVT) simulation. The system was maintained at 310 K for 1 ns equilibration with the CA constrained and another 2 ns equilibration without any constraints in NVT system. Finally, the system was switched to a fixed atom number, pressure, and temperature (NPT) simulation and all constraints were removed for the 100 ns production run and three independent runs were performed.
Evolutionary conservation analysis
Multiple sequence alignments (MSA) among different species were constructed to analyze the evolutionary conservation of the relevant wild type (WT) amino-acid residues. The EED protein sequence from 14 different species was collected from UniProt [18], including Homo sapiens, Mus musculus, Bos taurus, Danio rerio, Gallus gallus, Xenopus tropicalis, Heterocephalus glaber, Macaca mulatta, Callithrix jacchus, Hydra vulgaris, Tarsius syrichta, Salmo salar, Mesocricetus auratus, and Neovison vison. All sequences were then submitted to the T-Coffee server for MSA [19].
Prediction of folding and binding free energy change of mutant proteins
The structure of the EED–EZH2 complex resulting from the MD minimization steps was used for subsequent analyses. Changes in folding and binding free energy were predicted using several publicly available webservers including SAAFEC [20], mCSM [21], SDM [22], DUET [21], I-Mutant [23], PoPMuSiC [24], SAAMBE [25], BeAtMuSiC [26], and Mutabind [27].
Results
Patient 1
Clinical findings
Patient 1, a 10-year-old female at the time of consultation, was born at 39 weeks by Cesarean section because she was large for gestational age (4763 grams). She had an atrial septal defect that closed naturally. She walked at 14 months but was always described as poorly coordinated and clumsy. The Wechsler Intelligence Scale for Children-Fourth Edition revealed a full-scale IQ of 60, with verbal score of 93 and performance component of 55. Accelerated growth continued postnatally; bone age was 10 years at the chronological age of 7 years. She complained of chronic headaches. Brain magnetic resonance imaging (MRI) showed a cyst of the septum pellucidum which was surgically treated with shunting. The corpus callosum was foreshortened. There was partial congenital fusion of the second and third cervical vertebral bodies. MRI of the lumbar spine showed prominence of the epidural fat, causing mild canal stenosis. Physical exam at 10 years of age showed generalized overgrowth and macrocephaly with height 158.4 cm, weight 64.9 kg, and head circumference 59 cm. She wore men’s size 12 shoes (US/Canada sizing). Craniofacial features included macrocephaly with flattened occiput, sparse hair but prominent brows and long eyelashes, widely spaced eyes with an inner canthal distance of 4 cm (97th centile), and exotropia (Fig. 1a) [1, 2]. Her facial profile was flat with a depressed nasal bridge and flattened, broad nasal tip. Philtrum was deeply grooved, and palate was high and arched. Chin was pointed and showed midline dimple and horizontal crease below the mouth. Hands showed fleshy palms, tapered fingers, camptodactyly, and broad thumbs with prominent fingertip pads on the thumbs (Fig. 1a) [3, 4]. The diagnosis of Weaver syndrome was made based on her clinical features.

a Clinical features in Patient 1 included the following: flat facial profile, depressed nasal bridge, preauricular pit (A1), deeply grooved philtrum, pointed chin with horizontal crease, broad nasal tip, myopia, hypertelorism, long eyelashes, right exotropia (A2), tapered fingers, camptodactyly, broad thumbs (A3-4), broad halluces, short fourth and fifth toes, blunted second toes, thin and deep-set nails (A5). b Clinical features in Patient 2 included the following: thickening of the zygomatic arches, prominent jaw, broad nasal tip (B1), scoliosis, decreased adipose and muscle mass in extremities (B2), large fleshy hands, thick fingers (B4), large and wide feet, and dysplastic nails (B3). c Clinical features in Patient 3 included upslanting palpebral fissures, raised nasal bridge, flat facial profile, prognathism, and prominent chin crease (C1–2, 6), broad thumbs and great toes (C3–5), long tapered fingers (C3–4), and dysplastic enamel with large secondary teeth (C6)
WES findings
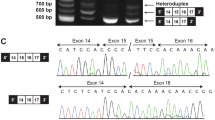
Whole-exome sequencing revealed a de novo or paternal (father not available for testing) missense change in the EED gene, c.773 A > T (p.[His258Leu]; NM_003797.3). This variant was absent from the public variation databases, gnomAD and ExAC, and was not reported in the Human Gene Mutation Database (HGMD). Additionally, this variant was predicted to be deleterious by the PolyPhen-2 (HumVar = 0.990), SIFT (score = 0.0), MutationTaster (probability > 0.999), and PROVEAN (score = −10.522) algorithms. Because the inheritance pattern of this variant could not be determined, it was classified as a variant of uncertain clinical significance. The affected amino acid residue (258) was located in the fourth WD domain of the EED protein. This variant has been submitted to the Leiden Open Variation EED shared database under Individual ID# 00134093.
Patient 2
Clinical findings
Patient 2, a 15-year-old female at the time of consultation, was born at 39 weeks weighing 3,515 grams and measuring 50.8 cm. At birth she was noted to have large hands and feet, as well as congenital hip dislocation. She had surgeries for intestinal malrotation at one month and treatment for Hirschsprung disease at two months of age. Additional procedures included multiple unsuccessful surgeries to correct ptosis, as well as an adenoidectomy and placement of ear tubes. She had an unspecified heart defect that resolved spontaneously. She was noted to have overgrowth of her hands, feet, and thigh bones in addition to bilateral hip dysplasia. She had decreased muscle bulk and adipose tissue on her arms and legs but had increased adipose tissue on the superior trunk (Fig. 1b) [2]. She had coarse facial features including a prominent jaw, broad nasal tip, thickening of zygomatic arches and orbital bones, and large thick ears (Fig. 1b) [1, 2]. Hands were fleshy with thick fingers and dysplastic nails (Fig. 1b) [4]. Patient 2 walked at 10 months of age but was slow in developing speech and language and did not say her first words until 2–3 years of age. Toilet training was accomplished at 4–5 years of age. She required special education classes in school. Psychological testing at age 18 years using the Weschler Adult Intelligence Scale (WAIS), 4th edition, showed a full-scale intellectual quotient score of 44. Her Wide Range Achievement Test (WRAT) revealed that she was functioning at the first to second grade level in both reading and math. She was classified as having moderate intellectual disability. The clinical findings suggested two possible disorders – an unknown lipodystrophy and an unknown overgrowth disorder. Microarray testing of this patient revealed a small 1q25.2 deletion that was also present in her unaffected mother, and chromosomal analysis revealed a pericentric inversion of chromosome 9, a recognized polymorphism.
WES findings
Whole-exome sequencing revealed a de novo missense change in the EED gene, c.581 A > G (p.[Asn194Ser]; NM_003797.3). This variant has been previously published and was present in HGMD (CM176505), but was absent from the public variation databases, gnomAD and ExAC, and was predicted to be deleterious by the PolyPhen-2 (HumVar = 1.0), SIFT (score = 0.0), MutationTaster (probability > 0.99), and PROVEAN (score = −4.872) algorithms. This variant was classified as likely pathogenic. The affected amino acid residue (194) was located in the third WD domain of the EED protein. This variant has been submitted to the Leiden Open Variation EED shared database under Individual ID# 00134094.
Patient 3
Clinical findings
Patient 3, a nine year eleven-month-old male at the time of consultation, was born at 40 weeks to a 28-year-old mother. Pregnancy was complicated by insulin-dependent gestational diabetes. He was born by Cesarean section because he was large for gestational age (5,159.6 grams, 59.7 cm long, 36.8 cm head circumference). He did not have any neonatal complications, though was noted to have dysmorphic craniofacial features, and was discharged from the newborn nursery on day of life number one. At six months of age he was suspected to have an overgrowth syndrome, his dysmorphic features remained present, his skin was noted to be doughy, and he was hypotonic. Early developmental milestones were reportedly achieved on time (rolling over both ways prior to five months, sitting up without support at five months) but thereafter milestones were somewhat delayed, with first words around 13 months and walking independently around 18 months. His developmental delays persisted and later in childhood he was diagnosed with intellectual disability. At nine years of age he was in a special needs classroom with an individualized education plan (IEP), participated in occupational therapy (goals included fastening buttons, tying shoes and other activities of daily living), physical therapy (for gait ataxia and tendency to lean forwards while walking and seated), and speech therapy (for articulation). His medical history included bruxism, snoring, umbilical hernia repair at 9 years of age, poor exercise tolerance, dysplastic enamel of primary teeth, large and crossed secondary teeth, frequent headaches, gynecomastia, right adrenal gland calcifications, and partial cortisol deficiency. Additionally, he had some minor vertebral abnormalities including a fusion of the posterior process of L2-L3, spondylolyses and partial spondylolisthesis. His dysmorphic facial features consisted of architecturally abnormal pinna with dysplastic helices and protruding ears, upslanting palpebral fissures with suggestion of ptosis, raised nasal bridge with flat facial profile and prognathism with deep central crease of the chin (Fig. 1c) [1, 2, 6, 7]. His hands and feet were large, with fragile, soft nails, broad thumbs and great toes, but otherwise long and tapered fingers. In addition, the second fingers are longer than third fingers, bilaterally (Fig. 1c) [3,4,5]. Altogether, the clinical findings were thought to be syndromic in origin, likely related to an overgrowth syndrome.
NGS overgrowth panel findings
Sequencing of the 16 genes comprising the Overgrowth/Macrocephaly NGS panel at Greenwood Genetic Center revealed a de novo or paternal (father not available for testing) missense change in the EED gene, c.772 C > T (p.[His258Tyr]; NM_003797.3). This variant has been previously published and was present in HGMD (CM1612171), but was absent from the public variation databases, gnomAD and ExAC, and was predicted to be deleterious by the PolyPhen-2 (HumVar = 0.994), SIFT (score = 0.0), MutationTaster (probability > 0.999), and PROVEAN (score = −5.767) algorithms. This variant was classified as likely pathogenic. The affected amino acid residue (258) was located in the fourth WD domain of the EED protein. This variant has been submitted to the Leiden Open Variation EED shared database under Individual ID# 00164661.
Prevalence of EED-associated overgrowth in the Greenwood Genetic Center patient cohort
At the time of analysis, 251 probands had been analyzed via WES at the Greenwood Genetic Center making the prevalence of EED-associated overgrowth in this population 2/251 (~0.8%). For the macrocephaly/overgrowth NGS panel, 37 samples had been analyzed when the 3rd patient was reported, of which only 12 were run on an updated panel including the EED gene. Therefore, the prevalence would be 1/12 (8.3%) for this panel. If the two types of analyses are combined, the prevalence becomes 3/263 (~1%). It is important to note, however, that this is a small sample size, each patient was referred for testing due to a suspected genetic condition (i.e. this prevalence is among patients, not the general population), and not every patient was referred for overgrowth symptoms. The prevalence in the cohort of samples ascertained at the Greenwood Genetic Center is likely to change over time as more samples are analyzed.
Evolutionary conservation analysis
MSA among different species revealed that the mutation sites are 100% conserved across the 14 species, indicating a low mutation tolerance of the WT residues (Supplementary Figure 2).
Results of in silico analysis of missense variants from this report and prior reports on structure and function of the PRC2 complex
p.Asn194Ser
The PRC2 consists of four core proteins: EZH2, EED, SUZ12 and RBAP46/RBAP48. EZH2 plays an important role in histone methylation while binding of EED to H3K27me3 allosterically activates the EZH2 protein. The p.Asn194Ser missense variant is located in the H3K27me3 binding pocket of the EED protein (Supplementary Figure 3A). During the simulation, we analyzed the hydrogen bond networks for Asn194. To quantify the effect on stability of the hydrogen bonds, we calculated ratio of hydrogen-bonding time to the total simulation time. As shown in Supplementary Table 1, three stable hydrogen bonds were found and substitution to Ser194 is expected to disturb the local hydrogen bond network, which results in reduced protein stability as indicated by the folding free energy predictions in Supplementary Table 2. In addition, the geometry of the binding pocket is altered by the mutation, potentially affecting the binding of H3K27me3. Thus, the mutation N194S is expected to alter allosteric regulation of the EZH2 protein.
p.Arg236Gly and p.Arg236Thr
Arg236 is located at the binding interface of the SRM in the compact state and is critical to hydrogen-bonding in the minimized structure (Supplementary Figure 3B). In the 10 ns simulation, Arg236 formed three salt bridges with the SRM (Supplementary Table 4). Thus, the WT arginine at position 236 is structurally important and any substitution would be expected to be deleterious. Indeed, the binding free energy calculation predicted that both the R236G and R236T variants largely destabilized SRM-EED interactions, thus affecting the compact state of PRC2 complex.
p.His258Leu and p.His258Tyr
His258 is also located at the SRM binding interface. This residue formed hydrogen bonds with the SRM domain in the minimized structure (Supplementary Figure 3C). However, His258 had fewer interactions with the SRM compared to Arg236 in the MD simulation. Combined with the outcome of the free energy calculations (Supplementary Tables 2 and 3), which indicated that p.His258Leu and p.His258Tyr have minor effects on both stability and binding, these results suggest that missense mutations at residue 258 may affect SRM-EED interactions but are likely to be milder than those associated with missense variation at residue 236.
p.Arg302Gly and p.Arg302Tyr
Arg302 is also located at the binding interface of the EED/EZH2–SRM. In the minimized structure, Arg302 forms hydrogen bonds with the SRM of EZH2 (Supplementary Figure 3D). In addition, Glu125 of the SRM is close to the Arg302 residue in the minimized structure, indicating the possibility of salt bridge formation. This is further supported by MD simulation as shown in Supplementary Table 4. Thus, in combination with the binding free energy predictions (Supplementary Table 3), these results suggest that p.Arg302Gly and p.Arg302Tyr affects SRM-EED binding.
Discussion
In vitro studies demonstrate that when EED is bound by an antibody specific to the EZH2 binding site, the methyltransferase activity of the PRC2 complex is inhibited in a dose-dependent manner [28]. EED contains seven WD40 repeat domains [29] that form a seven-bladed β propeller, and disruption of EED WD domains one through five, specifically, by single nucleotide variants results in blocking the interaction of EED and EZH2 [30]. EED has a C-terminal domain that specifically binds H3K27me3 residues thereby targeting the PRC2 complex to chromatin domains and allowing for further methylation of these regions [31, 32]. Additionally, mutations in this region abolish methyltransferase activity of the complex [32]. Recent structural studies have shown that the beta propeller structure of the EED WD40 domains interacts directly with the stimulatory response motif (SRM) of EZH2. This interaction, plus the binding of EED to H3K27me3, leads to the restructuring and stabilization of the EZH2 SET domain and allowing for the HMT activity of the holoenzyme [7, 33]. De novo pathogenic alterations in the EED gene, located on chromosome 11q14.2, have recently been reported in seven unrelated patients with phenotypic features similar to Weaver syndrome. The seven reported patients all had overgrowth phenotypes including varying levels of intellectual disability and developmental delay [9,10,11,12,13, 34].
All the variants reported here were absent from the public SNP databases and were predicted to be damaging by the PolyPhen-2, SIFT, MutationTaster, and PROVEAN in silico algorithms. Patient 1 had a missense change (p.[His258Leu]) that altered the amino acid at the same position as that of Patient 3 (p.[His258Tyr]) and as that of the patient reported by Cohen et al. [10] (p.[His258Tyr]), but the resulting amino acid change is distinct. Likewise, the variant described by Cooney et al. [11] (p.[Arg302Gly]), occurred at the same amino acid position as that reported in the second patient reported by Cohen, et al. [15] (p.[Arg302Ser]), and the missense variant reported by Imagawa et al. [12] (p.[Arg236Thr]), affected the same amino acid residue as the missense variant reported in the second patient reported by Tatton-Brown et al. [34] (p.[Arg236Gly]). Lastly, Patient 2 had the same missense change (p.Asn194Ser) as the first patient reported by Tatton-Brown et al. [34] (p.[Asn194Ser]). The only reported pathogenic variant outside these four amino acid residues is the c.917_919delinsCGG (p.[Arg306_Asn307delinsThrAsp]) indel reported in a single patient by Smigiel et al. [13]. This variant is located in the WD5 domain in the EED protein; the variants affecting amino acid residue 194 are located in the WD3 domain, the variants affecting residues 236 and 258 are located in the WD4 domain, and the variants affecting amino acid 302 are located proximal to the WD4 domain. Both exonic deletions as well as single nucleotide variants in the WD domain region have been shown to affect EED–EZH2 interactions as well as PRC2 function [30, 35]. Indeed, in mouse, the WD domain and histone binding regions, alone, have been shown to be sufficient for H3K27 methyltransferase activity in vitro [29]. In vitro studies using mutagenesis of mouse Eed and a yeast two-hybrid system demonstrated that missense changes at the amino acids flanking the 194 position (that correspond to amino acids 193 and 196 in the human EED protein) abolish binding of EED to EZH2 [36]. In combination with the observation that several of the patients share pathogenic variants at the same amino acid residues, the fact that all these EED variants alter amino acids in or near WD40 domains 3, 4, and 5 in the EED protein suggests that these domains are potential hotspots for mutation (that is, functionally important amino residues at which disease-causing variation is recurrent).
Dysmorphic features in common among the majority of the seven previously reported patients and the three new patients (i.e. features in 6 of the 10 patients) reported here include overgrowth, macrocephaly, large hands and feet, large/long ears, advanced bone age, prominent chin or chin crease, retrognathia, and hypertelorism (Table 1). Other common findings included developmental delay, intellectual disability, congenital heart abnormalities, and hernias (especially of the umbilicus). Unique findings in the three patients reported here included upslanting palpebral fissures, hip dysplasia, high arched palate, capillary hemangioma, thin/fragile skin, Hirschsprung disease, bruxism, lipodystrophy, Erlenmeyer flask deformity, septum pellucidum cyst, and hypoglycemia. It is possible these additional clinical features result from a separate diagnosis for which the causative variant or variants have yet to be identified, but expansion of the EED-associated overgrowth phenotype is likely.
In silico analysis of the apparently pathogenic missense variants suggests that, overall, p.Asn194Ser, p.Arg236Gly, p.Arg236Tyr, p.Arg302Gly, and p.Arg302Ser are likely to be the most deleterious variants. p.Asn194Ser is predicted to destabilize the H3K27me3 binding site and p.Arg236Gly, p.Arg236Tyr, p.Arg302Gly, and p.Arg302Ser are predicted to interfere with EED/EZH2–SRM binding. The p.His258Leu and p.His258Tyr variants are predicted to have an adverse effect on EZH2-EED interactions, but this effect is likely to be milder than those predicted for the other variants. It is unclear whether these predictions represent a genotype-phenotype correlation. Lee et al. [33], recently resolved the interactions between EED and EZH2 at the amino acid residue level and our results appear to recapitulate those findings for the variants in common between the two studies. Specifically, Lee et al. found that residues His258, Ser259, and Arg302, are residues important to anchoring EED to the SRM domain of EZH2, and residue Tyr365 is a residue located in the aromatic cage that specifically binds H3K27me3. In vitro functional studies of EED mutant proteins, p.His258Tyr, p.Ser259Phe, p.Arg302Ser, p.Arg302Gly, and p.Tyr365Ala, led to reduced and/or abolished activation of the PRC2 complex MHT activity with p.Arg302Gly showing the most dramatic deleterious effect, similar to the in silico findings reported here [33]. Therefore, the amino acid residues critical to the interaction of the EZH2–SRM domain and EED, as well as the EED amino acid residues critical to binding the H3K27me3 peptide on the nucleosome, likely represent mutational hotspots. We could speculate that other residues involved in the EED–EZH2/SRM domain interaction could also be mutation hotspots (EZH2 residues H129, D136, H158, and R161; EED residue Y308) (see Supplementary Figure 7). Interestingly, pathogenic changes at these residues have yet to be reported except for p.Asp136His in EZH2 (ClinVar) and the maximum allele frequency of presumably benign changes at these residues as reported by gnomAD are the following: EZH2 p.His129Tyr – 0.019 MAF and EZH2 p.His158His – 0.045%MAF. Sequence changes of any kind at any of these residues are rare. This is illustrated by the ExAC Z-scores of 2.69 and 5.45 for EED and EZH2, respectively (indicating moderate and high constraint for missense changes), and pLI scores of 1.0 for both genes (indicating little or no tolerance for loss-of-function changes).
In summary, we have identified three additional patients with syndromic overgrowth and apparently deleterious variants in the EED gene. The addition of these cases to the current literature not only expands the phenotypic features of EED-associated overgrowth, but also suggests mutation hotspots and the mechanism by which each might cause disruption of the PRC2 complex function.
The Greenwood Genetic Center receives revenue from diagnostic testing performed in the GGC Molecular Diagnostic Laboratory.
Web Resources
Online Mendelian Inheritance of Man: www.OMIM.org. Leiden Open Variation Database: www.LOVD.nl. Protein Data Bank (PDB): www.rcsb.org. PolyPhen-2: http://genetics.bwh.harvard.edu/pph2/. Mutation Taster: http://www.mutationtaster.org/. Sorting Intolerant From Tolerant (SIFT): http://sift.bii.a-star.edu.sg/. PROVEAN PROTEIN: http://provean.jcvi.org/seq_submit.php. Human Genetics Mutation Database (HGMD): https://portal.biobase-international.com/hgmd/pro/start.php. Exome Aggregation Consortium (ExAC): http://exac.broadinstitute.org/. genome Aggregation Database (gnomAD): http://gnomad.broadinstitute.org/. UniProt: http://www.uniprot.org. SAFEEC Web server: http://compbio.clemson.edu/SAAFEC/. mSCM: http://biosig.unimelb.edu.au/mcsm_na/. SDM: http://marid.bioc.cam.ac.uk/sdm2. DUET: http://biosig.unimelb.edu.au/duet/help. I-Mutant: http://folding.biofold.org/i-mutant/i-mutant2.0.html. PoPMuSiC: http://babylone.ulb.ac.be/popmusic. SAAMBE: http://compbio.clemson.edu/saambe_webserver/. BeAtMuSiC: http://babylone.ulb.ac.be/beatmusic/. MutaBind: https://www.ncbi.nlm.nih.gov/research/mutabind/index.fcgi/. T-Coffee MSA: http://tcoffee.crg.cat/. NAMD: http://www.ks.uiuc.edu/Research/namd/2.11/ug/node5.html. VMD psfgen plugin: http://www.ks.uiuc.edu/Research/vmd/plugins/psfgen/
Accession Numbers
The accession number for the c.773 A > T (p.[His258Leu]) sequence variant reported in this paper is LOVD: ID# 00134093. The accession number for the c.581 A > G (p.[Asn194Ser]) sequence variant reported in this paper is LOVD: ID# 00134094. The accession number for the c.772 C > T (p.[His258Tyr]) sequence variant reported in this paper is LOVD: ID# 00164661
References
O’Meara MM, Simon JA. Inner workings and regulatory inputs that control Polycomb repressive complex 2. Chromosoma. 2012;121:221–34.
Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–43.
Bracken AP, Helin K. Polycomb group proteins: navigators of lineage pathways led astray in cancer. Nat Rev Cancer. 2009;9:773–84.
Bachmann IM, Halvorsen OJ, Collett K, Stefansson IM, Straume O, Haukaas SA, et al. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J Clin Oncol. 2006;24:268–73.
Pasini D, Bracken AP, Jensen MR, Lazzerini Denchi E, Helin K. Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J. 2004;23:4061–71.
Ciferri C, Lander GC, Maiolica A, Herzog F, Aebersold R, Nogales E. Molecular architecture of human polycomb repressive complex 2. eLife. 2012;1:e00005.
Jiao L, Liu X. Structural basis of histone H3K27 trimethylation by an active polycomb repressive complex 2. Science. 2015;350:aac4383.
Justin N, Zhang Y, Tarricone C, Martin SR, Chen S, Underwood E, et al. Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat Commun. 2016;7:11316.
Cohen ASA, Tuysuz B, Shen Y, Bhalla SK, Jones SJM, Gibson WT. A novel mutation in EED associated with overgrowth. J Hum Genet. 2015;60:339–42.
Cohen AS. Gibson WT EED-associated overgrowth in a second male patient. J Hum Genet. 2016;61:831–4.
Cooney E, Bi W, Schlesinger AE, Vinson S, Potocki L, Novel EED. Mutation in patient with Weaver syndrome. Am J Med Genet A. 2017;173:541–5.
Imagawa E, Higashimoto K, Sakai Y, Numakura C, Okamoto N, Matsunaga S, et al. Mutations in genes encoding polycomb repressive complex 2 subunits cause Weaver syndrome. Hum Mutat. 2017;38:637–48.
Smigiel R, Biernacka A, Biela M, Murcia-Pienkowski V, Szmida E, Gasperowicz P, et al. Novel de novo mutation affecting two adjacent aminoacids in the EED gene in a patient with Weaver syndrome. J Hum Genet. 2018;63:517–20.
Kasinath V, Faini M, Poepsel S, Reif D, Feng XA, Stjepanovic G, et al. Structures of human PRC2 with its cofactors AEBP2 and JARID2. Science. 2018;359:940–4.
Xiang J, Honig, B. Jackal: A protein structure modeling package. New York: Columbia University and Howard Hughes Medical Institute; 2002.
Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–802.
Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–8.
UniProt Consortium.UniProt: a hub for protein information. Nucleic Acids Res. 2015;43:D204–12.
Magis C, Taly JF, Bussotti G, Chang JM, Di PT, Erb I, et al. T-Coffee: tree-based consistency objective function for alignment evaluation. Methods Mol Biol. 2014;1079:117–29.
Getov I, Petukh M, Alexov E. SAAFEC: predicting the effect of single point mutations on protein folding free energy using a knowledge-modified MM/PBSA approach. Int J Mol Sci. 2016;17:512.
Pires DEV, Ascher DB, Blundell TL. mCSM: predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics. 2014;30:335–42.
Worth CL, Preissner R, Blundell TL. SDM--a server for predicting effects of mutations on protein stability and malfunction. Nucleic Acids Res. 2011;39:W215–22.
Capriotti E, Fariselli P, Casadio R. I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005;33:W306–10.
Dehouck Y, Kwasigroch JM, Gilis D, Rooman M. PoPMuSiC 2.1: a web server for the estimation of protein stability changes upon mutation and sequence optimality. BMC Bioinformatics. 2011;12:151.
Petukh M, Dai L, Alexov E. SAAMBE: webserver to predict the charge of binding free energy caused by amino acids mutations. Int J Mol Sci. 2016;17:547.
Dehouck Y, Kwasigroch JM, Rooman M, Gilis D. BeAtMuSiC: prediction of changes in protein-protein binding affinity on mutations. Nucleic Acids Res. 2013;41:W333–9.
Li M, Simonetti FL, Goncearenco A, Panchenko AR. MutaBind estimates and interprets the effects of sequence variants on protein-protein interactions. Nucleic Acids Res. 2016;44):W494–501.
Cao Q, Wang X, Zhao M, Yang R, Malik R, Qiao Y, et al. The central role of EED in the orchestration of polycomb group complexes. Nat Commun. 2014;5:3127.
Montgomery ND, Yee D, Montgomery SA, Magnuson T. Molecular and functional mapping of EED motifs required for PRC2-dependent histone methylation. J Mol Biol. 2007;374:1145–57.
Denisenko O, Shnyreva M, Suzuki H, Bomsztyk K. Point mutations in the WD40 domain of Eed block its interaction with Ezh2. Mol Cell Biol. 1998;18:5634–42.
Xu C, Bian C, Yang W, Galka M, Ouyang H, Chen C, et al. Binding of different histone marks differentially regulates the activity and specificity of polycomb repressive complex 2 (PRC2). Proc Natl Acad Sci USA. 2010;107:19266–71.
Margueron R, Justin N, Ohno K, Sharpe ML, Son J, Drury WJ, et al. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature. 2009;461:762–7.
Lee C-H, Yu J-R, Kumar S, Jin Y, LeRoy G, Bhanu N, et al. Allosteric activation dictates PRC2 activity independent of its recruitment tochromatin. Mol Cell. 2018;70:422–34.e6.
Tatton-Brown K, Loveday C, Yost S, Clarke M, Ramsay E, Zachariou A, et al. Mutations in epigenetic regulation genes are a major cause of overgrowth with intellectual disability. Am J Hum Genet. 2017;100:725–36.
Ueda T, Sanada M, Matsui H, Yamasaki N, Honda Z-I, Shih L-Y, et al. EED mutants impair polycomb repressive complex 2 in myelodysplastic syndrome and related neoplasms. Leukemia . 2012;26:2557–60.
Sewalt RG, van der Vlag J, Gunster MJ, Hamer KM, den Blaauwen JL, Satijn DP, et al. Characterization of interactions between the mammalian polycomb-group proteins Enx1/EZH2 and EED suggests the existence of different mammalian polycomb-group protein complexes. Mol Cell Biol. 1998;18:3586–95.
Acknowledgements
We thank the patients and their families for their participation in this study. We also recognize the contributions of our laboratory staff and the clinical team at the Greenwood Genetic Center for their considerable efforts.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The Greenwood Genetic Center receives revenue from diagnostic testing performed in the GGC Molecular Diagnostic Laboratory.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Spellicy, C.J., Peng, Y., Olewiler, L. et al. Three additional patients with EED-associated overgrowth: potential mutation hotspots identified?. J Hum Genet 64, 561–572 (2019). https://doi.org/10.1038/s10038-019-0585-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-019-0585-5

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}